Role of Hedgehog Signaling in Breast Cancer: Pathogenesis and Therapeutics



Abstract
1. Introduction
2. Overview of Breast Cancer
3. The Hedgehog (Hh) Signaling Pathway
4. Involvement of Hh Signaling in BC: Molecular Mechanisms
4.1. Hh Signaling During Normal Mammary Gland Development
4.2. Canonical and Non-Canonical Hh Signaling in BC
4.3. Regulation of SHH in BC Cells
4.4. PTCH1 Expression in BC Cells
4.5. GLI1 Expression in BC
Truncated GLI1 in BC
4.6. LKB1 as a Negative Regulator of Hh Signaling in BC
4.7. Estrogen Receptor-Positive BC and Hh
4.8. Hh Signaling in TNBC
4.9. Hh Signaling in BC Stem Cells
4.10. Hh Signaling in Epithelial-Mesenchymal Transition and Metastasis in BC
4.10.1. EMT Contribution to Metastasis via the Hh Pathway
4.10.2. GLI1-Induced Metastasis Through CXCR4
4.11. Hh Signaling and BC Microenvironment
5. Correlation of Hh Markers with Clinical and Histopathological Parameters of BC Patients
5.1. Hh Signaling in Different BC Subtypes
5.2. Prognostic Value of Shh in BC
Prognostic Significance of Serum SHH Levels in BC Patients
5.3. PTCH1 and SMO Expression in BC Tumors
5.4. GLI1 Expression in BC Tumors
6. Targeting the Hh Pathway in BC: Past and Current Preclinical and Clinical Trials
6.1. Monoclonal Anti-Hh Protein Antibody, 5E1 mAb
6.2. SMO Inhibitors
6.2.1. Cyclopamine
6.2.2. GDC-0449 (Vismodegib)
6.2.3. LDE225 (Erismodegib, Sonidegib)
6.2.4. Itraconazole
6.3. GLI Antagonists
6.3.1. Hh Pathway “Specific” GLI Inhibitors
6.3.2. Indirect GLI Inhibitors
6.4. SMO Inhibitors vs. GLI Antagonists
6.5. Phytochemicals Targeting HH
6.5.1. Genistein
6.5.2. Pterostilbene
6.5.3. Curcumin
6.5.4. Resveratrol
6.5.5. Nitidine Chloride
6.5.6. Metformin
6.6. Alternative Strategies to Target Hh Pathway in BC
6.6.1. Statins
6.6.2. Vitamin D
7. HH Signaling and Resistance Mechanisms in BC
7.1. The Hh Pathway and Tamoxifen Resistance
7.2. The Hh Pathway and Chemoresistance in TNBC
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; Anderson, B.O.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016. JAMA Oncol. 2018, 4, 1553. [Google Scholar]
- Ovcaricek, T.; Frkovic, S.; Matos, E.; Mozina, B.; Borstnar, S. Triple negative breast cancer-prognostic factors and survival. Radiol. Oncol. 2011, 45, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Riaz, S.K.; Khan, J.S.; Shah, S.T.A.; Wang, F.; Ye, L.; Jiang, W.G.; Malik, M.F.A. Involvement of hedgehog pathway in early onset, aggressive molecular subtypes and metastatic potential of breast cancer. Cell Commun. Signal. 2018, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Monkkonen, T.; Lewis, M.T. New paradigms for the Hedgehog signaling network in mammary gland development and breast Cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Habib, J.G.; O’Shaughnessy, J.A. The hedgehog pathway in triple-negative breast cancer. Cancer Med. 2016, 5, 2989–3006. [Google Scholar] [CrossRef]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated oncogene homolog 1 (GLI1)expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef]
- Goel, H.L.; Pursell, B.; Chang, C.; Shaw, L.M.; Mao, J.; Simin, K.; Kumar, P.; Vander Kooi, C.W.; Shultz, L.D.; Greiner, D.L.; et al. GLI1 regulates a novel neuropilin-2/α6β1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol. Med. 2013, 5, 488–508. [Google Scholar] [CrossRef]
- Bieche, I.; Lidereau, R. Genetic alterations in breast cancer. Genes Chromosom. Cancer 1995, 14, 227–251. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Mao, J.; Zhang, Q.; Li, L. Overexpression of Hedgehog signaling molecules and its involvement in triple-negative breast cancer. Oncol. Lett. 2011, 2, 995–1001. [Google Scholar] [PubMed]
- Simpson, P.T.; Reis-Filho, J.S.; Lakhani, S.R. Breast pathology: Beyond morphology. Semin. Diagn. Pathol. 2010, 27, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Blenkiron, C.; Goldstein, L.D.; Thorne, N.P.; Spiteri, I.; Chin, S.-F.; Dunning, M.J.; Barbosa-Morais, N.L.; Teschendorff, A.E.; Green, A.R.; Ellis, I.O.; et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007, 8, R214. [Google Scholar] [CrossRef]
- Sørlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef]
- Moulder, S.L.; Craft, B.S.; Hortobagyi, G.N. Inhibition of receptor tyrosine kinases in combination with chemotherapy for the treatment of breast cancer. Anticancer. Agents Med. Chem. 2008, 8, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Lee, H.J.; Lulla, A.; Hernandez, L.; Gokare, P.; Lim, B. Current treatment of early breast cancer: Adjuvant and neoadjuvant therapy. F1000Research 2014, 3, 198. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, P. Medscape Breast Cancer Treatment & Management. Available online: https://emedicine.medscape.com/article/1947145-treatment#showall (accessed on 6 March 2019).
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J.; et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006, 66, 1630–1639. [Google Scholar] [CrossRef]
- Houghton, P.J. Everolimus. Clin. Cancer Res. 2010, 16, 1368–1372. [Google Scholar] [CrossRef]
- Marotti, J.D.; de Abreu, F.B.; Wells, W.A.; Tsongalis, G.J. Triple-Negative Breast Cancer. Am. J. Pathol. 2017, 187, 2133–2138. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Anders, C.K.; Carey, L.A. Biology, Metastatic Patterns, and Treatment of Patients with Triple-Negative Breast Cancer. Clin. Breast Cancer 2009, 9, S73–S81. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Jerusalem, G.; Collignon, J.; Schroeder, H.; Lousberg, L. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer Targets Ther. 2016, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A.; Gianni, L. Triple-Negative Breast Cancer: An Unmet Medical Need. Oncologist 2011, 16, 1–11. [Google Scholar] [CrossRef]
- Colleoni, M.; Sun, Z.; Price, K.N.; Karlsson, P.; Forbes, J.F.; Thürlimann, B.; Gianni, L.; Castiglione, M.; Gelber, R.D.; Coates, A.S.; et al. Annual Hazard Rates of Recurrence for Breast Cancer During 24 Years of Follow-Up: Results From the International Breast Cancer Study Group Trials I to V. J. Clin. Oncol. 2016, 34, 927. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef]
- Hong, R.; Ma, F.; Xu, B.; Li, Q.; Zhang, P.; Yuan, P.; Wang, J.; Fan, Y.; Cai, R. Efficacy of platinum-based chemotherapy in triple-negative breast cancer patients with metastases confined to the lungs. Anticancer. Drugs 2014, 25, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Guestini, F.; McNamara, K.M.; Ishida, T.; Sasano, H. Triple negative breast cancer chemosensitivity and chemoresistance: Current advances in biomarkers indentification. Expert Opin. Ther. Targets 2016, 20, 705–720. [Google Scholar] [CrossRef]
- Riddle, R.D.; Johnson, R.L.; Laufer, E.; Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993, 75, 1401–1416. [Google Scholar] [CrossRef]
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503. [Google Scholar] [CrossRef]
- Machold, R.; Hayashi, S.; Rutlin, M.; Muzumdar, M.D.; Nery, S.; Corbin, J.G.; Gritli-Linde, A.; Dellovade, T.; Porter, J.A.; Rubin, L.L.; et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron 2003, 39, 937–950. [Google Scholar] [CrossRef]
- Bhardwaj, G.; Murdoch, B.; Wu, D.; Baker, D.P.; Williams, K.P.; Chadwick, K.; Ling, L.E.; Karanu, F.N.; Bhatia, M. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat. Immunol. 2001, 2, 172–180. [Google Scholar] [CrossRef]
- Alexandre, C.; Jacinto, A.; Ingham, P.W. Transcriptional activation of hedgehog target genes in Drosophila is mediated directly by the cubitus interruptus protein, a member of the GLI family of zinc finger DNA-binding proteins. Genes Dev. 1996, 10, 2003–2013. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, J.M.; Kinzler, K.W.; Wong, A.J.; Bigner, S.H.; Kao, F.T.; Law, M.L.; Seuanez, H.N.; O’Brien, S.J.; Vogelstein, B. The GLI-Kruppel family of human genes. Mol. Cell. Biol. 1988, 8, 3104–3113. [Google Scholar] [CrossRef]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fallon, J.F.; Beachy, P.A. Hedgehog-Regulated Processing of Gli3 Produces an Anterior/Posterior Repressor Gradient in the Developing Vertebrate Limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar]
- Sasai, N.; Briscoe, J. Primary cilia and graded Sonic Hedgehog signaling. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 753–772. [Google Scholar] [CrossRef]
- Riobo, N.A.; Haines, G.M.; Emerson, C.P. Protein Kinase C-δ and Mitogen-Activated Protein/Extracellular Signal–Regulated Kinase-1 Control GLI Activation in Hedgehog Signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Toftgård, R. Non-Canonical Activation of GLI Transcription Factors: Implications for Targeted Anti-Cancer Therapy. Cell Cycle 2007, 6, 2458–2463. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical Hedgehog signaling. Vitam. Horm. 2012, 88, 55–72. [Google Scholar] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Gailani, M.R.; Ståhle-Bäckdahl, M.; Leffell, D.J.; Glyn, M.; Zaphiropoulos, P.G.; Undén, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; Toftgård, R.; et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet. 1996, 14, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Raffel, C.; Jenkins, R.B.; Frederick, L.; Hebrink, D.; Alderete, B.; Fults, D.W.; James, C.D. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997, 57, 842–845. [Google Scholar]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar]
