Genetic Diversity and Differentiation at Structurally Varying MHC Haplotypes and Microsatellites in Bottlenecked Populations of Endangered Crested Ibis


Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. MHC Genotyping
2.3. Haplotype Analysis
2.4. Microsatellite Genotyping
2.5. Sequence Analysis
2.6. Population Analysis
3. Results
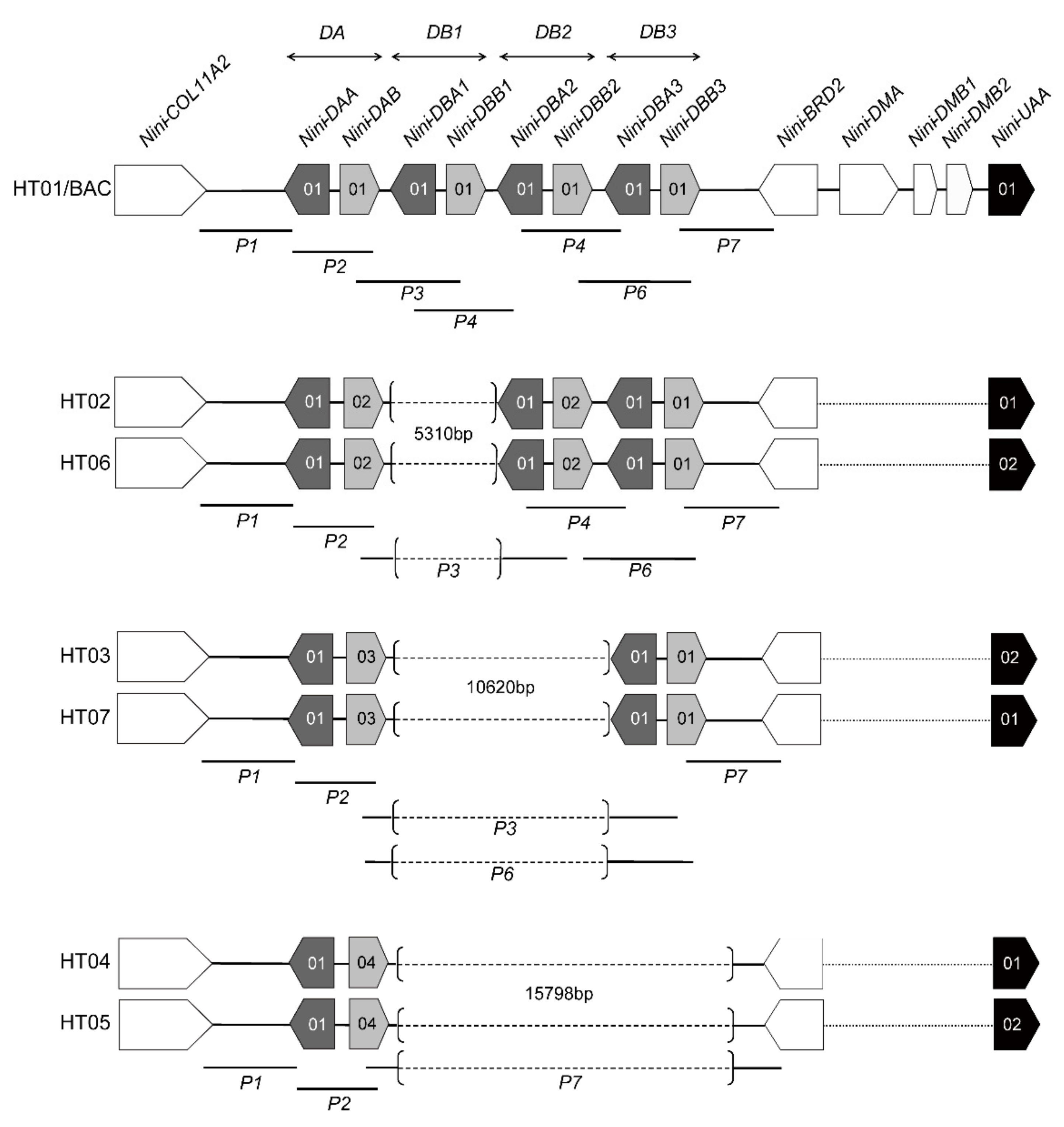
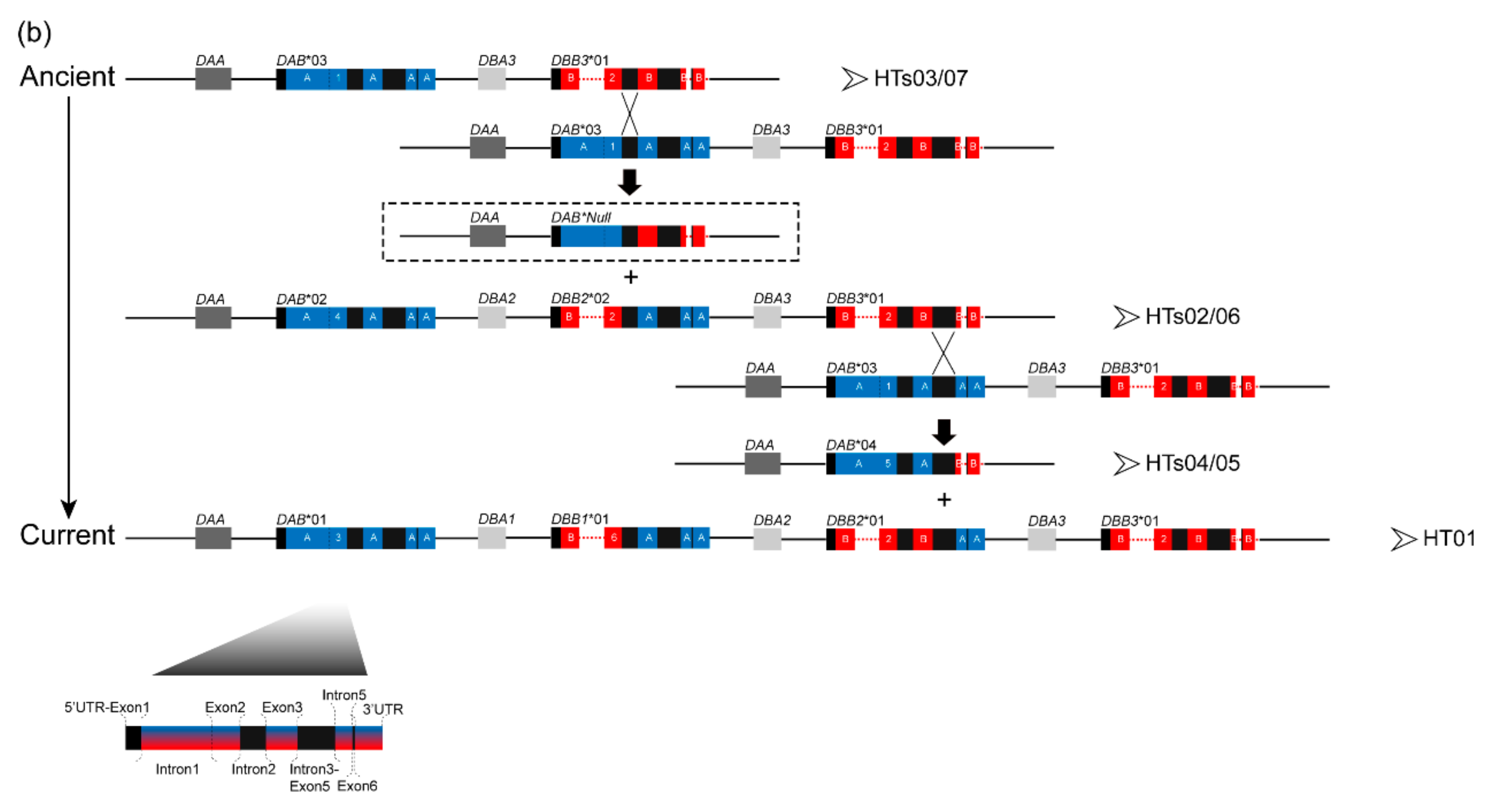
3.1. Characterization of Multilocus MHC Haplotypes
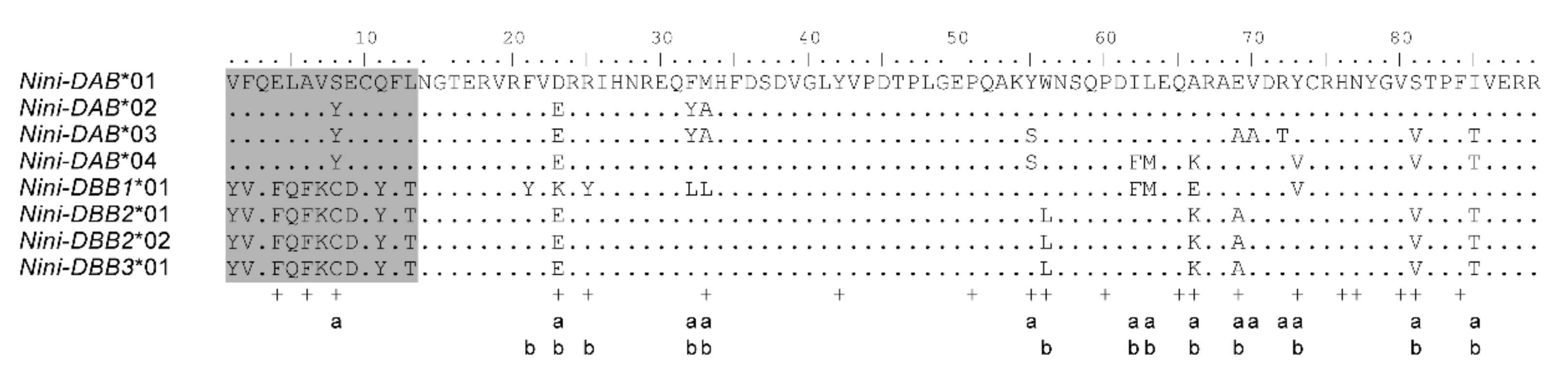
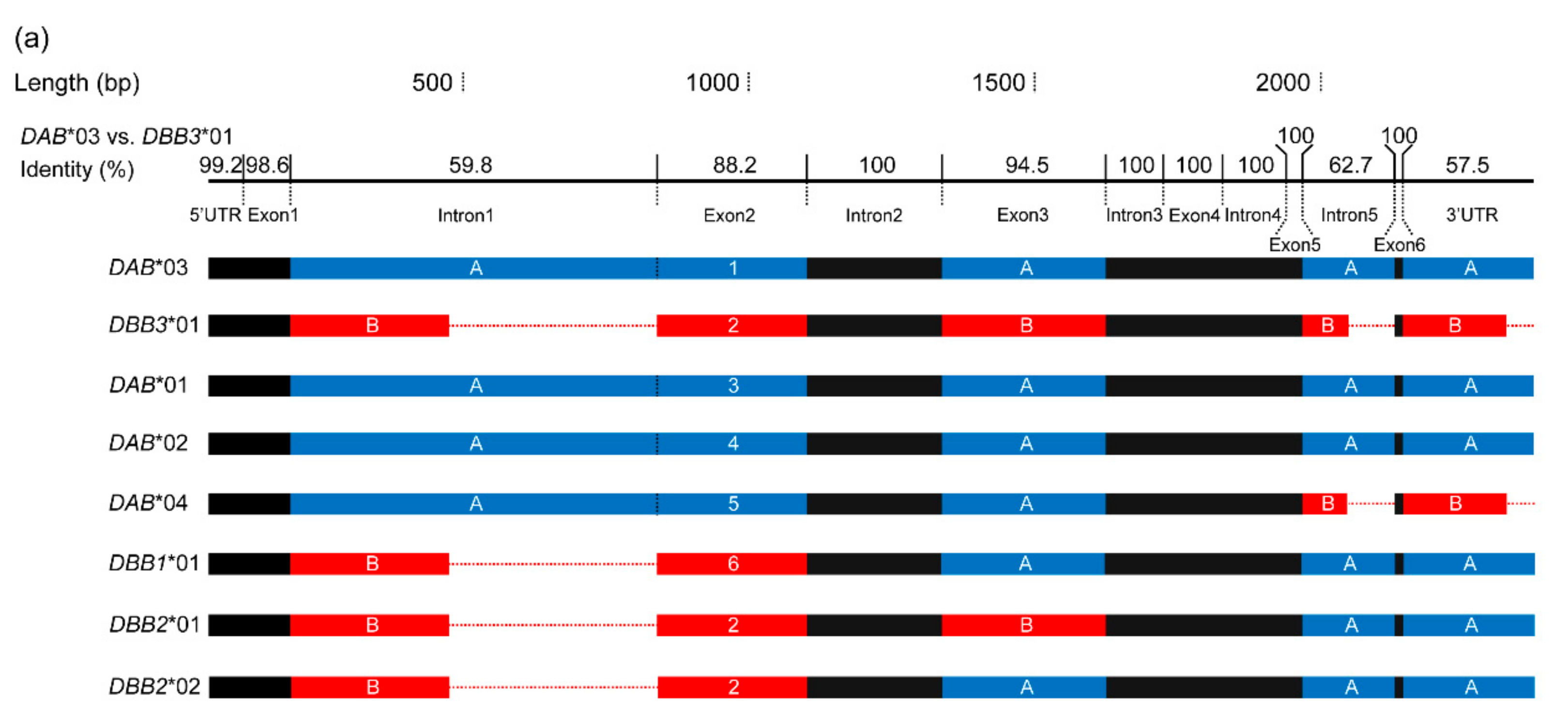
3.2. Positive Selection and Recombination on Nini-MHC-Iiβ
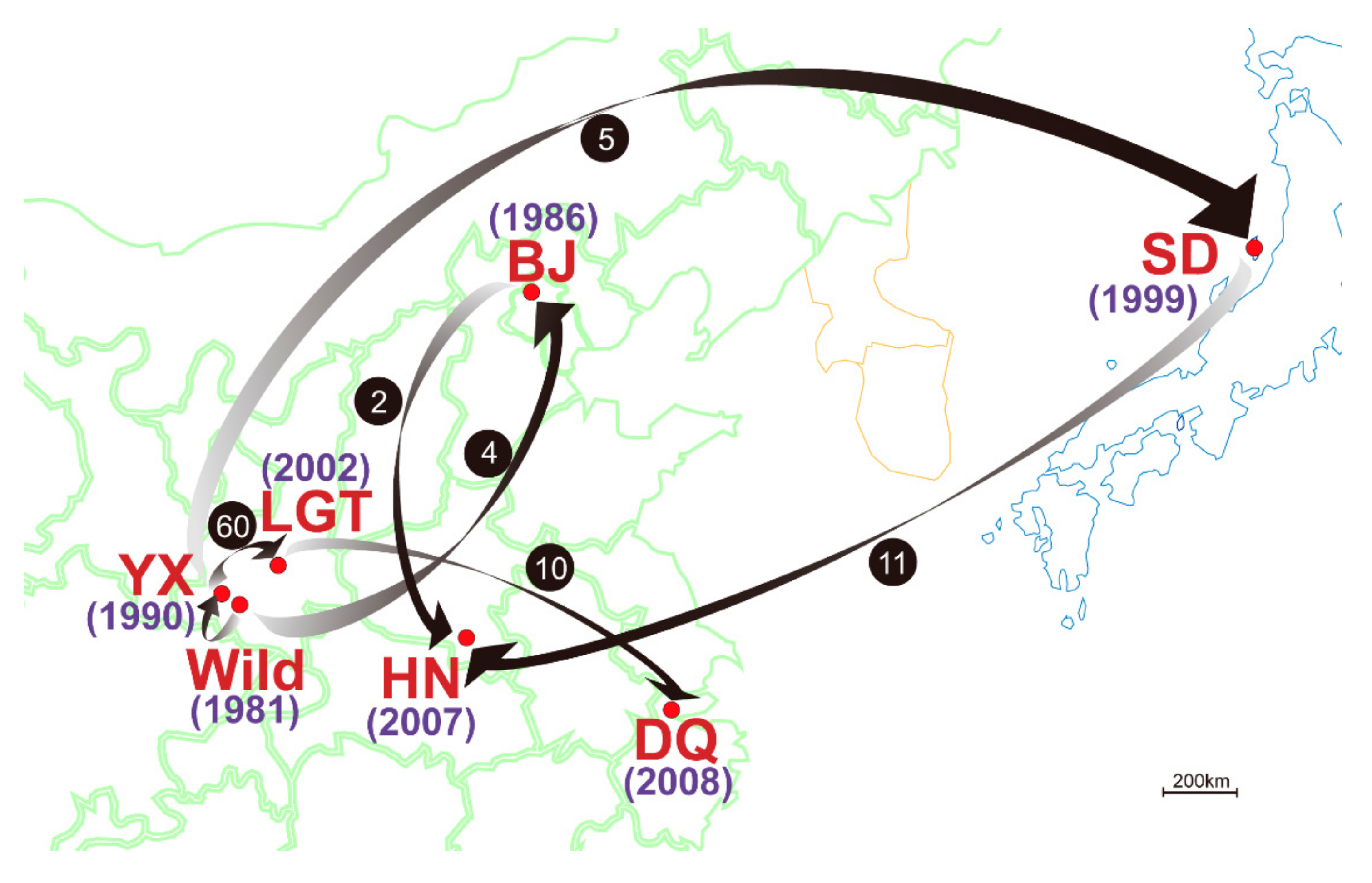
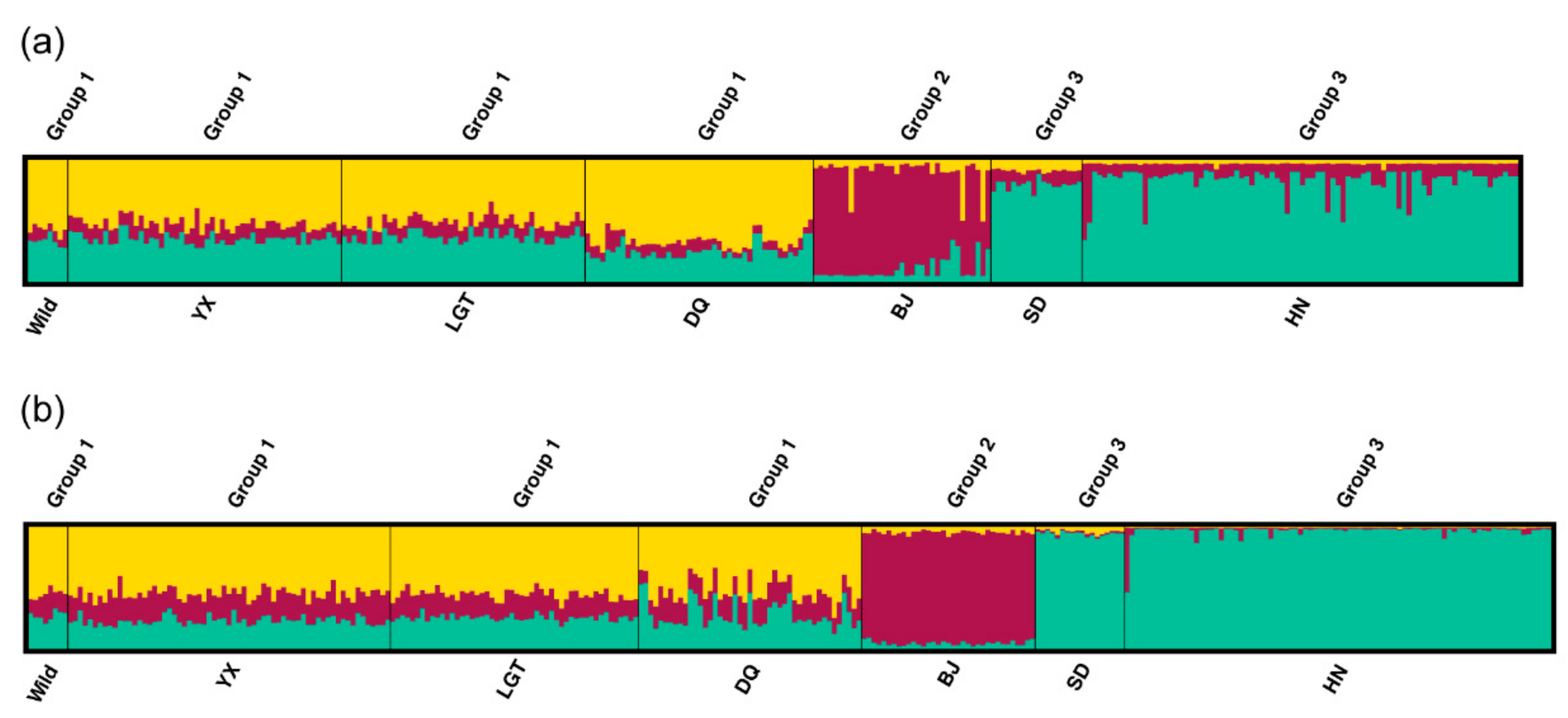
3.3. Genetic Variation within Populations
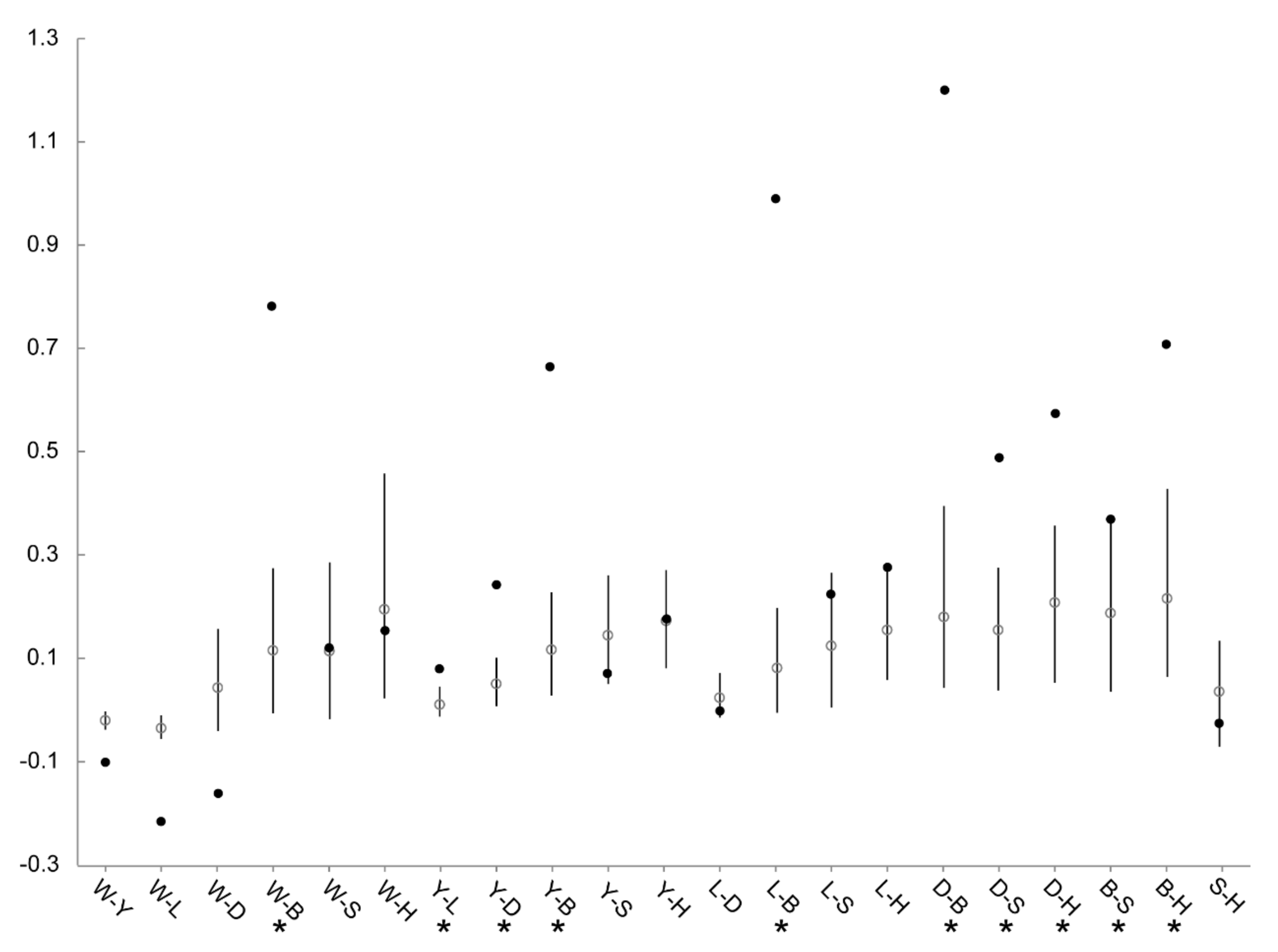
3.4. Patterns of Population Differentiation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Radwan, J.; Biedrzycka, A.; Babik, W. Does reduced MHC diversity decrease viability of vertebrate populations? Biol. Conserv. 2010, 143, 537–544. [Google Scholar] [CrossRef]
- Mills, L.S. Conservation of Wildlife Populations: Demography, Genetics, and Management, 1st ed.; Blackwell Publishing: Malden, MA, USA, 2007. [Google Scholar]
- Crandall, K.A.; Bininda-Emonds, O.R.P.; Mace, G.M.; Wayne, R.K. Considering evolutionary processes in conservation biology. Trends Ecol. Evol. 2000, 15, 290–295. [Google Scholar] [CrossRef]
- Alcaide, M. On the relative roles of selection and genetic drift in shaping MHC variation. Mol. Ecol. 2010, 19, 3842–3844. [Google Scholar] [CrossRef] [PubMed]
- Potts, W.K.; Wakeland, E.K. Evolution of diversity at the major histocompatibility complex. Trends Ecol. Evol. 1990, 5, 181–187. [Google Scholar] [CrossRef]
- Klein, J. Natural History of the Major Histocompatibility Complex; John Wiley and Sons: New York, NY, USA, 1986; pp. 685–687. [Google Scholar]
