Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous?


Abstract

1. Hypoxia and the Hypoxia-Inducible Factor (HIF) Pathway
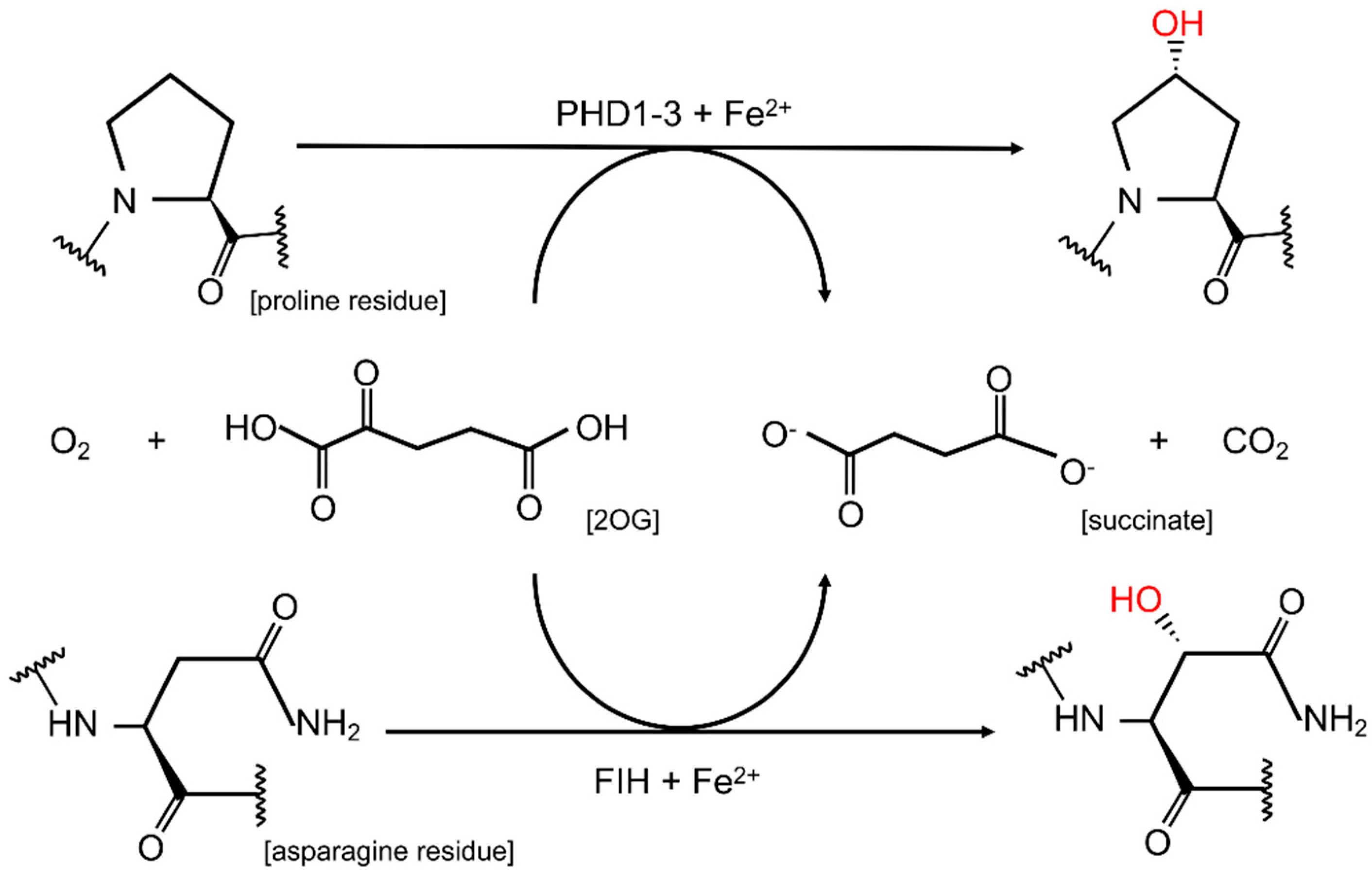
2. HIF Hydroxylation
3. Other Prolyl Hydroxylases
4. Methods Used to Detect Protein Hydroxylation
4.1. Enzymatic or Kinetic Assays
4.2. In Silico Screening
4.3. Mass Spectrometry
4.4. Von Hippel-Lindau Tumor Suppressor (VHL) Capture Assay
4.5. Immunoprecipitation and Other Antibody-Based Techniques
5. Evidence for HIF Hydroxylation
6. Evidence for Non-HIF Hydroxylation Targets
6.1. NF-κB
6.2. p53
6.3. FOXO3a
6.4. MAPK6
6.5. Cep192
6.6. ZHX2
7. Hydroxylation of Non-HIF Targets Mediated by Factor-Inhibiting HIF (FIH)
7.1. OTUB1
7.2. p105 and IκBα
7.3. RIPK4
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef]
- Semenza, G.L. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood 2009, 114, 2015–2019. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Keogh, C.E.; Crean, D.; Taylor, C.T. The role of HIF in immunity and inflammation. Mol. Aspects Med. 2016, 47–48, 24–34. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Kaidi, A.; Williams, A.C.; Paraskeva, C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 2007, 9, 210–217. [Google Scholar] [CrossRef]
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Colgan, S.P. Hypoxia and gastrointestinal disease. J. Mol. Med. Berl. Ger. 2007, 85, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Shibasaki, F. Hypoxia-inducible factor as an angiogenic master switch. Front. Pediatr. 2015, 3, 33. [Google Scholar] [CrossRef]
- Zurlo, G.; Guo, J.; Takada, M.; Wei, W.; Zhang, Q. New Insights into Protein Hydroxylation and Its Important Role in Human Diseases. Biochim. Biophys. Acta 2016, 1866, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.R.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. elegans EGL-9 and Mammalian Homologs Define a Family of Dioxygenases that Regulate HIF by Prolyl Hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Mole, D.R.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Regulation of HIF by the von Hippel-Lindau tumour suppressor: Implications for cellular oxygen sensing. IUBMB Life 2001, 52, 43–47. [Google Scholar]
- Yeh, T.-L.; Leissing, T.M.; Abboud, M.I.; Thinnes, C.C.; Atasoylu, O.; Holt-Martyn, J.P.; Zhang, D.; Tumber, A.; Lippl, K.; Lohans, C.T.; et al. Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem. Sci. 2017, 8, 7651–7668. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar]
- Wong, B.W.; Kuchnio, A.; Bruning, U.; Carmeliet, P. Emerging novel functions of the oxygen-sensing prolyl hydroxylase domain enzymes. Trends Biochem. Sci. 2013, 38, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, V.; Parinandi, N.L.; Adluri, R.S.; Goldman, J.W.; Hussain, N.; Sanchez, J.A.; Maulik, N. Molecular Mechanisms of Action and Therapeutic Uses of Pharmacological Inhibitors of HIF–Prolyl 4-Hydroxylases for Treatment of Ischemic Diseases. Antioxid. Redox Signal. 2014, 20, 2631–2665. [Google Scholar] [CrossRef]
- Lieb, M.E.; Menzies, K.; Moschella, M.C.; Ni, R.; Taubman, M.B. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem. Cell Biol. Biochim. Biol. Cell. 2002, 80, 421–426. [Google Scholar] [CrossRef]
- Metzen, E.; Berchner-Pfannschmidt, U.; Stengel, P.; Marxsen, J.H.; Stolze, I.; Klinger, M.; Huang, W.Q.; Wotzlaw, C.; Hellwig-Bürgel, T.; Jelkmann, W.; et al. Intracellular localisation of human HIF-1 alpha hydroxylases: Implications for oxygen sensing. J. Cell Sci. 2003, 116, 1319–1326. [Google Scholar] [CrossRef]
- Tambuwala, M.M.; Cummins, E.P.; Lenihan, C.R.; Kiss, J.; Stauch, M.; Scholz, C.C.; Fraisl, P.; Lasitschka, F.; Mollenhauer, M.; Saunders, S.P.; et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology 2010, 139, 2093–2101. [Google Scholar] [CrossRef]
- Strowitzki, M.J.; Kirchberg, J.; Tuffs, C.; Schiedeck, M.; Ritter, A.S.; Biller, M.; Harnoss, J.M.; Lasitschka, F.; Schmidt, T.; Radhakrishnan, P.; et al. Loss of Prolyl-Hydroxylase 1 Protects against Biliary Fibrosis via Attenuated Activation of Hepatic Stellate Cells. Am. J. Pathol. 2018, 188, 2826–2838. [Google Scholar] [CrossRef]