- Singh, S.; Wang, Z.; Liang Fei, D.; Black, K.E.; Goetz, J.A.; Tokhunts, R.; Giambelli, C.; Rodriguez-Blanco, J.; Long, J.; Lee, E.; et al. Hedgehog-Producing Cancer Cells Respond to and Require Autocrine Hedgehog Activity. Cancer Res. 2011, 71, 4454–4463. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, J.; He, J.; Zheng, Y.; Li, H.; Lu, Y.; Qian, J.; Lin, P.; Weber, D.M.; Yang, J.; et al. A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood 2014, 124, 2061–2071. [Google Scholar] [CrossRef]
- Li, X.; Wang, Z.; Ma, Q.; Xu, Q.; Liu, H.; Duan, W.; Lei, J.; Ma, J.; Wang, X.; Lv, S.; et al. Sonic Hedgehog Paracrine Signaling Activates Stromal Cells to Promote Perineural Invasion in Pancreatic Cancer. Clin. Cancer Res. 2014, 20, 4326–4338. [Google Scholar] [CrossRef]
- Chan, I.S.; Guy, C.D.; Chen, Y.; Lu, J.; Swiderska-Syn, M.; Michelotti, G.A.; Karaca, G.; Xie, G.; Kruger, L.; Syn, W.-K.; et al. Paracrine Hedgehog Signaling Drives Metabolic Changes in Hepatocellular Carcinoma. Cancer Res. 2012, 72, 6344–6350. [Google Scholar] [CrossRef]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS Activates Hedgehog Signaling Pathway in Pancreatic Cancer Cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Seto, M.; Ohta, M.; Asaoka, Y.; Ikenoue, T.; Tada, M.; Miyabayashi, K.; Mohri, D.; Tanaka, Y.; Ijichi, H.; Tateishi, K.; et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol. Carcinog. 2009, 48, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Hennighausen, L.; Robinson, G.W. Signaling Pathways in Mammary Gland Development. Dev. Cell 2001, 1, 467–475. [Google Scholar] [CrossRef]
- Gallego, M.I.; Beachy, P.A.; Hennighausen, L.; Robinson, G.W. Differential Requirements for Shh in Mammary Tissue and Hair Follicle Morphogenesis. Dev. Biol. 2002, 249, 131–139. [Google Scholar] [CrossRef]
- Kouros-Mehr, H.; Werb, Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev. Dyn. 2006, 235, 3404–3412. [Google Scholar] [CrossRef]
- Lewis, M.T.; Ross, S.; Strickland, P.A.; Sugnet, C.W.; Jimenez, E.; Hui, C.; Daniel, C.W. The Gli2 Transcription Factor Is Required for Normal Mouse Mammary Gland Development. Dev. Biol. 2001, 238, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Michno, K.; Boras-Granic, K.; Mill, P.; Hui, C.; Hamel, P.A. Shh expression is required for embryonic hair follicle but not mammary gland development. Dev. Biol. 2003, 264, 153–165. [Google Scholar] [CrossRef]
- Velanovich, V.; Yood, M.U.; Bawle, U.; Nathanson, S.D.; Strand, V.F.; Talpos, G.B.; Szymanski, W.; Lewis, F.R. Racial differences in the presentation and surgical management of breast cancer. Surgery 1999, 125, 375–379. [Google Scholar] [CrossRef]
- Hatsell, S.J.; Cowin, P. Gli3-mediated repression of Hedgehog targets is required for normal mammary development. Development 2006, 133, 3661–3670. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Balenci, L.; Okolowsky, N.; Muller, W.J.; Hamel, P.A. Mammary epithelial-restricted expression of activated c-src rescues the block to mammary gland morphogenesis due to the deletion of the C-terminus of Patched-1. Dev. Biol. 2012, 370, 187–197. [Google Scholar] [CrossRef]
- Moraes, R.C.; Zhang, X.; Harrington, N.; Fung, J.Y.; Wu, M.-F.; Hilsenbeck, S.G.; Allred, D.C.; Lewis, M.T. Constitutive activation of smoothened (SMO) in mammary glands of transgenic mice leads to increased proliferation, altered differentiation and ductal dysplasia. Development 2007, 134, 1231–1242. [Google Scholar] [CrossRef]
- Visbal, A.P.; LaMarca, H.L.; Villanueva, H.; Toneff, M.J.; Li, Y.; Rosen, J.M.; Lewis, M.T. Altered differentiation and paracrine stimulation of mammary epithelial cell proliferation by conditionally activated Smoothened. Dev. Biol. 2011, 352, 116–127. [Google Scholar] [CrossRef]
- Okolowsky, N.; Furth, P.A.; Hamel, P.A. Oestrogen receptor-alpha regulates non-canonical Hedgehog-signalling in the mammary gland. Dev. Biol. 2014, 391, 219–229. [Google Scholar] [CrossRef]
- Harvey, M.C.; Fleet, A.; Okolowsky, N.; Hamel, P.A. Distinct Effects of the mesenchymal dysplasia Gene Variant of Murine Patched-1 Protein on Canonical and Non-canonical Hedgehog Signaling Pathways. J. Biol. Chem. 2014, 289, 10939–10949. [Google Scholar] [CrossRef]
- McDermott, K.M.; Liu, B.Y.; Tlsty, T.D.; Pazour, G.J. Primary Cilia Regulate Branching Morphogenesis during Mammary Gland Development. Curr. Biol. 2010, 20, 731–737. [Google Scholar] [CrossRef]
- Xie, J.; Johnson, R.L.; Zhang, X.; Bare, J.W.; Waldman, F.M.; Cogen, P.H.; Menon, A.G.; Warren, R.S.; Chen, L.C.; Scott, M.P.; et al. Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res. 1997, 57, 2369–2372. [Google Scholar] [PubMed]
- Oro, A.E.; Higgins, K.M.; Hu, Z.; Bonifas, J.M.; Epstein, E.H.; Scott, M.P. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science 1997, 276, 817–821. [Google Scholar] [CrossRef]
- Jiao, X.; Wood, L.D.; Lindman, M.; Jones, S.; Buckhaults, P.; Polyak, K.; Sukumar, S.; Carter, H.; Kim, D.; Karchin, R.; et al. Somatic mutations in the notch, NF-KB, PIK3CA, and hedgehog pathways in human breast cancers. Genes Chromosom. Cancer 2012, 51, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog Signaling Pathway is a New Therapeutic Target for Patients with Breast Cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Katano, M. Hedgehog signaling pathway as a therapeutic target in various types of cancer. Cancer Sci. 2011, 102, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hui, C. Hedgehog Signaling in Development and Cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef]
- Chari, N.S.; McDonnell, T.J. The Sonic Hedgehog Signaling Network in Development and Neoplasia. Adv. Anat. Pathol. 2007, 14, 344–352. [Google Scholar] [CrossRef]
- O’Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.A.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog Overexpression Is Associated with Stromal Interactions and Predicts for Poor Outcome in Breast Cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef]
- Chang, H.; Li, Q.; Moraes, R.C.; Lewis, M.T.; Hamel, P.A. Activation of Erk by sonic hedgehog independent of canonical hedgehog signalling. Int. J. Biochem. Cell Biol. 2010, 42, 1462–1471. [Google Scholar] [CrossRef]
- Chinchilla, P.; Xiao, L.; Kazanietz, M.G.; Riobo, N.A. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 2010, 9, 570–579. [Google Scholar] [CrossRef]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Manning, D.R.; Riobo, N.A. Sonic Hedgehog activates the GTPases Rac1 and RhoA in a Gli-independent manner through coupling of smoothened to Gi proteins. Sci. Signal. 2011, 4, pt7. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gradilone, S.A.; Smoot, R.L.; Mertens, J.C.; Bronk, S.F.; Sirica, A.E.; Gores, G.J. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J. Hepatol. 2014, 60, 599–605. [Google Scholar] [CrossRef]
- Fiaschi, M.; Rozell, B.; Bergström, A.; Toftgård, R.; Kleman, M.I. Targeted expression of GLI1 in the mammary gland disrupts pregnancy-induced maturation and causes lactation failure. J. Biol. Chem. 2007, 282, 36090–36101. [Google Scholar] [CrossRef]
- Fiaschi, M.; Rozell, B.; Bergstrom, A.; Toftgard, R. Development of Mammary Tumors by Conditional Expression of GLI1. Cancer Res. 2009, 69, 4810–4817. [Google Scholar] [CrossRef]
- Harris, L.G.; Pannell, L.K.; Singh, S.; Samant, R.S.; Shevde, L.A. Increased vascularity and spontaneous metastasis of breast cancer by hedgehog signaling mediated upregulation of cyr61. Oncogene 2012, 31, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Nakamura, M.; Yamaguchi, H.; Yamanaka, N.; Akiyoshi, T.; Koga, K.; Yamaguchi, K.; Tsuneyoshi, M.; Tanaka, M.; Katano, M. Nuclear Factor-κB Contributes to Hedgehog Signaling Pathway Activation through Sonic Hedgehog Induction in Pancreatic Cancer. Cancer Res. 2006, 66, 7041–7049. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Wang, L.-H.; Wen, Y.-Y.; Song, M.; Li, B.-L.; Chen, X.-L.; Xu, M.; An, S.-X.; Zhao, J.; Lu, Y.-Y.; et al. Expression and regulation mechanisms of Sonic Hedgehog in breast cancer. Cancer Sci. 2010, 101, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Chariot, A. NF-κB, stem cells and breast cancer: The links get stronger. Breast Cancer Res. 2011, 13, 214. [Google Scholar] [CrossRef]
- Prasad, S.; Ravindran, J.; Aggarwal, B.B. NF-κB and cancer: How intimate is this relationship. Mol. Cell. Biochem. 2010, 336, 25–37. [Google Scholar] [CrossRef]
- Kasperczyk, H.; Baumann, B.; Debatin, K.-M.; Fulda, S. Characterization of sonic hedgehog as a novel NF-κB target gene that promotes NF-κB-mediated apoptosis resistance and tumor growth in vivo. FASEB J. 2009, 23, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, A.; Kameda, C.; Xu, R.; Tanaka, H.; Tasaka, T.; Chikazawa, N.; Suzuki, H.; Morisaki, T.; Kubo, M.; Onishi, H.; et al. Nuclear factor kappaB-activated monocytes contribute to pancreatic cancer progression through the production of Shh. Cancer Immunol. Immunother. 2010, 59, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-P.; Hsu, S.-H.; Feng, H.-C.; Huang, R.-F.S. Folate deprivation enhances invasiveness of human colon cancer cells mediated by activation of sonic hedgehog signaling through promoter hypomethylation and cross action with transcription nuclear factor-kappa B pathway. Carcinogenesis 2012, 33, 1158–1168. [Google Scholar] [CrossRef]
- Wang, L.-H.; Choi, Y.-L.; Hua, X.-Y.; Shin, Y.-K.; Song, Y.-J.; Youn, S.-J.; Yun, H.-Y.; Park, S.-M.; Kim, W.-J.; Kim, H.-J.; et al. Increased expression of sonic hedgehog and altered methylation of its promoter region in gastric cancer and its related lesions. Mod. Pathol. 2006, 19, 675–683. [Google Scholar] [CrossRef]