- Piertney, S.B.; Oliver, M.K. The evolutionary ecology of the major histocompatibility complex. Heredity 2006, 96, 7–21. [Google Scholar] [CrossRef]
- Hedrick, P.W. Pathogen resistance and genetic variation at MHC loci. Evolution 2002, 56, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Minias, P.; Pikus, E.; Whittingham, L.A.; Dunn, P.O. A global analysis of selection at the avian MHC. Evolution 2018, 72, 1278–1293. [Google Scholar] [CrossRef] [PubMed]
- Winternitz, J.C.; Minchey, S.G.; Garamszegi, L.Z.; Huang, S.; Stephens, P.R.; Altizer, S. Sexual selection explains more functional variation in the mammalian major histocompatibility complex than parasitism. Proc. R. Soc. B: Biol. Sci. 2013, 280, 20131605. [Google Scholar] [CrossRef] [PubMed]
- Spurgin, L.G.; Richardson, D.S. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc. R. Soc. B: Biol. Sci. 2010, 277, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Radwan, J.; Kawalko, A.; Wojcik, J.M.; Babik, W. MHC-DRB3 variation in a free-living population of the European bison, Bison bonasus. Mol. Ecol. 2007, 16, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Babik, W.; Pabijan, M.; Radwan, J. Contrasting patterns of variation in MHC loci in the Alpine newt. Mol. Ecol. 2008, 17, 2339–2355. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, C.; Richard, M.; Garnier, S.; Chastel, O.; Julliard, R.; Zoorob, R.; Sorci, G. Diversifying selection on MHC class I in the house sparrow (Passer domesticus). Mol. Ecol. 2009, 18, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, R.; Sæther, S.A.; Jacobsson, P.; Fiske, P.; Sahlman, T.; Grahn, M.; Kålås, J.A.; Höglund, J. Spatial pattern of MHC class II variation in the great snipe (Gallinago media). Mol. Ecol. 2007, 16, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, C.; Wilson, A.B.; Zhao, N.; Li, X.; Zhu, W.; Gao, X.; Liu, X.; Li, Y. Pathogen richness and abundance predict patterns of adaptive MHC variation in insular amphibians. Mol. Ecol. 2017, 26, 4671–4685. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.V.S. HLA associations with malaria in Africa: Some implications for MHC evolution. In Molecular Evolution of the Major Histocompatibility Complex; Klein, J., Klein, D., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 403–420. [Google Scholar]
- Alcaide, M.; Edwards, S.V.; Negro, J.J.; Serrano, D.; Tella, J.L. Extensive polymorphism and geographical variation at a positively selected MHC class II B gene of the lesser kestrel (Falco naumanni ). Mol. Ecol. 2008, 17, 2652–2665. [Google Scholar] [CrossRef] [PubMed]
- Burri, R.; Salamin, N.; Studer, R.A.; Roulin, A.; Fumagalli, L. Adaptive divergence of ancient gene duplicates in the avian MHC class II beta. Mol. Biol. Evol. 2010, 27, 2360–2374. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.C.; Lan, H.; Sun, L.; Deng, Y.L.; Tang, K.Y.; Wan, Q.H. Genomic organization of the crested ibis MHC provides new insight into ancestral avian MHC structure. Sci. Rep. 2015, 5, 7963. [Google Scholar] [CrossRef]
- Gaigher, A.; Burri, R.; Gharib, W.; Taberlet, P.; Roulin, A.; Fumagalli, L. Family-assisted inference of the genetic architecture of major histocompatibility complex variation. Mol. Ecol. Resour. 2016, 16, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, M.; Munoz, J.; Martínez-de la Puente, J.; Soriguer, R.; Figuerola, J. Extraordinary MHC class II B diversity in a non-passerine, wild bird: The Eurasian Coot Fulica atra (Aves: Rallidae). Ecol. Evol. 2014, 4, 688–698. [Google Scholar] [CrossRef]