- Schneider, M.; Van Geyte, K.; Fraisl, P.; Kiss, J.; Aragonés, J.; Mazzone, M.; Mairbäurl, H.; De Bock, K.; Jeoung, N.H.; Mollenhauer, M.; et al. Loss or Silencing of the PHD1 Prolyl Hydroxylase Protects Livers of Mice Against Ischemia/Reperfusion Injury. Gastroenterology 2010, 138, 1143–1154.e2. [Google Scholar] [CrossRef]
- Kennel, K.B.; Burmeister, J.; Schneider, M.; Taylor, C.T. The PHD1 oxygen sensor in health and disease. J. Physiol. 2018, 596, 3899–3913. [Google Scholar] [CrossRef]
- Fitzpatrick, S.F.; Fábián, Z.; Schaible, B.; Lenihan, C.R.; Schwarzl, T.; Rodriguez, J.; Zheng, X.; Li, Z.; Tambuwala, M.M.; Higgins, D.G.; et al. Prolyl hydroxylase-1 regulates hepatocyte apoptosis in an NF-κB-dependent manner. Biochem. Biophys. Res. Commun. 2016, 474, 579–586. [Google Scholar] [CrossRef]
- Chen, N.; Qian, J.; Chen, J.; Yu, X.; Mei, C.; Hao, C.; Jiang, G.; Lin, H.; Zhang, X.; Zuo, L.; et al. Phase 2 studies of oral hypoxia-inducible factor prolyl hydroxylase inhibitor FG-4592 for treatment of anemia in China. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. - Eur. Ren. Assoc. 2017, 32, 1373–1386. [Google Scholar] [CrossRef]
- Fraisl, P.; Aragonés, J.; Carmeliet, P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat. Rev. Drug Discov. 2009, 8, 139–152. [Google Scholar]
- Dao, J.H.; Kurzeja, R.J.M.; Morachis, J.M.; Veith, H.; Lewis, J.; Yu, V.; Tegley, C.M.; Tagari, P. Kinetic characterization and identification of a novel inhibitor of hypoxia-inducible factor prolyl hydroxylase 2 using a time-resolved fluorescence resonance energy transfer-based assay technology. Anal. Biochem. 2009, 384, 213–223. [Google Scholar] [CrossRef]
- Haase, V.H. HIF-prolyl hydroxylases as therapeutic targets in erythropoiesis and iron metabolism. Hemodial. Int. Int. Symp. Home Hemodial. 2017, 21, S110–S124. [Google Scholar] [CrossRef]
- Van Welden, S.; Selfridge, A.C.; Hindryckx, P. Intestinal hypoxia and hypoxia-induced signalling as therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 596–611. [Google Scholar] [CrossRef]
- Arsenault, P.R.; Heaton-Johnson, K.J.; Li, L.-S.; Song, D.; Ferreira, V.S.; Patel, N.; Master, S.R.; Lee, F.S. Identification of prolyl hydroxylation modifications in mammalian cell proteins. Proteomics 2015, 15, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Vranka, J.A.; Sakai, L.Y.; Bächinger, H.P. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 2004, 279, 23615–23621. [Google Scholar] [CrossRef] [PubMed]
- Hudson, D.M.; Eyre, D.R. Collagen prolyl 3-hydroxylation: A major role for a minor post-translational modification? Connect. Tissue Res. 2013, 54, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Pihlajaniemi, T.; Myllylä, R.; Kivirikko, K.I. Prolyl 4-hydroxylase and its role in collagen synthesis. J. Hepatol. 1991, 13, S2–S7. [Google Scholar] [CrossRef]
- Bickel, M.; Baringhaus, K.H.; Gerl, M.; Günzler, V.; Kanta, J.; Schmidts, L.; Stapf, M.; Tschank, G.; Weidmann, K.; Werner, U. Selective inhibition of hepatic collagen accumulation in experimental liver fibrosis in rats by a new prolyl 4-hydroxylase inhibitor. Hepatol. Baltim. Md. 1998, 28, 404–411. [Google Scholar] [CrossRef]
- Nietfeld, J.J.; Kemp, A. Properties of prolyl 4-hydroxylase containing firmly-bound iron. Biochim. Biophys. Acta 1980, 613, 349–358. [Google Scholar] [CrossRef]
- Kivirikko, K.I.; Pihlajaniemi, T. Collagen hydroxylases and the protein disulfide isomerase subunit of prolyl 4-hydroxylases. Adv. Enzymol. Relat. Areas Mol. Biol. 1998, 72, 325–398. [Google Scholar]
- Hirsilä, M.; Koivunen, P.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef]
- Hirsilä, M.; Koivunen, P.; Xu, L.; Seeley, T.; Kivirikko, K.I.; Myllyharju, J. Effect of desferrioxamine and metals on the hydroxylases in the oxygen sensing pathway. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1308–1310. [Google Scholar] [CrossRef]