- Duan, Z.-H.; Wang, H.-C.; Zhao, D.-M.; Ji, X.-X.; Song, M.; Yang, X.-J.; Cui, W. Cooperatively transcriptional and epigenetic regulation of sonic hedgehog overexpression drives malignant potential of breast cancer. Cancer Sci. 2015, 106, 1084–1091. [Google Scholar] [CrossRef]
- Wolf, I.; Bose, S.; Desmond, J.C.; Lin, B.T.; Williamson, E.A.; Karlan, B.Y.; Koeffler, H.P. Unmasking of epigenetically silenced genes reveals DNA promoter methylation and reduced expression of PTCH in breast cancer. Breast Cancer Res. Treat. 2007, 105, 139–155. [Google Scholar] [CrossRef]
- Sims-Mourtada, J.; Yang, D.; Tworowska, I.; Larson, R.; Smith, D.; Tsao, N.; Opdenaker, L.; Mourtada, F.; Woodward, W. Detection of Canonical Hedgehog Signaling in Breast Cancer by 131-Iodine-Labeled Derivatives of the Sonic Hedgehog Protein. J. Biomed. Biotechnol. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Lu, Y.; Teng, K.-Y.; Nuovo, G.; Li, X.; Shapiro, C.L.; Majumder, S. Hedgehog Signaling Is a Novel Therapeutic Target in Tamoxifen-Resistant Breast Cancer Aberrantly Activated by PI3K/AKT Pathway. Cancer Res. 2012, 72, 5048–5059. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Y.; Fan, C.; Gao, P.; Wang, X.; Wei, G.; Wei, J. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Mol. Cancer 2014, 13, 137. [Google Scholar] [CrossRef]
- Thomas, Z.I.; Gibson, W.; Sexton, J.Z.; Aird, K.M.; Ingram, S.M.; Aldrich, A.; Lyerly, H.K.; Devi, G.R.; Williams, K.P. Targeting GLI1 expression in human inflammatory breast cancer cells enhances apoptosis and attenuates migration. Br. J. Cancer 2011, 104, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Nguyen, M.P.; Padalecki, S.S.; Grubbs, B.G.; Merkel, A.R.; Oyajobi, B.O.; Matrisian, L.M.; Mundy, G.R.; Sterling, J.A. TGF-β Promotion of Gli2-Induced Expression of Parathyroid Hormone-Related Protein, an Important Osteolytic Factor in Bone Metastasis, Is Independent of Canonical Hedgehog Signaling. Cancer Res. 2011, 71, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Kameda, C.; Tanaka, H.; Yamasaki, A.; Nakamura, M.; Koga, K.; Sato, N.; Kubo, M.; Kuroki, S.; Tanaka, M.; Katano, M. The Hedgehog pathway is a possible therapeutic target for patients with estrogen receptor-negative breast cancer. Anticancer Res. 2009, 29, 871–879. [Google Scholar] [PubMed]
- ten Haaf, A.; Bektas, N.; von Serenyi, S.; Losen, I.; Arweiler, E.C.; Hartmann, A.; Knüchel, R.; Dahl, E. Expression of the glioma-associated oncogene homolog (GLI) 1in human breast cancer is associated with unfavourable overall survival. BMC Cancer 2009, 9, 298. [Google Scholar] [CrossRef]
- Cao, X.; Geradts, J.; Dewhirst, M.W.; Lo, H.-W. Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene 2012, 31, 104–115. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1–AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Wang, K.; Cheng, X.; Qu, X.; Jiang, B.; Li, Z.; Luo, J.; Shao, Z.; Duan, T. LKB1 Inhibits Breast Cancer Partially through Repressing the Hedgehog Signaling Pathway. PLoS ONE 2013, 8, e67431. [Google Scholar] [CrossRef]
- Fillmore, C.M.; Gupta, P.B.; Rudnick, J.A.; Caballero, S.; Keller, P.J.; Lander, E.S.; Kuperwasser, C. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21737–21742. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Zhang, X.-T.; Wang, M.-L.; Zheng, H.-Y.; Liu, L.-J.; Wang, Z.-Y. ER-α36-Mediated Rapid Estrogen Signaling Positively Regulates ER-Positive Breast Cancer Stem/Progenitor Cells. PLoS ONE 2014, 9, e88034. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, J.; Kanomata, N.; Yamashita, T.; Shimo, T.; Moriya, T. Antitumor and anticancer stem cell activities of eribulin mesylate and antiestrogens in breast cancer cells. Breast Cancer 2016, 23, 425–436. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.S.; Farnie, G.; Howell, S.J.; Clarke, R.B. Breast Cancer Stem Cells and Their Role in Resistance to Endocrine Therapy. Horm. Cancer 2011, 2, 91–103. [Google Scholar] [CrossRef]
- Souzaki, M.; Kubo, M.; Kai, M.; Kameda, C.; Tanaka, H.; Taguchi, T.; Tanaka, M.; Onishi, H.; Katano, M. Hedgehog signaling pathway mediates the progression of non-invasive breast cancer to invasive breast cancer. Cancer Sci. 2011, 102, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Elsawaf, Z.; Sinn, H.-P.; Rom, J.; Bermejo, J.L.; Schneeweiss, A.; Aulmann, S. Biological subtypes of triple-negative breast cancer are associated with distinct morphological changes and clinical behaviour. Breast 2013, 22, 986–992. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Weisman, P.S.; Ng, C.K.Y.; Brogi, E.; Eisenberg, R.E.; Won, H.H.; Piscuoglio, S.; De Filippo, M.R.; Ioris, R.; Akram, M.; Norton, L.; et al. Genetic alterations of triple negative breast cancer by targeted next-generation sequencing and correlation with tumor morphology. Mod. Pathol. 2016, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C.; Kishimoto, H.; Fuchs, R.K.; Mehrotra, S.; Bhat-Nakshatri, P.; Turner, C.H.; Goulet, R.; Badve, S.; Nakshatri, H. CD44+/CD24-breast cancer cells exhibit enhanced invasive properties: An early step necessary for metastasis. Breast Cancer Res. 2006, 8, R59. [Google Scholar] [CrossRef] [PubMed]
- Croker, A.K.; Goodale, D.; Chu, J.; Postenka, C.; Hedley, B.D.; Hess, D.A.; Allan, A.L. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell. Mol. Med. 2009, 13, 2236–2252. [Google Scholar] [CrossRef] [PubMed]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of Breast Cancer Stem Cells Identified by Aldehyde Dehydrogenase 1 Expression with Resistance to Sequential Paclitaxel and Epirubicin-Based Chemotherapy for Breast Cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Liu, R.; Wang, X.; Chen, G.Y.; Dalerba, P.; Gurney, A.; Hoey, T.; Sherlock, G.; Lewicki, J.; Shedden, K.; Clarke, M.F. The Prognostic Role of a Gene Signature from Tumorigenic Breast-Cancer Cells. N. Engl. J. Med. 2007, 356, 217–226. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.-M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde Dehydrogenase 1-Positive Cancer Stem Cells Mediate Metastasis and Poor Clinical Outcome in Inflammatory Breast Cancer. Clin. Cancer Res. 2010, 16, 45–55. [Google Scholar] [CrossRef]
- Leth-Larsen, R.; Terp, M.G.; Christensen, A.G.; Elias, D.; Kühlwein, T.; Jensen, O.N.; Petersen, O.W.; Ditzel, H.J. Functional heterogeneity within the CD44 high human breast cancer stem cell-like compartment reveals a gene signature predictive of distant metastasis. Mol. Med. 2012, 18, 1109–1121. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and In vitro Propagation of Tumorigenic Breast Cancer Cells with Stem/Progenitor Cell Properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef]
- Dontu, G.; Wicha, M.S. Survival of Mammary Stem Cells in Suspension Culture: Implications for Stem Cell Biology and Neoplasia. J. Mammary Gland Biol. Neoplasia 2005, 10, 75–86. [Google Scholar] [CrossRef]
- Idowu, M.O.; Kmieciak, M.; Dumur, C.; Burton, R.S.; Grimes, M.M.; Powers, C.N.; Manjili, M.H. CD44+/CD24−/low cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum. Pathol. 2012, 43, 364–373. [Google Scholar] [CrossRef]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef]
- Menzl, I.; Lebeau, L.; Pandey, R.; Hassounah, N.B.; Li, F.W.; Nagle, R.; Weihs, K.; McDermott, K.M. Loss of primary cilia occurs early in breast cancer development. Cilia 2014, 3, 7. [Google Scholar] [CrossRef]
- Di Mauro, C.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Formisano, L.; De Falco, S.; Cicatiello, V.; et al. Hedgehog signalling pathway orchestrates angiogenesis in triple-negative breast cancers. Br. J. Cancer 2017, 116, 1425–1435. [Google Scholar] [CrossRef]
- Kwon, Y.-J.; Hurst, D.R.; Steg, A.D.; Yuan, K.; Vaidya, K.S.; Welch, D.R.; Frost, A.R. Gli1 enhances migration and invasion via up-regulation of MMP-11 and promotes metastasis in ERα negative breast cancer cell lines. Clin. Exp. Metastasis 2011, 28, 437–449. [Google Scholar] [CrossRef]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q.; et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef]
- Das, S.; Samant, R.S.; Shevde, L.A. Nonclassical Activation of Hedgehog Signaling Enhances Multidrug Resistance and Makes Cancer Cells Refractory to Smoothened-targeting Hedgehog Inhibition. J. Biol. Chem. 2013, 288, 11824–11833. [Google Scholar] [CrossRef] [PubMed]
- Sterling, J.A.; Oyajobi, B.O.; Grubbs, B.; Padalecki, S.S.; Munoz, S.A.; Gupta, A.; Story, B.; Zhao, M.; Mundy, G.R. The Hedgehog Signaling Molecule Gli2 Induces Parathyroid Hormone-Related Peptide Expression and Osteolysis in Metastatic Human Breast Cancer Cells. Cancer Res. 2006, 66, 7548–7553. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; André, J.; Verrecchia, F.; Mauviel, A. Cloning of the Human GLI2 Promoter. J. Biol. Chem. 2009, 284, 31523–31531. [Google Scholar] [CrossRef]
- Pratap, J.; Wixted, J.J.; Gaur, T.; Zaidi, S.K.; Dobson, J.; Gokul, K.D.; Hussain, S.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Runx2 Transcriptional Activation of Indian Hedgehog and a Downstream Bone Metastatic Pathway in Breast Cancer Cells. Cancer Res. 2008, 68, 7795–7802. [Google Scholar] [CrossRef]