- Eimes, J.A.; Bollmer, J.L.; Whittingham, L.A.; Johnson, J.A.; Van Oosterhout, C.; Dunn, P.O. Rapid loss of MHC class II variation in a bottlenecked population is explained by drift and loss of copy number variation. J. Evol. Biol. 2011, 24, 1847–1856. [Google Scholar] [CrossRef]
- Tsuji, H.; Taniguchi, Y.; Ishizuka, S.; Matsuda, H.; Yamada, T.; Naito, K.; Iwaisaki, H. Structure and polymorphisms of the major histocompatibility complex in the Oriental stork, Ciconia boyciana. Sci. Rep. 2017, 7, 42864. [Google Scholar] [CrossRef]
- Hosomichi, K.; Shiina, T.; Suzuki, S.; Tanaka, M.; Shimizu, S.; Iwamoto, S.; Hara, H.; Yoshida, Y.; Kulski, J.K.; Inoko, H.; et al. The major histocompatibility complex (Mhc) class IIB region has greater genomic structural flexibility and diversity in the quail than the chicken. BMC Genom. 2006, 7, 322. [Google Scholar] [CrossRef] [PubMed]
- Agudo, R.; Alcaide, M.; Rico, C.; Lemus, J.A.; Blanco, G.; Hiraldo, F.; Donázar, J.A. Major histocompatibility complex variation in insular populations of the Egyptian vulture: Inferences about the roles of genetic drift and selection. Mol. Ecol. 2011, 20, 2329–2340. [Google Scholar] [CrossRef] [PubMed]
- Sin, Y.W.; Annavi, G.; Dugdale, H.L.; Newman, C.; Burke, T.; MacDonald, D.W. Pathogen burden, co-infection and major histocompatibility complex variability in the European badger (Meles meles). Mol. Ecol. 2014, 23, 5072–5088. [Google Scholar] [CrossRef] [PubMed]
- The IUCN Red List of Threatened Species (Version 2018-1). Available online: www.iucnredlist.org (accessed on 12 June 2018).
- Ding, C.Q. Research on the Crested Ibis; Shanghai Scientific and Technological Education Publishing: Shanghai, China, 2004; p. 388. [Google Scholar]
- He, L.P.; Wan, Q.H.; Fang, S.G.; Xi, Y.M. Development of novel microsatellite loci and assessment of genetic diversity in the endangered Crested Ibis, Nipponia Nippon. Conserv. Genet. 2006, 7, 157–160. [Google Scholar] [CrossRef]