- Manresa, M.C.; Tambuwala, M.M.; Radhakrishnan, P.; Harnoss, J.M.; Brown, E.; Cavadas, M.A.; Keogh, C.E.; Cheong, A.; Barrett, K.E.; Cummins, E.P.; et al. Hydroxylases regulate intestinal fibrosis through the suppression of ERK-mediated TGF-β1 signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G1076–G1090. [Google Scholar] [CrossRef]
- Matsumura, Y.; Sakaida, I.; Uchida, K.; Kimura, T.; Ishihara, T.; Okita, K. Prolyl 4-hydroxylase inhibitor (HOE 077) inhibits pig serum-induced rat liver fibrosis by preventing stellate cell activation. J. Hepatol. 1997, 27, 185–192. [Google Scholar] [CrossRef]
- Vasta, J.D.; Andersen, K.A.; Deck, K.M.; Nizzi, C.P.; Eisenstein, R.S.; Raines, R.T. Selective Inhibition of Collagen Prolyl 4-Hydroxylase in Human Cells. ACS Chem. Biol. 2016, 11, 193–199. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, K.S.; McNeill, L.A.; Riordan, M.V.; Tian, Y.-M.; Bullock, A.N.; Welford, R.W.; Elkins, J.M.; Oldham, N.J.; Bhattacharya, S.; Gleadle, J.M.; et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol. Chem. 2002, 277, 26351–26355. [Google Scholar] [CrossRef]
- Zheng, X.; Zhai, B.; Koivunen, P.; Shin, S.J.; Lu, G.; Liu, J.; Geisen, C.; Chakraborty, A.A.; Moslehi, J.J.; Smalley, D.M.; et al. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev. 2014, 28, 1429–1444. [Google Scholar] [CrossRef]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NF-κB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef]
- Ullah, K.; Rosendahl, A.-H.; Izzi, V.; Bergmann, U.; Pihlajaniemi, T.; Mäki, J.M.; Myllyharju, J. Hypoxia-inducible factor prolyl-4-hydroxylase-1 is a convergent point in the reciprocal negative regulation of NF-κB and p53 signaling pathways. Sci. Rep. 2017, 7, 17220. [Google Scholar] [CrossRef]
- Deschoemaeker, S.; Di Conza, G.; Lilla, S.; Martín-Pérez, R.; Mennerich, D.; Boon, L.; Hendrikx, S.; Maddocks, O.D.K.; Marx, C.; Radhakrishnan, P.; et al. PHD1 regulates p53-mediated colorectal cancer chemoresistance. EMBO Mol. Med. 2015, 7, 1350–1365. [Google Scholar] [CrossRef]
- Rodriguez, J.; Herrero, A.; Li, S.; Rauch, N.; Quintanilla, A.; Wynne, K.; Krstic, A.; Acosta, J.C.; Taylor, C.; Schlisio, S.; et al. PHD3 Regulates p53 Protein Stability by Hydroxylating Proline 359. Cell Rep. 2018, 24, 1316–1329. [Google Scholar] [CrossRef]
- Rodriguez, J.; Pilkington, R.; Garcia Munoz, A.; Nguyen, L.K.; Rauch, N.; Kennedy, S.; Monsefi, N.; Herrero, A.; Taylor, C.T.; von Kriegsheim, A. Substrate-Trapped Interactors of PHD3 and FIH Cluster in Distinct Signaling Pathways. Cell Rep. 2016, 14, 2745–2760. [Google Scholar] [CrossRef] [PubMed]
- Moser, S.C.; Bensaddek, D.; Ortmann, B.; Maure, J.-F.; Mudie, S.; Blow, J.J.; Lamond, A.I.; Swedlow, J.R.; Rocha, S. PHD1 links cell-cycle progression to oxygen sensing through hydroxylation of the centrosomal protein Cep192. Dev. Cell 2013, 26, 381–392. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, T.; Simon, J.; Takada, M.; Saito, R.; Fan, C.; Liu, X.-D.; Jonasch, E.; Xie, L.; Chen, X.; et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 2018, 361, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Rodriguez, J.; Pickel, C.; Burr, S.; Fabrizio, J.-A.; Nolan, K.A.; Spielmann, P.; Cavadas, M.A.S.; Crifo, B.; Halligan, D.N.; et al. FIH Regulates Cellular Metabolism through Hydroxylation of the Deubiquitinase OTUB1. PLoS Biol. 2016, 14, e1002347. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Lancaster, D.E.; Stolze, I.P.; Hewitson, K.S.; McDonough, M.A.; Coleman, M.L.; Coles, C.H.; Yu, X.; Hay, R.T.; Ley, S.C.; et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc. Natl. Acad. Sci. USA 2006, 103, 14767–14772. [Google Scholar] [CrossRef]
- Gorres, K.L.; Raines, R.T. Direct and continuous assay for prolyl 4-hydroxylase. Anal. Biochem. 2009, 386, 181–185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rhoads, R.E.; Udenfriend, S. Decarboxylation of alpha-ketoglutarate coupled to collagen proline hydroxylase. Proc. Natl. Acad. Sci. USA 1968, 60, 1473–1478. [Google Scholar] [CrossRef]