- Heller, E.; Hurchla, M.A.; Xiang, J.; Su, X.; Chen, S.; Schneider, J.; Joeng, K.-S.; Vidal, M.; Goldberg, L.; Deng, H.; et al. Hedgehog Signaling Inhibition Blocks Growth of Resistant Tumors through Effects on Tumor Microenvironment. Cancer Res. 2012, 72, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Samant, R.S.; Shevde, L.A. Hedgehog Signaling Induced by Breast Cancer Cells Promotes Osteoclastogenesis and Osteolysis. J. Biol. Chem. 2011, 286, 9612–9622. [Google Scholar] [CrossRef]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, R10. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.; Szczepny, A.; Watkins, D.; Cain, J. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Hou, Y.; Yang, G.; Zhang, H.; Tu, G.; Du, Y.; Wen, S.; Xu, L.; Tang, X.; Tang, S.; et al. LncRNA-Hh Strengthen Cancer Stem Cells Generation in Twist-Positive Breast Cancer via Activation of Hedgehog Signaling Pathway. Stem Cells 2016, 34, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer Stem Cells: An Old Idea—A Paradigm Shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Kopper, L.; Hajdú, M. Tumor stem cells. Pathol. Oncol. Res. 2004, 10, 69–73. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pagès, M.; Sotgia, F.; Lisanti, M.P. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog-GLI signalling in breast cancer cells. Oncotarget 2015, 6, 10728–10745. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.H.; Chepko, G. Mammary epithelial stem cells. Microsc. Res. Tech. 2001, 52, 190–203. [Google Scholar] [CrossRef]
- Stingl, J.; Eirew, P.; Ricketson, I.; Shackleton, M.; Vaillant, F.; Choi, D.; Li, H.I.; Eaves, C.J. Purification and unique properties of mammary epithelial stem cells. Nature 2006, 439, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog Signaling and Bmi-1 Regulate Self-renewal of Normal and Malignant Human Mammary Stem Cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.; Langhans, S.A. Crosstalk of Oncogenic Signaling Pathways during Epithelial–Mesenchymal Transition. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, W. Epithelial–mesenchymal transition in human cancer: Comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol. Ther. 2015, 150, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, J.; Matsuda, M.; Furuse, M.; Tsukita, S. Regulation of tight junctions during the epithelium-mesenchyme transition: Direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci. 2003, 116, 1959–1967. [Google Scholar] [CrossRef]
- Kokkinos, M.I.; Wafai, R.; Wong, M.K.; Newgreen, D.F.; Thompson, E.W.; Waltham, M. Vimentin and Epithelial-Mesenchymal Transition in Human Breast Cancer—Observations in vitro and in vivo. Cells Tissues Organs 2007, 185, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Hollier, B.G.; Tinnirello, A.A.; Werden, S.J.; Evans, K.W.; Taube, J.H.; Sarkar, T.R.; Sphyris, N.; Shariati, M.; Kumar, S.V.; Battula, V.L.; et al. FOXC2 Expression Links Epithelial-Mesenchymal Transition and Stem Cell Properties in Breast Cancer. Cancer Res. 2013, 73, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, H.; Tan, G.; Gao, W.; Cheng, L.; Jiang, X.; Yu, L.; Tan, Y. FOXM1 promotes the epithelial to mesenchymal transition by stimulating the transcription of Slug in human breast cancer. Cancer Lett. 2013, 340, 104–112. [Google Scholar] [CrossRef]
- Ke, Z.; Caiping, S.; Qing, Z.; Xiaojing, W. Sonic hedgehog–Gli1 signals promote epithelial–mesenchymal transition in ovarian cancer by mediating PI3K/AKT pathway. Med. Oncol. 2015, 32, 368. [Google Scholar] [CrossRef]
- Yamamichi, F.; Shigemura, K.; Behnsawy, H.M.; Meligy, F.Y.; Huang, W.-C.; Li, X.; Yamanaka, K.; Hanioka, K.; Miyake, H.; Tanaka, K.; et al. Sonic hedgehog and androgen signaling in tumor and stromal compartments drives epithelial–mesenchymal transition in prostate cancer. Scand. J. Urol. 2014, 48, 523–532. [Google Scholar] [CrossRef][Green Version]
- Lei, J.; Ma, J.; Ma, Q.; Li, X.; Liu, H.; Xu, Q.; Duan, W.; Sun, Q.; Xu, J.; Wu, Z.; et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol. Cancer 2013, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Li, H.; Che, J.; Zhang, Y.; Tseng, H.-H.K.; Jin, J.Q.; Luh, T.M.; Giroux-Leprieur, E.; Mo, M.; Zheng, Q.; et al. Hedgehog/Gli promotes epithelial-mesenchymal transition in lung squamous cell carcinomas. J. Exp. Clin. Cancer Res. 2014, 33, 34. [Google Scholar] [CrossRef]
- Lei, J.; Fan, L.; Wei, G.; Chen, X.; Duan, W.; Xu, Q.; Sheng, W.; Wang, K.; Li, X. Gli-1 is crucial for hypoxia-induced epithelial-mesenchymal transition and invasion of breast cancer. Tumor Biol. 2015, 36, 3119–3126. [Google Scholar] [CrossRef]
- García-Zaragoza, E.; Pérez-Tavarez, R.; Ballester, A.; Lafarga, V.; Jiménez-Reinoso, A.; Ramírez, Á.; Murillas, R.; Gallego, M.I. Intraepithelial paracrine Hedgehog signaling induces the expansion of ciliated cells that express diverse progenitor cell markers in the basal epithelium of the mouse mammary gland. Dev. Biol. 2012, 372, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Im, S.; Choi, H.J.; Yoo, C.; Jung, J.-H.; Jeon, Y.-W.; Suh, Y.J.; Kang, C.S. Hedgehog Related Protein Expression in Breast Cancer: Gli-2 Is Associated with Poor Overall Survival. Korean J. Pathol. 2013, 47, 116. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Neelakantan, D.; Drasin, D.J.; Ford, H.L. Intratumoral heterogeneity: Clonal cooperation in epithelial-to-mesenchymal transition and metastasis. Cell Adh. Migr. 2015, 9, 265–276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Inaguma, S.; Riku, M.; Ito, H.; Tsunoda, T.; Ikeda, H.; Kasai, K. GLI1 orchestrates CXCR4/CXCR7 signaling to enhance migration and metastasis of breast cancer cells. Oncotarget 2015, 6, 33648–33657. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Singh, A.K.; Arya, R.K.; Trivedi, A.K.; Sanyal, S.; Baral, R.; Dormond, O.; Briscoe, D.M.; Datta, D. Chemokine receptor trio: CXCR3, CXCR4 and CXCR7 crosstalk via CXCL11 and CXCL12. Cytokine Growth Factor Rev. 2013, 24, 41–49. [Google Scholar] [CrossRef]
- Zhang, Z.; Ni, C.; Chen, W.; Wu, P.; Wang, Z.; Yin, J.; Huang, J.; Qiu, F. Expression of CXCR4 and breast cancer prognosis: A systematic review and meta-analysis. BMC Cancer 2014, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.P.; Luker, K.E.; Garbow, J.R.; Prior, J.L.; Jackson, E.; Piwnica-Worms, D.; Luker, G.D. CXCR4 Regulates Growth of Both Primary and Metastatic Breast Cancer. Cancer Res. 2004, 64, 8604–8612. [Google Scholar] [CrossRef]
- Gao, Y.; Li, C.; Nie, M.; Lu, Y.; Lin, S.; Yuan, P.; Sun, X. CXCR4 as a novel predictive biomarker for metastasis and poor prognosis in colorectal cancer. Tumor Biol. 2014, 35, 4171–4175. [Google Scholar] [CrossRef]
- Ko, S.-Y.; Park, C.-J.; Park, S.-H.; Cho, Y.-U.; Jang, S.; Seo, E.; Kim, N.; Kim, D.-Y.; Koh, K.N.; Im, H.J.; et al. High CXCR4 and low VLA-4 expression predicts poor survival in adults with acute lymphoblastic leukemia. Leuk. Res. 2014, 38, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-F.; Liu, S.-Y.; Min, X.-Y.; Ji, Y.-Y.; Wang, N.; Liu, D.; Ma, N.; Li, Z.-F.; Li, K. The Prognostic Value of CXCR4 in Ovarian Cancer: A Meta-Analysis. PLoS ONE 2014, 9, e92629. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.J.; Bosch, R.; Dieguez-Gonzalez, R.; Novelli, S.; Mozos, A.; Gallardo, A.; Pavón, M.Á.; Céspedes, M.V.; Grañena, A.; Alcoceba, M.; et al. CXCR4 expression enhances diffuse large B cell lymphoma dissemination and decreases patient survival. J. Pathol. 2015, 235, 445–455. [Google Scholar] [CrossRef]
- Wu, J.; Wu, X.; Liang, W.; Chen, C.; Zheng, L.; An, H. Clinicopathological and prognostic significance of chemokine receptor CXCR4 overexpression in patients with esophageal cancer: A meta-analysis. Tumor Biol. 2014, 35, 3709–3715. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, L.; Sánchez-Mateos, P.; Cabañas, C. CXCR7 impact on CXCL12 biology and disease. Trends Mol. Med. 2013, 19, 12–22. [Google Scholar] [CrossRef]
- Saze, Z.; Terashima, M.; Kogure, M.; Ohsuka, F.; Suzuki, H.; Gotoh, M. Activation of the Sonic Hedgehog Pathway and Its Prognostic Impact in Patients with Gastric Cancer. Dig. Surg. 2012, 29, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef]
- Mori, Y.; Okumura, T.; Tsunoda, S.; Sakai, Y.; Shimada, Y. Gli-1 Expression Is Associated with Lymph Node Metastasis and Tumor Progression in Esophageal Squamous Cell Carcinoma. Oncology 2006, 70, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, J.; Latuske, E.; Kohler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef]
- Ciucci, A.; De Stefano, I.; Vellone, V.G.; Lisi, L.; Bottoni, C.; Scambia, G.; Zannoni, G.F.; Gallo, D. Expression of the Glioma-Associated Oncogene Homolog 1 (Gli1) in Advanced Serous Ovarian Cancer Is Associated with Unfavorable Overall Survival. PLoS ONE 2013, 8, e60145. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, J.A.; Chappell, D.L.; Harrington, B.K.; Agrawal, K.; Andritsos, L.A.; Flynn, J.M.; Jones, J.A.; Paulaitis, M.E.; Bolon, B.; Johnson, A.J.; et al. Lymphocyte cytosolic protein 1 is a chronic lymphocytic leukemia membrane-associated antigen critical to niche homing. Blood 2013, 122, 3308–3316. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.W.; Ronnov-Jessen, L.; Howlett, A.R.; Bissell, M.J. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA 1992, 89, 9064–9068. [Google Scholar] [CrossRef]
- Bartling, B.; Hofmann, H.-S.; Silber, R.-E.; Simm, A. Differential impact of fibroblasts on the efficient cell death of lung cancer cells induced by paclitaxel and cisplatin. Cancer Biol. Ther. 2008, 7, 1250–1261. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martinez-Outschoorn, U.; Sotgia, F.; Lisanti, M.P. Tumor Microenvironment and Metabolic Synergy in Breast Cancers: Critical Importance of Mitochondrial Fuels and Function. Semin. Oncol. 2014, 41, 195–216. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Awad, P.; Campisi, J.; Desprez, P.-Y. Epithelial-Mesenchymal Transition Induced by Senescent Fibroblasts. Cancer Microenviron. 2012, 5, 39–44. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Ko, Y.-H.; Goldberg, A.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; et al. Understanding the metabolic basis of drug resistance. Cell Cycle 2011, 10, 2521–2528. [Google Scholar] [CrossRef]