- He, X.L.; Ding, C.Q.; Han, J.L. Lack of structural variation but extensive length polymorphisms and heteroplasmic length variations in the mitochondrial DNA control region of highly inbred crested ibis, Nipponia nippon. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Fang, Q.; Barnett, R.; Li, C.; Han, S.; Kuhlwilm, M.; Zhou, L.; Pan, H.; Deng, Y.; Chen, G.; et al. The genomic footprints of the fall and recovery of the crested ibis. Curr. Biol. 2019, 29, 340–349.e347. [Google Scholar] [CrossRef]
- Fan, G.L.; Yang, Z.Q.; Gao, G.G.; Bin, J.J.; Qiao, H.L.; Huang, Z.X.; Hou, Y.F. The histopathological observations of young crested ibis infected with Escherichia coli. Chinese. J. Zool. 2004, 39, 44–46. [Google Scholar]
- Xi, Y.M.; Wood, C.; Lu, B.Z.; Zhang, Y.M. Prevalence of a septicemia disease in the crested ibis (Nipponia nippon) in China. Avian Dis. 2007, 51, 614–617. [Google Scholar] [CrossRef]
- Chen, S.L.; Hao, H.F.; Liu, Q.T.; Wang, R.; Zhang, P.; Wang, X.L.; Du, E.Q.; Yang, Z.Q. Phylogenetic and pathogenic analyses of two virulent Newcastle disease viruses isolated from Crested Ibis (Nipponia nippon) in China. Virus Genes. 2013, 46, 447–453. [Google Scholar] [CrossRef]
- Zhang, X.; Qiao, J.Y.; Wu, X.M.; Ma, Q.Y.; Hu, H.; Wang, J.; Che, L.F. Ascaris spp. and Capillaria caudinflata infections in captive-bred crested ibis (Nipponia nippon ) in China. Zoo Biol. 2015, 34, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; He, S.; Mao, D.; Liu, Y.; Xiong, Z.; Fu, D.; Li, B.; Wei, S.; Xu, X.; Li, S.; et al. Signatures of crested Ibis MHC revealed by recombination screening and short-reads assembly strategy. PLoS ONE 2016, 11, e0168744. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Matsumoto, K.; Matsuda, H.; Yamada, T.; Sugiyama, T.; Homma, K.; Kaneko, Y.; Yamagishi, S.; Iwaisaki, H. Structure and polymorphism of the major histocompatibility complex class II region in the Japanese crested ibis, Nipponia nippon. PLoS ONE 2014, 9, e108506. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Wan, Q.H.; Zhang, P.; Ni, X.W.; Wu, H.L.; Chen, Y.Y.; Kuang, Y.Y.; Ge, Y.F.; Fang, S.G. A novel HURRAH protocol reveals high numbers of monomorphic MHC class II loci and two asymmetric multi-locus haplotypes in the Père David’s deer. PLoS ONE 2011, 6, e14518. [Google Scholar] [CrossRef]
- Zhu, Y.; Sun, D.D.; Ge, Y.F.; Yu, B.; Chen, Y.Y.; Wan, Q.H. Isolation and characterization of class I MHC genes in the giant panda (Ailuropoda melanoleuca). Chin. Sci. Bull. 2013, 58, 2140–2147. [Google Scholar] [CrossRef][Green Version]
- Lan, H.; Chen, H.; Chen, L.C.; Wang, B.B.; Sun, L.; Ma, M.Y.; Fang, S.G.; Wan, Q.H. The first report of a Pelecaniformes defensin cluster: Characterization of β-defensin genes in the crested ibis based on BAC libraries. Sci. Rep. 2014, 4, 6923. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Stephens, M.; Donnelly, P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003, 73, 1162–1169. [Google Scholar] [CrossRef]
- Ji, Y.J.; Liu, Y.D.; Ding, C.Q.; Zhang, D.X. Eight polymorphic microsatellite loci for the critically endangered crested ibis, Nipponia nippon (Ciconiiformes: Threskiornithidae). Mol. Ecol. Notes. 2004, 4, 615–617. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes. 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.J.; McVean, G. Estimating diversifying selection and functional constraint in the presence of recombination. Genetics 2006, 172, 1411–1425. [Google Scholar] [CrossRef]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices (ver 2.9.3.2). Available online: http://www2.unil.ch/popgen/softwares/fstat.htm (accessed on 1 February 2018).