- Zhang, J.H.; Qi, R.C.; Chen, T.; Chung, T.D.; Stern, A.M.; Hollis, G.F.; Copeland, R.A.; Oldenburg, K.R. Development of a carbon dioxide-capture assay in microtiter plate for aspartyl-beta-hydroxylase. Anal. Biochem. 1999, 271, 137–142. [Google Scholar] [CrossRef] [PubMed]
- McNeill, L.A.; Bethge, L.; Hewitson, K.S.; Schofield, C.J. A fluorescence-based assay for 2-oxoglutarate-dependent oxygenases. Anal. Biochem. 2005, 336, 125–131. [Google Scholar] [CrossRef]
- Gorres, K.L.; Edupuganti, R.; Krow, G.R.; Raines, R.T. Conformational preferences of substrates for human prolyl 4-hydroxylase. Biochemistry 2008, 47, 9447–9455. [Google Scholar] [CrossRef][Green Version]
- Juva, K.; Prockop, D.J. Modified procedure for the assay of H-3-or C-14-labeled hydroxyproline. Anal. Biochem. 1966, 15, 77–83. [Google Scholar] [CrossRef]
- Koivunen, P.; Tiainen, P.; Hyvärinen, J.; Williams, K.E.; Sormunen, R.; Klaus, S.J.; Kivirikko, K.I.; Myllyharju, J. An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J. Biol. Chem. 2007, 282, 30544–30552. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, Q.; Mooney, S.M.; Lee, F.S. Sequence determinants in hypoxia-inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J. Biol. Chem. 2002, 277, 39792–39800. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Chicooree, N.; Unwin, R.D.; Griffiths, J.R. The application of targeted mass spectrometry-based strategies to the detection and localization of post-translational modifications. Mass Spectrom. Rev. 2015, 34, 595–626. [Google Scholar]
- Cockman, M.E.; Webb, J.D.; Kramer, H.B.; Kessler, B.M.; Ratcliffe, P.J. Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol. Cell. Proteomics MCP 2009, 8, 535–546. [Google Scholar] [CrossRef]
- Tian, Y.-M.; Yeoh, K.K.; Lee, M.K.; Eriksson, T.; Kessler, B.M.; Kramer, H.B.; Edelmann, M.J.; Willam, C.; Pugh, C.W.; Schofield, C.J.; et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J. Biol. Chem. 2011, 286, 13041–13051. [Google Scholar] [CrossRef]
- Lee, S.-H.; Moon, J.H.; Cho, E.A.; Ryu, S.E.; Lee, M.K. Monoclonal antibody-based screening assay for factor inhibiting hypoxia-inducible factor inhibitors. J. Biomol. Screen. 2008, 13, 494–503. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 and human disease: One highly involved factor. Genes Dev. 2000, 14, 1983–1991. [Google Scholar]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Bergeron, D.L.; Runge, A.; Adelman, D.M.; Gohil, M.; Simon, M.C. HIF-dependent hematopoietic factors regulate the development of the embryonic vasculature. Dev. Cell 2006, 11, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Maltepe, E.; Schmidt, J.V.; Baunoch, D.; Bradfield, C.A.; Simon, M.C. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature 1997, 386, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008, 9, 285–296. [Google Scholar] [CrossRef]
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Safran, M.; Kaelin, W.G. HIF hydroxylation and the mammalian oxygen-sensing pathway. J. Clin. Invest. 2003, 111, 779–783. [Google Scholar] [CrossRef]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Chan, D.A.; Sutphin, P.D.; Yen, S.-E.; Giaccia, A.J. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol. Cell. Biol. 2005, 25, 6415–6426. [Google Scholar] [CrossRef]
- Warboys, C.M.; de Luca, A.; Amini, N.; Luong, L.; Duckles, H.; Hsiao, S.; White, A.; Biswas, S.; Khamis, R.; Chong, C.K.; et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 985–995. [Google Scholar] [CrossRef]
- Hutchison, G.J.; Valentine, H.R.; Loncaster, J.A.; Davidson, S.E.; Hunter, R.D.; Roberts, S.A.; Harris, A.L.; Stratford, I.J.; Price, P.M.; West, C.M.L. Hypoxia-inducible factor 1alpha expression as an intrinsic marker of hypoxia: Correlation with tumor oxygen, pimonidazole measurements, and outcome in locally advanced carcinoma of the cervix. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 8405–8412. [Google Scholar] [CrossRef]