- Spivak-Kroizman, T.R.; Hostetter, G.; Posner, R.; Aziz, M.; Hu, C.; Demeure, M.J.; Von Hoff, D.; Hingorani, S.R.; Palculict, T.B.; Izzo, J.; et al. Hypoxia Triggers Hedgehog-Mediated Tumor-Stromal Interactions in Pancreatic Cancer. Cancer Res. 2013, 73, 3235–3247. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Koga, K.; Nakamura, M.; Nakashima, H.; Akiyoshi, T.; Kubo, M.; Sato, N.; Kuroki, S.; Nomura, M.; Tanaka, M.; Katano, M. Novel link between estrogen receptor alpha and hedgehog pathway in breast cancer. Anticancer Res. 2008, 28, 731–740. [Google Scholar] [PubMed]
- Noman, A.S.; Uddin, M.; Rahman, M.Z.; Nayeem, M.J.; Alam, S.S.; Khatun, Z.; Wahiduzzaman, M.; Sultana, A.; Rahman, M.L.; Ali, M.Y.; et al. Overexpression of sonic hedgehog in the triple negative breast cancer: Clinicopathological characteristics of high burden breast cancer patients from Bangladesh. Sci. Rep. 2016, 6, 18830. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y.; Azatyan, A.; Rahman, M.F.-U.; Zhao, C.; Zhu, J.; Dahlman-Wright, K.; Zaphiropoulos, P.G.; Diao, Y.; Azatyan, A.; Ferdous-Ur Rahman, M.; et al. Blockade of the Hedgehog pathway downregulates estrogen receptor alpha signaling in breast cancer cells. Oncotarget 2016, 7, 71580–71593. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [CrossRef]
- Jeng, K.-S.; Yu, M.-C.; Hsiau, H.-I.; Chang, F.-Y.; Sheen, I.-S.; Jeng, W.-J. High expression of Sonic Hedgehog signaling pathway genes indicates a risk of recurrence of breast carcinoma. Onco. Targets. Ther. 2013, 7, 79. [Google Scholar] [CrossRef]
- Li, Y.; Yang, W.; Yang, Q.; Zhou, S. Nuclear localization of GLI1 and elevated expression of FOXC2 in breast cancer is associated with the basal-like phenotype. Histol. Histopathol. 2012, 27, 475–484. [Google Scholar]
- Noman, A.S.; Uddin, M.; Chowdhury, A.A.; Nayeem, M.J.; Raihan, Z.; Rashid, M.I.; Azad, A.K.; Rahman, M.L.; Barua, D.; Sultana, A.; et al. Serum sonic hedgehog (SHH) and interleukin-(IL-6) as dual prognostic biomarkers in progressive metastatic breast cancer. Sci. Rep. 2017, 7, 1796. [Google Scholar] [CrossRef]
- Azim, H.A.; Partridge, A.H. Biology of breast cancer in young women. Breast Cancer Res. 2014, 16, 427. [Google Scholar] [CrossRef]
- Bieche, I.; Lerebours, F.; Tozlu, S.; Espie, M.; Marty, M.; Lidereau, R. Molecular Profiling of Inflammatory Breast Cancer: Identification of a Poor-Prognosis Gene Expression Signature. Clin. Cancer Res. 2004, 10, 6789–6795. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Frolova, N.; Sadlonova, A.; Novak, Z.; Steg, A.; Page, G.P.; Welch, D.R.; Lobo-Ruppert, S.M.; Ruppert, J.M.; Johnson, M.R.; et al. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 2006, 5, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Bachelot, T.; Ray-Coquard, I.; Menetrier-Caux, C.; Rastkha, M.; Duc, A.; Blay, J.-Y. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br. J. Cancer 2003, 88, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Köhler, B.; Schönicke, A.; Scharwächter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Chang-Claude, J.; Dunning, A.; Schnitzbauer, U.; Galmbacher, P.; Tee, L.; Wjst, M.; Chalmers, J.; Zemzoum, I.; Harbeck, N.; Pharoah, P.D.P.; et al. Thepatched polymorphism Pro1315Leu (C3944T) may modulate the association between use of oral contraceptives and breast cancer risk. Int. J. Cancer 2003, 103, 779–783. [Google Scholar] [CrossRef]
- Hu, Z.; Bonifas, J.M.; Aragon, G.; Kopelovich, L.; Liang, Y.; Ohta, S.; Israel, M.A.; Bickers, D.R.; Aszterbaum, M.; Epstein, E.H. Evidence for lack of enhanced hedgehog target gene expression in common extracutaneous tumors. Cancer Res. 2003, 63, 923–928. [Google Scholar] [PubMed]
- O’Toole, S.A.; Swarbrick, A.; Sutherland, R.L. The Hedgehog signalling pathway as a therapeutic target in early breast cancer development. Expert Opin. Ther. Targets 2009, 13, 1095–1103. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and Safety of Vismodegib in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog–Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Kim, J.J.; Tang, J.Y.; Gong, R.; Kim, J.J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a Commonly Used Antifungal that Inhibits Hedgehog Pathway Activity and Cancer Growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot Clinical Trial of Hedgehog Pathway Inhibitor GDC-0449 (Vismodegib) in Combination with Gemcitabine in Patients with Metastatic Pancreatic Adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Berlin, J.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L.; et al. A Randomized Phase II Trial of Vismodegib versus Placebo with FOLFOX or FOLFIRI and Bevacizumab in Patients with Previously Untreated Metastatic Colorectal Cancer. Clin. Cancer Res. 2013, 19, 258–267. [Google Scholar] [CrossRef]
- Belani, C.P.; Dahlberg, S.E.; Rudin, C.M.; Fleisher, M.; Chen, H.X.; Takebe, N.; Velasco, M.R.; Tester, W.J.; Sturtz, K.; Hann, C.L.; et al. Vismodegib or cixutumumab in combination with standard chemotherapy for patients with extensive-stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E1508). Cancer 2016, 122, 2371–2378. [Google Scholar] [CrossRef]
- Maun, H.R.; Wen, X.; Lingel, A.; de Sauvage, F.J.; Lazarus, R.A.; Scales, S.J.; Hymowitz, S.G. Hedgehog Pathway Antagonist 5E1 Binds Hedgehog at the Pseudo-active Site. J. Biol. Chem. 2010, 285, 26570–26580. [Google Scholar] [CrossRef]
- Das, S.; Tucker, J.A.; Khullar, S.; Samant, R.S.; Shevde, L.A. Hedgehog signaling in tumor cells facilitates osteoblast-enhanced osteolytic metastases. PLoS ONE 2012, 7, e34374. [Google Scholar] [CrossRef]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef]
- Che, J.; Zhang, F.-Z.; Zhao, C.-Q.; Hu, X.-D.; Fan, S.-J. Cyclopamine is a novel Hedgehog signaling inhibitor with significant anti-proliferative, anti-invasive and anti-estrogenic potency in human breast cancer cells. Oncol. Lett. 2013, 5, 1417–1421. [Google Scholar] [CrossRef]
- Chai, F.; Zhou, J.; Chen, C.; Xie, S.; Chen, X.; Su, P.; Shi, J. The Hedgehog inhibitor cyclopamine antagonizes chemoresistance of breast cancer cells. Onco. Targets. Ther. 2013, 6, 1643–1647. [Google Scholar]
- Boamah, E.; Ibrahim, Q.; Kwinji, L.; Patel, R.; Ajayi, D.; Danquah, M. EGFR inhibitors in combination with cyclopamine as chemotherapeutic strategy for treating breast cancer. Synergy 2015, 2, 7–18. [Google Scholar] [CrossRef]
- Sabol, M.; Trnski, D.; Uzarevic, Z.; Ozretic, P.; Musani, V.; Rafaj, M.; Cindric, M.; Levanat, S. Combination of Cyclopamine and Tamoxifen Promotes Survival and Migration of MCF-7 Breast Cancer Cells—Interaction of Hedgehog-Gli and Estrogen Receptor Signaling Pathways. PLoS ONE 2014, 9, e114510. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Wang, W.; Liu, D.; Zhao, Y.; He, J.; Wang, X.; Dai, Z.; Zhang, H.; Li, X. Targeting of sonic hedgehog-Gli signaling: A potential therapeutic target for patients with breast cancer. Oncol. Lett. 2016, 12, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Harrington, N.; Moraes, R.C.; Wu, M.-F.; Hilsenbeck, S.G.; Lewis, M.T. Cyclopamine inhibition of human breast cancer cell growth independent of Smoothened (Smo). Breast Cancer Res. Treat. 2009, 115, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Sahebjam, S.; Siu, L.L.; Razak, A.A. The Utility of Hedgehog Signaling Pathway Inhibition for Cancer. Oncologist 2012, 17, 1090–1099. [Google Scholar] [CrossRef]
- Mahindroo, N.; Punchihewa, C.; Fujii, N. Hedgehog-Gli Signaling Pathway Inhibitors as Anticancer Agents. J. Med. Chem. 2009, 52, 3829–3845. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.W.; Hong, R. Significance of the hedgehog pathway-associated proteins Gli-1 and Gli-2 and the epithelial-mesenchymal transition-associated proteins Twist and E-cadherin in hepatocellular carcinoma. Oncol. Lett. 2016, 12, 1753–1762. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A Phase I, Multicenter, Open-Label, First-in-Human, Dose-Escalation Study of the Oral Smoothened Inhibitor Sonidegib (LDE225) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef]
- Jalili, A.; Mertz, K.D.; Romanov, J.; Wagner, C.; Kalthoff, F.; Stuetz, A.; Pathria, G.; Gschaider, M.; Stingl, G.; Wagner, S.N. NVP-LDE225, a Potent and Selective SMOOTHENED Antagonist Reduces Melanoma Growth In Vitro and In Vivo. PLoS ONE 2013, 8, e69064. [Google Scholar] [CrossRef]
- Stathis, A.; Hess, D.; von Moos, R.; Homicsko, K.; Griguolo, G.; Joerger, M.; Mark, M.; Ackermann, C.J.; Allegrini, S.; Catapano, C.V.; et al. Phase I trial of the oral smoothened inhibitor sonidegib in combination with paclitaxel in patients with advanced solid tumors. Invest. New Drugs 2017, 35, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Borrego, M.; Jimenez, B.; Antolín, S.; García-Saenz, J.A.; Corral, J.; Jerez, Y.; Trigo, J.; Urruticoechea, A.; Colom, H.; Gonzalo, N.; et al. A phase Ib study of sonidegib (LDE225), an oral small molecule inhibitor of smoothened or Hedgehog pathway, in combination with docetaxel in triple negative advanced breast cancer patients: GEICAM/2012–12 (EDALINE) study. Invest. New Drugs 2019, 37, 98–108. [Google Scholar] [CrossRef]
- Pounds, R.; Leonard, S.; Dawson, C.; Kehoe, S. Repurposing itraconazole for the treatment of cancer. Oncol. Lett. 2017, 14, 2587–2597. [Google Scholar] [CrossRef]
- UniProt UniProtKB-Q16850 (CP51A_HUMAN). Available online: https://www.uniprot.org/uniprot/Q16850 (accessed on 29 March 2019).