- Piry, S.; Luikart, G.; Cornuet, J.M. Bottleneck: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes. 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Langella, O. POPULATIONS (version 1.2.31). Available online: http://bioinformatics.org/populations/ (accessed on 3 May 2018).
- Balloux, F.; Goudet, J. Statistical properties of population differentiation estimators under stepwise mutation in a finite island model. Mol. Ecol. 2002, 11, 771–783. [Google Scholar] [CrossRef]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes. 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Hedrick, P.W. A standardized genetic differentiation measure. Evolution 2005, 59, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Jardetzky, T.S.; Brown, J.H.; Gorga, J.C.; Stern, L.J.; Urban, R.; Chi, Y.I.; Stauffacher, C.; Strominger, J.L.; Wiley, D.C. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 1994, 368, 711–718. [Google Scholar] [CrossRef]
- Stern, L.J.; Brown, J.H.; Jardetzky, T.S.; Gorga, J.C.; Urban, R.G.; Strominger, J.L.; Wiley, D.C. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature 1994, 368, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Hastings, P.J.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009, 10, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Minias, P.; Pikus, E.; Whittingham, L.A.; Dunn, P.O. Evolution of copy number at the MHC varies across the avian tree of life. Genome Biol Evol. 2018, 11, 17–28. [Google Scholar] [CrossRef]
- Nei, M.; Gu, X.; Sitnikova, T. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc. Natl. Acad. Sci. USA. 1997, 94, 7799–7806. [Google Scholar] [CrossRef]
- Schrider, D.R.; Hahn, M.W. Gene copy-number polymorphism in nature. Proc. R. Soc. B: Biol. Sci. 2010, 277, 3213–3221. [Google Scholar] [CrossRef]
- Ploshnitsa, A.I.; Goltsman, M.E.; Macdonald, D.W.; Kennedy, L.J.; Sommer, S. Impact of historical founder effects and a recent bottleneck on MHC variability in Commander Arctic foxes (Vulpes lagopus). Ecol. Evol. 2012, 2, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Ejsmond, M.J.; Radwan, J. MHC diversity in bottlenecked populations: A simulation model. Conserv. Genet. 2011, 12, 129–137. [Google Scholar] [CrossRef]
- Aguilar, A.; Roemer, G.; Debenham, S.; Binns, M.; Garcelon, D.; Wayne, R.K. High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. Proc. Natl. Acad. Sci. USA 2004, 101, 3490–3494. [Google Scholar] [CrossRef]
- Miller, K.A.; Nelson, N.J.; Smith, H.G.; Moore, J.A. How do reproductive skew and founder group size affect genetic diversity in reintroduced populations? Mol. Ecol. 2009, 18, 3792–3802. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Zhang, Z.; He, W.; Yue, B.; Zhang, A.; Zhang, L.; Hou, R.; Wang, C.; Watanabe, T. Microsatellite variability reveals the necessity for genetic input from wild giant pandas (Ailuropoda melanoleuca) into the captive population. Mol. Ecol. 2009, 18, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Winton, C.L.; Plante, Y.; Hind, P.; McMahon, R.; Hegarty, M.J.; McEwan, N.R.; Davies-Morel, M.C.; Morgan, C.M.; Powell, W.; Nash, D.M. Comparative genetic diversity in a sample of pony breeds from the UK and North America: A case study in the conservation of global genetic resources. Ecol. Evol. 2015, 5, 3507–3522. [Google Scholar] [CrossRef]
- Zhang, Q.; Ji, Y.; Zeng, Z. Influence of the founder effect on genetic diversity of translocated populations: An example from Hainan Eld’s Deer. Chinese. J. Zool. 2007, 42, 54. [Google Scholar]
- Vonholdt, B.M.; Stahler, D.R.; Bangs, E.E.; Smith, D.W.; Jimenez, M.D.; Mack, C.M.; Niemeyer, C.C.; Pollinger, J.P.; Wayne, R.K. A novel assessment of population structure and gene flow in grey wolf populations of the Northern Rocky Mountains of the United States. Mol. Ecol. 2010, 19, 4412–4427. [Google Scholar] [CrossRef] [PubMed]
- Hinkson, K.M.; Henry, N.L.; Hensley, N.M.; Richter, S.C. Initial founders of captive populations are genetically representative of natural populations in critically endangered dusky gopher frogs, Lithobates sevosus. Zoo Biol. 2016, 35, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, B.; Cheng, C.; Xiong, Z.; Liu, Q.; Lai, J.; Carey, H.V.; Zhang, Q.; Zheng, H.; Wei, S. Genomic signatures of near-extinction and rebirth of the crested ibis and other endangered bird species. Genome. Biol. 2014, 15, 557. [Google Scholar] [CrossRef]