- Snell, C.E.; Turley, H.; McIntyre, A.; Li, D.; Masiero, M.; Schofield, C.J.; Gatter, K.C.; Harris, A.L.; Pezzella, F. Proline-hydroxylated hypoxia-inducible factor 1α (HIF-1α) upregulation in human tumours. PLoS ONE 2014, 9, e88955. [Google Scholar] [CrossRef]
- Cummins, E.P.; Taylor, C.T. Hypoxia-responsive transcription factors. Pflugers Arch. 2005, 450, 363–371. [Google Scholar] [CrossRef]
- Cummins, E.P.; Keogh, C.E. Respiratory gases and the regulation of transcription. Exp. Physiol. 2016, 101, 986–1002. [Google Scholar] [CrossRef]
- Taylor, C.T.; Cummins, E.P. The role of NF-kappaB in hypoxia-induced gene expression. Ann. N. Y. Acad. Sci. 2009, 1177, 178–184. [Google Scholar] [CrossRef]
- Koong, A.C.; Chen, E.Y.; Giaccia, A.J. Hypoxia causes the activation of nuclear factor kappa B through the phosphorylation of I kappa B alpha on tyrosine residues. Cancer Res. 1994, 54, 1425–1430. [Google Scholar]
- Cummins, E.P.; Seeballuck, F.; Keely, S.J.; Mangan, N.E.; Callanan, J.J.; Fallon, P.G.; Taylor, C.T. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 2008, 134, 156–165. [Google Scholar] [CrossRef]
- Hams, E.; Saunders, S.P.; Cummins, E.P.; O’Connor, A.; Tambuwala, M.T.; Gallagher, W.M.; Byrne, A.; Campos-Torres, A.; Moynagh, P.M.; Jobin, C.; et al. The hydroxylase inhibitor dimethyloxallyl glycine attenuates endotoxic shock via alternative activation of macrophages and IL-10 production by B1 cells. Shock Augusta Ga 2011, 36, 295–302. [Google Scholar] [CrossRef]
- Scholz, C.C.; Cavadas, M.A.S.; Tambuwala, M.M.; Hams, E.; Rodríguez, J.; von Kriegsheim, A.; Cotter, P.; Bruning, U.; Fallon, P.G.; Cheong, A.; et al. Regulation of IL-1β-induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 18490–18495. [Google Scholar] [CrossRef]
- Takeda, K.; Ichiki, T.; Narabayashi, E.; Inanaga, K.; Miyazaki, R.; Hashimoto, T.; Matsuura, H.; Ikeda, J.; Miyata, T.; Sunagawa, K. Inhibition of prolyl hydroxylase domain-containing protein suppressed lipopolysaccharide-induced TNF-alpha expression. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2132–2137. [Google Scholar] [CrossRef]
- Winning, S.; Splettstoesser, F.; Fandrey, J.; Frede, S. Acute hypoxia induces HIF-independent monocyte adhesion to endothelial cells through increased intercellular adhesion molecule-1 expression: The role of hypoxic inhibition of prolyl hydroxylase activity for the induction of NF-kappa B. J. Immunol. Baltim. Md 1950 2010, 185, 1786–1793. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, W.; Gao, Q.; Fan, L.; Qin, Y.; Zhou, H.; Li, M.; Fang, J. pVHL mediates K63-linked ubiquitination of IKKβ, leading to IKKβ inactivation. Cancer Lett. 2016, 383, 1–8. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef]
- Alarcón, R.; Koumenis, C.; Geyer, R.K.; Maki, C.G.; Giaccia, A.J. Hypoxia induces p53 accumulation through MDM2 down-regulation and inhibition of E6-mediated degradation. Cancer Res. 1999, 59, 6046–6051. [Google Scholar]
- Graeber, T.G.; Peterson, J.F.; Tsai, M.; Monica, K.; Fornace, A.J.; Giaccia, A.J. Hypoxia induces accumulation of p53 protein, but activation of a G1-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol. Cell. Biol. 1994, 14, 6264–6277. [Google Scholar] [CrossRef]
- Hammond, E.M.; Giaccia, A.J. The role of p53 in hypoxia-induced apoptosis. Biochem. Biophys. Res. Commun. 2005, 331, 718–725. [Google Scholar] [CrossRef]
- Chen, B.; Longtine, M.S.; Sadovsky, Y.; Nelson, D.M. Hypoxia downregulates p53 but induces apoptosis and enhances expression of BAD in cultures of human syncytiotrophoblasts. Am. J. Physiol.-Cell Physiol. 2010, 299, C968–C976. [Google Scholar] [CrossRef]