- Wang, X.; Wei, S.; Zhao, Y.; Shi, C.; Liu, P.; Zhang, C.; Lei, Y.; Zhang, B.; Bai, B.; Huang, Y.; et al. Anti-proliferation of breast cancer cells with itraconazole: Hedgehog pathway inhibition induces apoptosis and autophagic cell death. Cancer Lett. 2017, 385, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.; Silva, D.; Correia, A.; Vilanova, M.; Gärtner, F.; Vale, N. Study of New Therapeutic Strategies to Combat Breast Cancer Using Drug Combinations. Biomolecules 2018, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Xu, J.; Lu, J.; Bhat, S.; Sullivan, D.J.; Liu, J.O. Inhibition of Angiogenesis by the Antifungal Drug Itraconazole. ACS Chem. Biol. 2007, 2, 263–270. [Google Scholar] [CrossRef]
- Ademuyiwa, F.O.; Zhao, Q.; Perkins, S.M.; Gebregziabher, N.; Jones, D.R.; Vaughn, L.G.; Sledge, G.W.; Miller, K. A pilot trial of itraconazole pharmacokinetics in patients with metastatic breast cancer. J. Clin. Oncol. 2011, 29, e13565. [Google Scholar] [CrossRef]
- Malhi, V.; Colburn, D.; Williams, S.J.; Hop, C.E.C.A.; Dresser, M.J.; Chandra, P.; Graham, R.A. A clinical drug–drug interaction study to evaluate the effect of a proton-pump inhibitor, a combined P-glycoprotein/cytochrome 450 enzyme (CYP)3A4 inhibitor, and a CYP2C9 inhibitor on the pharmacokinetics of vismodegib. Cancer Chemother. Pharmacol. 2016, 78, 41–49. [Google Scholar] [CrossRef][Green Version]
- Ji, Y.; Rounds, T.; Crocker, A.; Sussman, B.; Hovey, R.C.; Kingsley, F.; Muss, H.B.; Garber, J.E.; Wood, M.E. The Effect of Atorvastatin on Breast Cancer Biomarkers in High-Risk Women. Cancer Prev. Res. 2016, 9, 379–384. [Google Scholar] [CrossRef]
- Agyeman, A.; Jha, B.K.; Mazumdar, T.; Houghton, J.A. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget 2014, 5, 4492–4503. [Google Scholar] [CrossRef]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, A.; Iovine, V.; Botta, B.; Quaglio, D.; D’Acquarica, I.; Ciogli, A.; Iazzetti, A.; Alfonsi, R.; Lospinoso Severini, L.; Infante, P.; et al. Chemical, computational and functional insights into the chemical stability of the Hedgehog pathway inhibitor GANT61. J. Enzyme Inhib. Med. Chem. 2018, 33, 349–358. [Google Scholar] [CrossRef]
- Jeng, K.-S.; Jeng, C.-J.; Sheen, I.-S.; Wu, S.-H.; Lu, S.-J.; Wang, C.-H.; Chang, C.-F. Glioma-Associated Oncogene Homolog Inhibitors Have the Potential of Suppressing Cancer Stem Cells of Breast Cancer. Int. J. Mol. Sci. 2018, 19, 1375. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog signaling pathway in cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, J.; Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T. Anti-cancer stem cell activity of a hedgehog inhibitor GANT61 in estrogen receptor-positive breast cancer cells. Cancer Sci. 2017, 108, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T.; Kurebayashi, J. Anti-cell growth and anti-cancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast Cancer 2017, 24, 683–693. [Google Scholar] [CrossRef]
- Li, B.; Fei, D.L.; Flaveny, C.A.; Dahmane, N.; Baubet, V.; Wang, Z.; Bai, F.; Pei, X.-H.; Rodriguez-Blanco, J.; Hang, B.; et al. Pyrvinium Attenuates Hedgehog Signaling Downstream of Smoothened. Cancer Res. 2014, 74, 4811–4821. [Google Scholar] [CrossRef]
- Makinodan, E.; Marneros, A.G. Protein kinase A activation inhibits oncogenic Sonic hedgehog signalling and suppresses basal cell carcinoma of the skin. Exp. Dermatol. 2012, 21, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, T.; Arai, M.A.; Koyano, T.; Kowithayakorn, T.; Ishibashi, M. Naturally Occurring Small-Molecule Inhibitors of Hedgehog/GLI-Mediated Transcription. ChemBioChem 2008, 9, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3–RENKCTD11 ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.-H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.-H.; Ayad, N.G.; et al. The BET Bromodomain Inhibitor I-BET151 Acts Downstream of Smoothened Protein to Abrogate the Growth of Hedgehog Protein-driven Cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef]
- Platanias, L.C. Biological responses to arsenic compounds. J. Biol. Chem. 2009, 284, 18583–18587. [Google Scholar] [CrossRef]
- Baj, G.; Arnulfo, A.; Deaglio, S.; Mallone, R.; Vigone, A.; De Cesaris, M.G.; Surico, N.; Malavasi, F.; Ferrero, E. Arsenic trioxide and breast cancer: Analysis of the apoptotic, differentiative and immunomodulatory effects. Breast Cancer Res. Treat. 2002, 73, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Kozono, S.; Lin, Y.-M.; Seo, H.-S.; Pinch, B.; Lian, X.; Qiu, C.; Herbert, M.K.; Chen, C.-H.; Tan, L.; Gao, Z.J.; et al. Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat. Commun. 2018, 9, 3069. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.-C.; Peaceman, D.; Özdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Invest. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Varghese, E.; Samuel, S.M.; Abotaleb, M.; Cheema, S.; Mamtani, R.; Büsselberg, D. The “Yin and Yang” of Natural Compounds in Anticancer Therapy of Triple-Negative Breast Cancers. Cancers 2018, 10, 346. [Google Scholar] [CrossRef] [PubMed]
- King, T.D.; Suto, M.J.; Li, Y. The wnt/β-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. J. Cell. Biochem. 2012, 113, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Speiser, J.J.; Erşahin, Ç.; Osipo, C. The Functional Role of Notch Signaling in Triple-Negative Breast Cancer. Vitam Horm. 2013, 93, 277–306. [Google Scholar]
- Poma, P.; Labbozzetta, M.; D’Alessandro, N.; Notarbartolo, M. NF-κB Is a Potential Molecular Drug Target in Triple-Negative Breast Cancers. Omi. A J. Integr. Biol. 2017, 21, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Gordon, V.; Banerji, S. Molecular Pathways: PI3K Pathway Targets in Triple-Negative Breast Cancers. Clin. Cancer Res. 2013, 19, 3738–3744. [Google Scholar] [CrossRef] [PubMed]
- Giltnane, J.M.; Balko, J.M. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar] [PubMed]
- Jamdade, V.S.; Sethi, N.; Mundhe, N.A.; Kumar, P.; Lahkar, M.; Sinha, N. Therapeutic targets of triple-negative breast cancer: A review. Br. J. Pharmacol. 2015, 172, 4228–4237. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Valachovicova, T.; Slivova, V.; Sliva, D. Cellular and physiological effects of soy flavonoids. Mini Rev. Med. Chem. 2004, 4, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Green, D.R.; Blaser, K.; Simon, H.U. Protein-tyrosine phosphorylation regulates apoptosis in human eosinophils and neutrophils. Proc. Natl. Acad. Sci. USA 1994, 91, 10868–10872. [Google Scholar] [CrossRef]
- Fan, P.; Fan, S.; Wang, H.; Mao, J.; Shi, Y.; Ibrahim, M.M.; Ma, W.; Yu, X.; Hou, Z.; Wang, B.; et al. Genistein decreases the breast cancer stem-like cell population through Hedgehog pathway. Stem Cell Res. Ther. 2013, 4, 146. [Google Scholar] [CrossRef]
- Pan, M.-H.; Chiou, Y.-S.; Chen, W.-J.; Wang, J.-M.; Badmaev, V.; Ho, C.-T. Pterostilbene inhibited tumor invasion via suppressing multiple signal transduction pathways in human hepatocellular carcinoma cells. Carcinogenesis 2009, 30, 1234–1242. [Google Scholar] [CrossRef]
- Wu, C.-H.; Hong, B.-H.; Ho, C.-T.; Yen, G.-C. Targeting Cancer Stem Cells in Breast Cancer: Potential Anticancer Properties of 6-Shogaol and Pterostilbene. J. Agric. Food Chem. 2015, 63, 2432–2441. [Google Scholar] [CrossRef]
- Shishodia, S.; Sethi, G.; Aggarwal, B.B. Curcumin: Getting Back to the Roots. Ann. N. Y. Acad. Sci. 2005, 1056, 206–217. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem. Pharmacol. 2006, 71, 1397–1421. [Google Scholar] [CrossRef]
- Shukla, P.K.; Khanna, V.K.; Ali, M.M.; Khan, M.Y.; Srimal, R.C. Anti-ischemic Effect of Curcumin in Rat Brain. Neurochem. Res. 2008, 33, 1036–1043. [Google Scholar] [CrossRef]
- Ströfer, M.; Jelkmann, W.; Depping, R. Curcumin Decreases Survival of Hep3B Liver and MCF-7 Breast Cancer Cells. Strahlentherapie und Onkol. 2011, 187, 393–400. [Google Scholar] [CrossRef]
- Lev-Ari, S.; Vexler, A.; Starr, A.; Ashkenazy-Voghera, M.; Greif, J.; Aderka, D.; Ben-Yosef, R. Curcumin Augments Gemcitabine Cytotoxic Effect on Pancreatic Adenocarcinoma Cell Lines. Cancer Invest. 2007, 25, 411–418. [Google Scholar] [CrossRef]
- Slusarz, A.; Shenouda, N.S.; Sakla, M.S.; Drenkhahn, S.K.; Narula, A.S.; MacDonald, R.S.; Besch-Williford, C.L.; Lubahn, D.B. Common Botanical Compounds Inhibit the Hedgehog Signaling Pathway in Prostate Cancer. Cancer Res. 2010, 70, 3382–3390. [Google Scholar] [CrossRef]
- Elamin, M.H.; Shinwari, Z.; Hendrayani, S.-F.; Al-Hindi, H.; Al-Shail, E.; Khafaga, Y.; Al-kofide, A.; Aboussekhra, A. Curcumin inhibits the Sonic Hedgehog signaling pathway and triggers apoptosis in medulloblastoma cells. Mol. Carcinog. 2010, 49, 302–314. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Bordoloi, D.; Harsha, C.; Banik, K.; Gupta, S.C.; Aggarwal, B.B. Curcumin mediates anticancer effects by modulating multiple cell signaling pathways. Clin. Sci. 2017, 131, 1781–1799. [Google Scholar] [CrossRef]