- Taylor, R.L., Jr. Major histocompatibility (B) complex control of responses against Rous sarcomas. Poult. Sci. 2004, 83, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Banat, G.R.; Tkalcic, S.; Dzielawa, J.A.; Jackwood, M.W.; Saggese, M.D.; Yates, L.; Kopulos, R.; Briles, W.E.; Collisson, E.W. Association of the chicken MHC B haplotypes with resistance to avian coronavirus. Dev. Comp. Immunol. 2013, 39, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.F.; Taylor, R.L., Jr.; Abplanalp, H. B-complex associated immunity to Salmonella enteritidis challenge in congenic chickens. Poult. Sci. 1998, 77, 1846–1851. [Google Scholar] [CrossRef]
- Deng, Y.L. Paternity Testing and Correlation Analysis Between MHC and Offspring Mortality of Deqing Captive Crested Ibises. Master’s Thesis, Zhejiang University, Zhejiang, China, 2015. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Year Established | Founder Size | Sample Size | Sampling Year | Population Size Around (Sampling Year) | Original Population of Founders |
|---|---|---|---|---|---|---|
| Wild | 1981 | 6 b | 8 | 1990–1993 | 17 (1993) | - |
| YX | 1990 | unknown | 54/65 * | 1997–2006 | 145 (2004) | Wild |
| LGT | 2002 | 60 | 48/50 * | 2006 | 185 (2004) | YX |
| DQ | 2008 | 10 | 45 | 2008–2012 | 45 (2012) | LGT |
| BJ a | 1986 | 4 | 35 | 2011 | 35 (2011) | Wild |
| SD | 1999 | 5 | 18 | 2011–2012 | 120 (2011) | YX |
| HN | 2007 | 13 | 86 | 2012 | 86 (2012) | 11 from SD, 2 from BJ |
| Population | Wild | YX | LGT | DQ | BJ | SD | HN | Total | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample size (MHC/microsatellite) | 8/8 | 54/65 | 48/50 | 45/45 | 35/35 | 18/18 | 86/86 | 294/307 | ||
| MHC | Homozygote 1 | 5 | 34 | 34 | 23 | 27 | 9 | 45 | 177 | |
| Heterozygote 2 | 3 | 20 | 14 | 22 | 8 | 9 | 41 | 117 | ||
| Haplotype frequency (%) | HT01 | 6.3 | 7.4 | 5.2 | 10.0 | 0.0 | 0.0 | 0.0 | 3.9 | |
| HT02 | 43.8 | 28.7 | 44.8 | 51.1 | 14.3 | 25.0 | 28.5 | 33.2 | ||
| HT03 | 25.0 | 26.9 | 21.9 | 18.9 | 60.0 | 36.1 | 32.0 | 30.8 | ||
| HT04 | 12.5 | 19.4 | 15.6 | 6.7 | 2.9 | 22.2 | 32.0 | 18.5 | ||
| HT05 | 12.5 | 17.6 | 11.5 | 12.2 | 8.6 | 5.6 | 1.2 | 9.0 | ||
| HT06 | 0.0 | 0.0 | 1.0 | 1.1 | 0.0 | 0.0 | 0.0 | 0.3 | ||
| HT07 | 0.0 | 0.0 | 0.0 | 0.0 | 14.3 | 11.1 | 6.4 | 4.3 | ||
| HTs02/06 | 43.8 | 28.7 | 45.8 | 52.2 | 14.3 | 25.0 | 28.5 | 33.5 | ||
| HTs03/07 | 25.0 | 26.9 | 21.9 | 18.9 | 74.3 | 47.2 | 38.4 | 35.1 | ||
| HTs04/05 | 25.0 | 37.0 | 27.1 | 18.9 | 11.5 | 27.8 | 33.2 | 27.5 | ||
| HO | 0.63 | 0.78 | 0.75 | 0.71 | 0.54* | 0.78 | 0.81 | 0.71 | ||
| HE | 0.76 | 0.78 | 0.72 | 0.68 | 0.60 | 0.76 | 0.71 | 0.75 | ||
| AR | 5.00 | 4.67 | 4.59 | 4.60 | 4.09 | 4.61 | 3.84 | 4.78 | ||
| Microsatellite | HO | 0.43 | 0.50 | 0.46 | 0.44 | 0.49 | 0.55 | 0.39 | 0.45 | |
| HE | 0.49 | 0.45 | 0.44 | 0.43 | 0.46 | 0.47 | 0.37 | 0.44 | ||
| AR | 2.27 | 2.22 | 2.22 | 2.14 | 2.14 | 2.17 | 2.13 | 2.23 | ||
| Locus | Grouping | Source of Variation | d.f. | SS | Variance Components | Percentage of Variation | Fixation Index | P Value |
|---|---|---|---|---|---|---|---|---|
| MHC | (Wild, LGT, DQ) (YX) (BJ) (SD, HN) | Among groups | 3 | 12.800 | 0.02996 | 7.81 | 0.07810 | 0.00484 |
| Among populations within groups | 3 | 0.633 | −0.00250 | −0.65 | −0.00706 | 0.81299 | ||
| Within populations | 581 | 206.899 | 0.35611 | 92.84 | 0.07159 | 0.00000 | ||
| Microsatellite | (Wild, YX, LGT, DQ) (BJ) (SD, HN) | Among groups | 2 | 49.130 | 0.10917 | 5.10 | 0.05102 | 0.00897 |
| Among populations within groups | 4 | 17.424 | 0.03245 | 1.52 | 0.01598 | 0.00002 | ||
| Within populations | 607 | 1212.932 | 1.99824 | 93.38 | 0.06618 | 0.00000 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, H.; Zhou, T.; Wan, Q.-H.; Fang, S.-G. Genetic Diversity and Differentiation at Structurally Varying MHC Haplotypes and Microsatellites in Bottlenecked Populations of Endangered Crested Ibis. Cells 2019, 8, 377. https://doi.org/10.3390/cells8040377
Lan H, Zhou T, Wan Q-H, Fang S-G. Genetic Diversity and Differentiation at Structurally Varying MHC Haplotypes and Microsatellites in Bottlenecked Populations of Endangered Crested Ibis. Cells. 2019; 8(4):377. https://doi.org/10.3390/cells8040377
Chicago/Turabian StyleLan, Hong, Tong Zhou, Qiu-Hong Wan, and Sheng-Guo Fang. 2019. "Genetic Diversity and Differentiation at Structurally Varying MHC Haplotypes and Microsatellites in Bottlenecked Populations of Endangered Crested Ibis" Cells 8, no. 4: 377. https://doi.org/10.3390/cells8040377
APA StyleLan, H., Zhou, T., Wan, Q.-H., & Fang, S.-G. (2019). Genetic Diversity and Differentiation at Structurally Varying MHC Haplotypes and Microsatellites in Bottlenecked Populations of Endangered Crested Ibis. Cells, 8(4), 377. https://doi.org/10.3390/cells8040377