- Sermeus, A.; Rebucci, M.; Fransolet, M.; Flamant, L.; Desmet, D.; Delaive, E.; Arnould, T.; Michiels, C. Differential effect of hypoxia on etoposide-induced DNA damage response and p53 regulation in different cell types: DNA DAMAGE AND p53 REGULATION BY HYPOXIA. J. Cell. Physiol. 2013, 228, 2365–2376. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Hillion, J.; Le Coniat, M.; Jonveaux, P.; Berger, R.; Bernard, O.A. AF6q21, a novel partner of the MLL gene in t(6; 11) (q21;q23), defines a forkhead transcriptional factor subfamily. Blood 1997, 90, 3714–3719. [Google Scholar]
- Borkhardt, A.; Repp, R.; Haas, O.A.; Leis, T.; Harbott, J.; Kreuder, J.; Hammermann, J.; Henn, T.; Lampert, F. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11) (q13; q23). Oncogene 1997, 14, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zviti, R.; Ha, C.; Wang, Z.V.; Hill, J.A.; Lin, F. Forkhead box O3 (FoxO3) regulates kidney tubular autophagy following urinary tract obstruction. J. Biol. Chem. 2017, 292, 13774–13783. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kang, H.; Zhang, Q.; D’Agati, V.D.; Al-Awqati, Q.; Lin, F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J. Clin. Invest. 2019, 130, 122256. [Google Scholar] [CrossRef]
- Mouchantaf, R.; Azakir, B.A.; McPherson, P.S.; Millard, S.M.; Wood, S.A.; Angers, A. The ubiquitin ligase itch is auto-ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X. J. Biol. Chem. 2006, 281, 38738–38747. [Google Scholar] [CrossRef]
- Xie, Y.; Avello, M.; Schirle, M.; McWhinnie, E.; Feng, Y.; Bric-Furlong, E.; Wilson, C.; Nathans, R.; Zhang, J.; Kirschner, M.W.; et al. Deubiquitinase FAM/USP9X interacts with the E3 ubiquitin ligase SMURF1 protein and protects it from ligase activity-dependent self-degradation. J. Biol. Chem. 2013, 288, 2976–2985. [Google Scholar] [CrossRef]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef]
- Schmidt, M.; Fernandez de Mattos, S.; van der Horst, A.; Klompmaker, R.; Kops, G.J.P.L.; Lam, E.W.-F.; Burgering, B.M.T.; Medema, R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell. Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef]
- Ferber, E.C.; Peck, B.; Delpuech, O.; Bell, G.P.; East, P.; Schulze, A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968–979. [Google Scholar] [CrossRef]
- Jensen, K.S.; Binderup, T.; Jensen, K.T.; Therkelsen, I.; Borup, R.; Nilsson, E.; Multhaupt, H.; Bouchard, C.; Quistorff, B.; Kjaer, A.; et al. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 2011, 30, 4554–4570. [Google Scholar] [CrossRef]
- Aragonés, J.; Schneider, M.; Van Geyte, K.; Fraisl, P.; Dresselaers, T.; Mazzone, M.; Dirkx, R.; Zacchigna, S.; Lemieux, H.; Jeoung, N.H.; et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat. Genet. 2008, 40, 170–180. [Google Scholar] [CrossRef]
- Strowitzki, M.J.; Radhakrishnan, P.; Pavicevic, S.; Scheer, J.; Kimmer, G.; Ritter, A.S.; Tuffs, C.; Volz, C.; Vondran, F.; Harnoss, J.M.; et al. High hepatic expression of PDK4 improves survival upon multimodal treatment of colorectal liver metastases. Br. J. Cancer 2019, 120, 675–688. [Google Scholar] [CrossRef]
- Wang, W.; Bian, K.; Vallabhaneni, S.; Zhang, B.; Wu, R.-C.; O’Malley, B.W.; Long, W. ERK3 promotes endothelial cell functions by upregulating SRC-3/SP1-mediated VEGFR2 expression. J. Cell. Physiol. 2014, 229, 1529–1537. [Google Scholar] [CrossRef]
- Coulombe, P.; Rodier, G.; Pelletier, S.; Pellerin, J.; Meloche, S. Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin-proteasome pathway defines a novel paradigm of mitogen-activated protein kinase regulation during cellular differentiation. Mol. Cell. Biol. 2003, 23, 4542–4558. [Google Scholar] [CrossRef]
- Hansen, C.A.; Bartek, J.; Jensen, S. A functional link between the human cell cycle-regulatory phosphatase Cdc14A and the atypical mitogen-activated kinase Erk3. Cell Cycle Georget. Tex. 2008, 7, 325–334. [Google Scholar] [CrossRef]
- Julien, C.; Coulombe, P.; Meloche, S. Nuclear export of ERK3 by a CRM1-dependent mechanism regulates its inhibitory action on cell cycle progression. J. Biol. Chem. 2003, 278, 42615–42624. [Google Scholar] [CrossRef]
- Kenneth, N.S.; Rocha, S. Regulation of gene expression by hypoxia. Biochem. J. 2008, 414, 19–29. [Google Scholar] [CrossRef]