- Pandolfi, L.; Bellini, M.; Vanna, R.; Morasso, C.; Zago, A.; Carcano, S.; Avvakumova, S.; Bertolini, J.A.; Rizzuto, M.A.; Colombo, M.; et al. H-Ferritin Enriches the Curcumin Uptake and Improves the Therapeutic Efficacy in Triple Negative Breast Cancer Cells. Biomacromolecules 2017, 18, 3318–3330. [Google Scholar] [CrossRef]
- Chen, W.; Li, L.; Zhang, X.; Liang, Y.; Pu, Z.; Wang, L.; Mo, J. Curcumin: A calixarene derivative micelle potentiates anti-breast cancer stem cells effects in xenografted, triple-negative breast cancer mouse models. Drug Deliv. 2017, 24, 1470–1481. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Xie, C.; Zhu, J.; Meng, Y.; Chen, Y.; Li, Y.; Jiang, Y.; Yang, X.; Wang, S.; et al. Sonic hedgehog and Wnt/β-catenin pathways mediate curcumin inhibition of breast cancer stem cells. Anticancer. Drugs 2018, 29, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Kapinova, A.; Stefanicka, P.; Kubatka, P.; Zubor, P.; Uramova, S.; Kello, M.; Mojzis, J.; Blahutova, D.; Qaradakhi, T.; Zulli, A.; et al. Are plant-based functional foods better choice against cancer than single phytochemicals? A critical review of current breast cancer research. Biomed. Pharmacother. 2017, 96, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Kubatka, P.; Kello, M.; Kajo, K.; Kruzliak, P.; Výbohová, D.; Mojžiš, J.; Adamkov, M.; Fialová, S.; Veizerová, L.; Zulli, A.; et al. Oregano demonstrates distinct tumour-suppressive effects in the breast carcinoma model. Eur. J. Nutr. 2017, 56, 1303–1316. [Google Scholar] [CrossRef]
- Kubatka, P.; Uramova, S.; Kello, M.; Kajo, K.; Kruzliak, P.; Mojzis, J.; Vybohova, D.; Adamkov, M.; Jasek, K.; Lasabova, Z.; et al. Antineoplastic effects of clove buds (Syzygium aromaticum L.) in the model of breast carcinoma. J. Cell. Mol. Med. 2017, 21, 2837–2851. [Google Scholar] [CrossRef] [PubMed]
- Pem, D.; Jeewon, R. Fruit and Vegetable Intake: Benefits and Progress of Nutrition Education Interventions- Narrative Review Article. Iran. J. Public Health 2015, 44, 1309–1321. [Google Scholar]
- Zhang, Y.-J.; Gan, R.-Y.; Li, S.; Zhou, Y.; Li, A.-N.; Xu, D.-P.; Li, H.-B. Antioxidant Phytochemicals for the Prevention and Treatment of Chronic Diseases. Molecules 2015, 20, 21138–21156. [Google Scholar] [CrossRef]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A. Cancer Prevention and Treatment with Resveratrol: From Rodent Studies to Clinical Trials. Cancer Prev. Res. 2009, 2, 409–418. [Google Scholar] [CrossRef]
- Jiang, Z.; Chen, K.; Cheng, L.; Yan, B.; Qian, W.; Cao, J.; Li, J.; Wu, E.; Ma, Q.; Yang, W. Resveratrol and cancer treatment: Updates. Ann. N. Y. Acad. Sci. 2017, 1403, 59–69. [Google Scholar] [CrossRef]
- Singh, C.K.; Ndiaye, M.A.; Ahmad, N. Resveratrol and cancer: Challenges for clinical translation. Biochim. Biophys. Acta-Mol. Basis Dis. 2015, 1852, 1178–1185. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Cantó, C. The molecular targets of resveratrol. Biochim. Biophys. Acta-Mol. Basis Dis. 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Tomé-Carneiro, J.; Larrosa, M.; González-Sarrías, A.; Tomás-Barberán, F.A.; García-Conesa, M.T.; Espín, J.C. Resveratrol and clinical trials: The crossroad from in vitro studies to human evidence. Curr. Pharm. Des. 2013, 19, 6064–6093. [Google Scholar] [CrossRef]
- Kim, C.-W.; Hwang, K.-A.; Choi, K.-C. Anti-metastatic potential of resveratrol and its metabolites by the inhibition of epithelial-mesenchymal transition, migration, and invasion of malignant cancer cells. Phytomedicine 2016, 23, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Sarkar, N.; Biswas, J.; Bishayee, A. Resveratrol for breast cancer prevention and therapy: Preclinical evidence and molecular mechanisms. Semin. Cancer Biol. 2016, 40–41, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Andreani, C.; Bartolacci, C.; Wijnant, K.; Crinelli, R.; Bianchi, M.; Magnani, M.; Hysi, A.; Iezzi, M.; Amici, A.; Marchini, C. Resveratrol fuels HER2 and ERα-positive breast cancer behaving as proteasome inhibitor. Aging 2017, 9, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Yamabe, N.; Zhu, B.T. Resveratrol attenuates the anticancer efficacy of paclitaxel in human breast cancer cells in vitro and in vivo. Eur. J. Cancer 2010, 46, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Choi, H.J.; Zhu, B.T. Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic. Biol. Med. 2010, 49, 800–813. [Google Scholar] [CrossRef]
- Fukui, M.; Yamabe, N.; Kang, K.S.; Zhu, B.T. Growth-stimulatory effect of resveratrol in human cancer cells. Mol. Carcinog. 2010, 49, 750–759. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Mattson, M.P.; Calabrese, V. Resveratrol commonly displays hormesis: Occurrence and biomedical significance. Hum. Exp. Toxicol. 2010, 29, 980–1015. [Google Scholar] [CrossRef]
- Li, Q.; Huyan, T.; Ye, L.-J.; Li, J.; Shi, J.-L.; Huang, Q.-S. Concentration-Dependent Biphasic Effects of Resveratrol on Human Natural Killer Cells in Vitro. J. Agric. Food Chem. 2014, 62, 10928–10935. [Google Scholar] [CrossRef]
- Szende, B.; Tyihak, E.; Kiraly-Veghely, Z. Dose-dependent effect of resveratrol on proliferation and apoptosis in endothelial and tumor cell cultures. Exp. Mol. Med. 2000, 32, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, P.; Satapathy, S.R.; Siddharth, S.; Das, D.; Nayak, A.; Kundu, C.N. Resveratrol and curcumin synergistically induces apoptosis in cigarette smoke condensate transformed breast epithelial cells through a p21Waf1/Cip1 mediated inhibition of Hh-Gli signaling. Int. J. Biochem. Cell Biol. 2015, 66, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, N.; Wang, X.; Cai, C.; Cun, J.; Li, Y.; Lv, S.; Yang, Q. Nitidine chloride induces apoptosis, cell cycle arrest, and synergistic cytotoxicity with doxorubicin in breast cancer cells. Tumor Biol. 2014, 35, 10201–10212. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, N.; Wang, X.; Li, Y.; Qi, W.; Zhang, H.; Li, Z.; Yang, Q. Hedgehog pathway is involved in nitidine chloride induced inhibition of epithelial-mesenchymal transition and cancer stem cells-like properties in breast cancer cells. Cell Biosci. 2016, 6, 44. [Google Scholar] [CrossRef]
- Fan, C.; Wang, Y.; Liu, Z.; Sun, Y.; Wang, X.; Wei, G.; Wei, J. Metformin exerts anticancer effects through the inhibition of the Sonic hedgehog signaling pathway in breast cancer. Int. J. Mol. Med. 2015, 36, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, M.; Meier, C.; Krahenbuhl, S.; Jick, S.S.; Meier, C.R. Long-Term Metformin Use Is Associated With Decreased Risk of Breast Cancer. Diabetes Care 2010, 33, 1304–1308. [Google Scholar] [CrossRef]
- Bosco, J.L.F.; Antonsen, S.; Sorensen, H.T.; Pedersen, L.; Lash, T.L. Metformin and Incident Breast Cancer among Diabetic Women: A Population-Based Case-Control Study in Denmark. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 101–111. [Google Scholar] [CrossRef]
- Kisfalvi, K.; Eibl, G.; Sinnett-Smith, J.; Rozengurt, E. Metformin Disrupts Crosstalk between G Protein-Coupled Receptor and Insulin Receptor Signaling Systems and Inhibits Pancreatic Cancer Growth. Cancer Res. 2009, 69, 6539–6545. [Google Scholar] [CrossRef]
- Hosono, K.; Endo, H.; Takahashi, H.; Sugiyama, M.; Sakai, E.; Uchiyama, T.; Suzuki, K.; Iida, H.; Sakamoto, Y.; Yoneda, K.; et al. Metformin Suppresses Colorectal Aberrant Crypt Foci in a Short-term Clinical Trial. Cancer Prev. Res. 2010, 3, 1077–1083. [Google Scholar] [CrossRef]
- Azoulay, L.; Dell’Aniello, S.; Gagnon, B.; Pollak, M.; Suissa, S. Metformin and the Incidence of Prostate Cancer in Patients with Type 2 Diabetes. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 337–344. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin Selectively Targets Cancer Stem Cells, and Acts Together with Chemotherapy to Block Tumor Growth and Prolong Remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef]
- Zhang, Y.; Bulkley, D.P.; Xin, Y.; Roberts, K.J.; Asarnow, D.E.; Sharma, A.; Myers, B.R.; Cho, W.; Cheng, Y.; Beachy, P.A. Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 2018, 175, 1352–1364. [Google Scholar] [CrossRef]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187. [Google Scholar] [CrossRef]
- Luchetti, G.; Sircar, R.; Kong, J.H.; Nachtergaele, S.; Sagner, A.; Byrne, E.F.; Covey, D.F.; Siebold, C.; Rohatgi, R. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. Elife 2016, 5, e20304. [Google Scholar] [CrossRef]
- Byrne, E.F.X.; Sircar, R.; Miller, P.S.; Hedger, G.; Luchetti, G.; Nachtergaele, S.; Tully, M.D.; Mydock-McGrane, L.; Covey, D.F.; Rambo, R.P.; et al. Structural basis of Smoothened regulation by its extracellular domains. Nature 2016, 535, 517–522. [Google Scholar] [CrossRef]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Scott, M.P. Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8408–8413. [Google Scholar] [CrossRef]
- Nachtergaele, S.; Mydock, L.K.; Krishnan, K.; Rammohan, J.; Schlesinger, P.H.; Covey, D.F.; Rohatgi, R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol. 2012, 8, 211–220. [Google Scholar] [CrossRef]