- Culver, C.; Sundqvist, A.; Mudie, S.; Melvin, A.; Xirodimas, D.; Rocha, S. Mechanism of hypoxia-induced NF-kappaB. Mol. Cell. Biol. 2010, 30, 4901–4921. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Kshitiz; Gilkes, D.M.; Rey, S.; Wong, C.C.; Luo, W.; Kim, D.-H.; Dang, C.V.; Levchenko, A.; Semenza, G.L. A nontranscriptional role for HIF-1α as a direct inhibitor of DNA replication. Sci. Signal. 2013, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Avidor-Reiss, T.; Gopalakrishnan, J. Building a centriole. Curr. Opin. Cell Biol. 2013, 25, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Stearns, T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 2011, 13, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhang, Z.; Liang, X.; Gao, L.; Zhang, X.; Zhao, D.; Liu, X.; Ma, H.; Guo, M.; Spear, B.T.; et al. Zinc fingers and homeoboxes 2 inhibits hepatocellular carcinoma cell proliferation and represses expression of Cyclins A and E. Gastroenterology 2012, 142, 1559–1570. [Google Scholar] [CrossRef]
- Nagel, S.; Schneider, B.; Meyer, C.; Kaufmann, M.; Drexler, H.G.; Macleod, R.A.F. Transcriptional deregulation of homeobox gene ZHX2 in Hodgkin lymphoma. Leuk. Res. 2012, 36, 646–655. [Google Scholar] [CrossRef]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef]
- Karunarathna, U.; Kongsema, M.; Zona, S.; Gong, C.; Cabrera, E.; Gomes, A.R.; Man, E.P.S.; Khongkow, P.; Tsang, J.W.-H.; Khoo, U.-S.; et al. OTUB1 inhibits the ubiquitination and degradation of FOXM1 in breast cancer and epirubicin resistance. Oncogene 2016, 35, 1433–1444. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, J.; Fu, X.; Du, W.; Zhou, L.; Meng, X.; Yu, H.; Lin, J.; Ye, W.; Liu, J.; et al. OTUB1 promotes metastasis and serves as a marker of poor prognosis in colorectal cancer. Mol. Cancer 2014, 13, 258. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, W.-N.; Wang, J.-G.; Chen, H. Colon cancer bears overexpression of OTUB1. Pathol. Res. Pract. 2014, 210, 770–773. [Google Scholar] [CrossRef]
- Iglesias-Gato, D.; Chuan, Y.-C.; Jiang, N.; Svensson, C.; Bao, J.; Paul, I.; Egevad, L.; Kessler, B.M.; Wikström, P.; Niu, Y.; et al. OTUB1 de-ubiquitinating enzyme promotes prostate cancer cell invasion in vitro and tumorigenesis in vivo. Mol. Cancer 2015, 14, 8. [Google Scholar] [CrossRef]
- Zampetaki, A.; Mitsialis, S.A.; Pfeilschifter, J.; Kourembanas, S. Hypoxia induces macrophage inflammatory protein-2 (MIP-2) gene expression in murine macrophages via NF-kappaB: The prominent role of p42/p44 and PI3 kinase pathways. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 1090–1092. [Google Scholar]
- Schmedtje, J.F.; Ji, Y.S.; Liu, W.L.; DuBois, R.N.; Runge, M.S. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J. Biol. Chem. 1997, 272, 601–608. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef]
- Sedgwick, S.G.; Smerdon, S.J. The ankyrin repeat: A diversity of interactions on a common structural framework. Trends Biochem. Sci. 1999, 24, 311–316. [Google Scholar] [CrossRef]
- Tanghe, G.; Urwyler-Rösselet, C.; De Groote, P.; Dejardin, E.; De Bock, P.-J.; Gevaert, K.; Vandenabeele, P.; Declercq, W. RIPK4 activity in keratinocytes is controlled by the SCFβ-TrCP ubiquitin ligase to maintain cortical actin organization. Cell. Mol. Life Sci. CMLS 2018, 75, 2827–2841. [Google Scholar] [CrossRef]
- Chen, L.; Haider, K.; Ponda, M.; Cariappa, A.; Rowitch, D.; Pillai, S. Protein kinase C-associated kinase (PKK), a novel membrane-associated, ankyrin repeat-containing protein kinase. J. Biol. Chem. 2001, 276, 21737–21744. [Google Scholar] [CrossRef]
- Adams, S.; Pankow, S.; Werner, S.; Munz, B. Regulation of NF-kappaB activity and keratinocyte differentiation by the RIP4 protein: Implications for cutaneous wound repair. J. Invest. Dermatol. 2007, 127, 538–544. [Google Scholar] [CrossRef]
- Qi, Z.-H.; Xu, H.-X.; Zhang, S.-R.; Xu, J.-Z.; Li, S.; Gao, H.-L.; Jin, W.; Wang, W.-Q.; Wu, C.-T.; Ni, Q.-X.; et al. RIPK4/PEBP1 axis promotes pancreatic cancer cell migration and invasion by activating RAF1/MEK/ERK signaling. Int. J. Oncol. 2018, 52, 1105–1116. [Google Scholar] [CrossRef]
- Muto, A.; Ruland, J.; McAllister-Lucas, L.M.; Lucas, P.C.; Yamaoka, S.; Chen, F.F.; Lin, A.; Mak, T.W.; Núñez, G.; Inohara, N. Protein kinase C-associated kinase (PKK) mediates Bcl10-independent NF-kappa B activation induced by phorbol ester. J. Biol. Chem. 2002, 277, 31871–31876. [Google Scholar] [CrossRef]
- Huang, X.; McGann, J.C.; Liu, B.Y.; Hannoush, R.N.; Lill, J.R.; Pham, V.; Newton, K.; Kakunda, M.; Liu, J.; Yu, C.; et al. Phosphorylation of Dishevelled by Protein Kinase RIPK4 Regulates Wnt Signaling. Science 2013, 339, 1441–1445. [Google Scholar] [CrossRef]
- Gong, Y.; Luo, X.; Yang, J.; Jiang, Q.; Liu, Z. RIPK4 promoted the tumorigenicity of nasopharyngeal carcinoma cells. Biomed. Pharmacother. Biomedecine Pharmacother. 2018, 108, 1–6. [Google Scholar] [CrossRef]
- X Wang, X.; Zhu, W.; Zhou, Y.; Xu, W.; Wang, H. RIPK4 is downregulated in poorly differentiated tongue cancer and is associated with migration/invasion and cisplatin-induced apoptosis. Int. J. Biol. Markers 2014, 29, e150–e159. [Google Scholar] [CrossRef]
- Heim, D.; Cornils, K.; Schulze, K.; Fehse, B.; Lohse, A.W.; Brümmendorf, T.H.; Wege, H. Retroviral insertional mutagenesis in telomerase-immortalized hepatocytes identifies RIPK4 as novel tumor suppressor in human hepatocarcinogenesis. Oncogene 2015, 34, 364–372. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. The origins of enzyme kinetics. FEBS Lett. 2013, 587, 2725–2730. [Google Scholar] [CrossRef]
- Ivan, M.; Haberberger, T.; Gervasi, D.C.; Michelson, K.S.; Günzler, V.; Kondo, K.; Yang, H.; Sorokina, I.; Conaway, R.C.; Conaway, J.W.; et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA 2002, 99, 13459–13464. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods to Screen, Detect and Verify Hydroxylation | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Target | Enzyme [Residue] | Publication | CO2 Capture Assay and Other | In Silico and Other | MS, MS/MS and Other | Immunoprecipitation (e.g., GST Pulldown Assay) | VHL Capture Assay | Substrate- Trapping | Physiological Relevance * |
| HIF-1α | PHD1–3 [Pro402, 564] | [43,44,45] | ✓ | ✓ | ✓ | ✓ | Co, D or Fe | in vitro | |
| HIF-1α | FIH [Asn803] | [46] | ✓ | ✓ | ✓ | ✓ | e.g., Co, Fe | in vitro | |
| PHDs | |||||||||
| IKKβ | PHD1 [Pro191?] | [47,48] | ✓ | ✓ | ✓ | DMOG | in vitro | ||
| p53 | PHD1 [Pro142] | [49,50] | ✓ | ✓ | ✓ | (DMOG) | in vitro, in vivo | ||
| p53 | PHD3 [Pro359] | [51] | ✓ | ✓ | DMOG | in vitro | |||
| FOXO3a | PHD1 [Pro426, 437] | [47] | ✓ | ✓ | ✓ | DMOG | in vitro, in vivo | ||
| MAPK6 | PHD3 [Pro25] | [52] | ✓ | ✓ | ✓ | DMOG/JNJ | in vitro | ||
| Cep192 | PHD1 [Pro1717] | [53] | ✓ | ✓ | ✓ | in vitro | |||
| ZHX2 | PHD? [Pro427, 440 and 464] | [54] | ✓ | ✓ | ✓ | e.g., DMOG | in vitro, in vivo | ||
| FIH | |||||||||
| OTUB1 | FIH [Asn22] | [55] | ✓ | ✓ | ✓ | ✓ | DMOG | in vitro | |
| p105 | FIH [ASN678] | [56] | ✓ | ✓ | ✓ | ✓ | DMOG | in vitro | |
| IκBα | FIH [Asn244 > 210] | [56] | ✓ | ✓ | ✓ | ✓ | DMOG | in vitro | |
| RIPK4 | FIH [Asn] | [52] | ✓ | ✓ | ✓ | DMOG | in vitro | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strowitzki, M.J.; Cummins, E.P.; Taylor, C.T. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 2019, 8, 384. https://doi.org/10.3390/cells8050384
Strowitzki MJ, Cummins EP, Taylor CT. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells. 2019; 8(5):384. https://doi.org/10.3390/cells8050384
Chicago/Turabian StyleStrowitzki, Moritz J., Eoin P. Cummins, and Cormac T. Taylor. 2019. "Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous?" Cells 8, no. 5: 384. https://doi.org/10.3390/cells8050384
APA StyleStrowitzki, M. J., Cummins, E. P., & Taylor, C. T. (2019). Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells, 8(5), 384. https://doi.org/10.3390/cells8050384