- Nedelcu, D.; Liu, J.; Xu, Y.; Jao, C.; Salic, A. Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat. Chem. Biol. 2013, 9, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Myers, B.R.; Sever, N.; Chong, Y.C.; Kim, J.; Belani, J.D.; Rychnovsky, S.; Bazan, J.F.; Beachy, P.A. Hedgehog Pathway Modulation by Multiple Lipid Binding Sites on the Smoothened Effector of Signal Response. Dev. Cell 2013, 26, 346–357. [Google Scholar] [CrossRef]
- Kimbung, S.; Chang, C.; Bendahl, P.-O.; Dubois, L.; Thompson, J.W.; McDonnell, D.P.; Borgquist, S. Impact of 27-hydroxylase (CYP27A1) and 27-hydroxycholesterol in breast cancer. Endocr. Relat. Cancer 2017, 24, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Minder, C.M.; Blumenthal, R.S.; Blaha, M.J. Statins for primary prevention of cardiovascular disease. Curr. Opin. Cardiol. 2013, 28, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Mueck, A.O.; Seeger, H.; Wallwiener, D. Effect of statins combined with estradiol on the proliferation of human receptor-positive and receptor-negative breast cancer cells. Menopause 2003, 10, 332–336. [Google Scholar] [CrossRef]
- Kumar, A.S.; Benz, C.C.; Shim, V.; Minami, C.A.; Moore, D.H.; Esserman, L.J. Estrogen Receptor-Negative Breast Cancer Is Less Likely to Arise among Lipophilic Statin Users. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Seeger, H.; Wallwiener, D.; Mueck, A. Statins Can Inhibit Proliferation of Human Breast Cancer Cells in Vitro. Exp. Clin. Endocrinol. Diabetes 2003, 111, 47–48. [Google Scholar] [CrossRef]
- Campbell, M.J.; Esserman, L.J.; Zhou, Y.; Shoemaker, M.; Lobo, M.; Borman, E.; Baehner, F.; Kumar, A.S.; Adduci, K.; Marx, C.; et al. Breast Cancer Growth Prevention by Statins. Cancer Res. 2006, 66, 8707–8714. [Google Scholar] [CrossRef] [PubMed]
- Pocobelli, G.; Newcomb, P.A.; Trentham-Dietz, A.; Titus-Ernstoff, L.; Hampton, J.M.; Egan, K.M. Statin use and risk of breast cancer. Cancer 2008, 112, 27–33. [Google Scholar] [CrossRef]
- Cauley, J.A.; Zmuda, J.M.; Lui, L.-Y.; Hillier, T.A.; Ness, R.B.; Stone, K.L.; Cummings, S.R.; Bauer, D.C. Lipid-Lowering Drug Use and Breast Cancer in Older Women: A Prospective Study. J. Women’s Heal. 2003, 12, 749–756. [Google Scholar] [CrossRef]
- Farwell, W.R.; Scranton, R.E.; Lawler, E.V.; Lew, R.A.; Brophy, M.T.; Fiore, L.D.; Gaziano, J.M. The Association Between Statins and Cancer Incidence in a Veterans Population. JNCI J. Natl. Cancer Inst. 2008, 100, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Karp, I.; Behlouli, H.; LeLorier, J.; Pilote, L. Statins and Cancer Risk. Am. J. Med. 2008, 121, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Blais, L.; Desgagné, A.; LeLorier, J. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors and the risk of cancer: A nested case-control study. Arch. Intern. Med. 2000, 160, 2363–2368. [Google Scholar] [CrossRef]
- Boudreau, D.M.; Gardner, J.S.; Malone, K.E.; Heckbert, S.R.; Blough, D.K.; Daling, J.R. The association between 3-hydroxy-3-methylglutaryl conenzyme A inhibitor use and breast carcinoma risk among postmenopausal women. Cancer 2004, 100, 2308–2316. [Google Scholar] [CrossRef]
- Browning, D.R.L.; Martin, R.M. Statins and risk of cancer: A systematic review and metaanalysis. Int. J. Cancer 2007, 120, 833–843. [Google Scholar] [CrossRef]
- Dale, K.M.; Coleman, C.I.; Henyan, N.N.; Kluger, J.; White, C.M. Statins and Cancer Risk. JAMA 2006, 295, 74. [Google Scholar] [CrossRef]
- Peto, R.; Emberson, J.; Landray, M.; Baigent, C.; Collins, R.; Clare, R.; Califf, R. Analyses of Cancer Data from Three Ezetimibe Trials. N. Engl. J. Med. 2008, 359, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.L.; Wells, B.J.; Smolak, M.J. Statins and cancer: A meta-analysis of case–control studies. Eur. J. Cancer Prev. 2008, 17, 259–268. [Google Scholar] [CrossRef]
- Undela, K.; Srikanth, V.; Bansal, D. Statin use and risk of breast cancer: A meta-analysis of observational studies. Breast Cancer Res. Treat. 2012, 135, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Sprague, J.R.; Wood, M.E. Statins and Breast Cancer Prevention: Time for Randomized Controlled Trials. J. Clin. Oncol. 2006, 24, 2129–2130. [Google Scholar] [CrossRef]
- Prowell, T.M.; Stearns, V.; Trock, B. Lipophilic Statins Merit Additional Study for Breast Cancer Chemoprevention. J. Clin. Oncol. 2006, 24, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Vinayak, S.; Schwartz, E.J.; Jensen, K.; Lipson, J.; Alli, E.; McPherson, L.; Fernandez, A.M.; Sharma, V.B.; Staton, A.; Mills, M.A.; et al. A clinical trial of lovastatin for modification of biomarkers associated with breast cancer risk. Breast Cancer Res. Treat. 2013, 142, 389–398. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Higgins, M.J.; Prowell, T.M.; Blackford, A.L.; Byrne, C.; Khouri, N.F.; Slater, S.A.; Jeter, S.C.; Armstrong, D.K.; Davidson, N.E.; Emens, L.A.; et al. A short-term biomarker modulation study of simvastatin in women at increased risk of a new breast cancer. Breast Cancer Res. Treat. 2012, 131, 915–924. [Google Scholar] [CrossRef]
- Garwood, E.R.; Kumar, A.S.; Baehner, F.L.; Moore, D.H.; Au, A.; Hylton, N.; Flowers, C.I.; Garber, J.; Lesnikoski, B.-A.; Hwang, E.S.; et al. Fluvastatin reduces proliferation and increases apoptosis in women with high grade breast cancer. Breast Cancer Res. Treat. 2010, 119, 137–144. [Google Scholar] [CrossRef]
- Succurro, E.; Arturi, F.; Grembiale, A.; Iorio, F.; Laino, I.; Andreozzi, F.; Sciacqua, A.; Hribal, M.L.; Perticone, F.; Sesti, G. Positive association between plasma IGF1 and high-density lipoprotein cholesterol levels in adult nondiabetic subjects. Eur. J. Endocrinol. 2010, 163, 75–80. [Google Scholar] [CrossRef]
- Colangelo, L.A.; Liu, K.; Gapstur, S.M. CARDIA Male Hormone Study Insulin-like Growth Factor-1, Insulin-like Growth Factor Binding Protein-3, and Cardiovascular Disease Risk Factors in Young Black Men and White Men: The CARDIA Male Hormone Study. Am. J. Epidemiol. 2004, 160, 750–757. [Google Scholar] [CrossRef][Green Version]
- Zhang, K.; Song, L. Association between Vitamin D Receptor Gene Polymorphisms and Breast Cancer Risk: A Meta-Analysis of 39 Studies. PLoS ONE 2014, 9, e96125. [Google Scholar] [CrossRef]
- McCullough, M.L.; Stevens, V.L.; Diver, W.R.; Feigelson, H.S.; Rodriguez, C.; Bostick, R.M.; Thun, M.J.; Calle, E.E. Vitamin D pathway gene polymorphisms, diet, and risk of postmenopausal breast cancer: A nested case-control study. Breast Cancer Res. 2007, 9, R9. [Google Scholar] [CrossRef]
- Abd-Elsalam, E.A.-E.; Ismaeil, N.A.; Abd-Alsalam, H.S. Vitamin D receptor gene polymorphisms and breast cancer risk among postmenopausal Egyptian women. Tumor Biol. 2015, 36, 6425–6431. [Google Scholar] [CrossRef] [PubMed]
- Zinser, G.; Packman, K.; Welsh, J. Vitamin D(3) receptor ablation alters mammary gland morphogenesis. Development 2002, 129, 3067–3076. [Google Scholar] [PubMed]
- Zinser, G.M.; Welsh, J. Effect of Vitamin D3 receptor ablation on murine mammary gland development and tumorigenesis. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 433–436. [Google Scholar] [CrossRef]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef]
- Alimirah, F.; Peng, X.; Gupta, A.; Yuan, L.; Welsh, J.; Cleary, M.; Mehta, R.G. Crosstalk between the vitamin D receptor (VDR) and miR-214 in regulating SuFu, a hedgehog pathway inhibitor in breast cancer cells. Exp. Cell Res. 2016, 349, 15–22. [Google Scholar] [CrossRef]
- Bijlsma, M.F.; Spek, C.A.; Zivkovic, D.; van de Water, S.; Rezaee, F.; Peppelenbosch, M.P. Repression of Smoothened by Patched-Dependent (Pro-)Vitamin D3 Secretion. PLoS Biol. 2006, 4, e232. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, R.; Neefjes, J.; Michalides, R. A Role for Estrogen Receptor Phosphorylation in the Resistance to Tamoxifen. Int. J. Breast Cancer 2011, 2011, 1–10. [Google Scholar] [CrossRef]
- Parl, F.F. Multiple mechanisms of estrogen receptor gene repression contribute to ER-negative breast cancer. Pharmacogenomics J. 2003, 3, 251–253. [Google Scholar] [CrossRef]
- Yang, X.; Phillips, D.L.; Ferguson, A.T.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001, 61, 7025–7029. [Google Scholar]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G Mutation in Estrogen Receptor-α: A Novel Mechanism for Acquired Endocrine Resistance in Breast Cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Ojo, D.; Wei, F.; Liu, Y.; Wang, E.; Zhang, H.; Lin, X.; Wong, N.; Bane, A.; Tang, D. Factors Promoting Tamoxifen Resistance in Breast Cancer via Stimulating Breast Cancer Stem Cell Expansion. Curr. Med. Chem. 2015, 22, 2360–2374. [Google Scholar] [CrossRef] [PubMed]
- Rondón-Lagos, M.; Villegas, V.; Rangel, N.; Sánchez, M.; Zaphiropoulos, P. Tamoxifen Resistance: Emerging Molecular Targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar] [CrossRef] [PubMed]
- Villegas, V.E.; Rondón-Lagos, M.; Annaratone, L.; Castellano, I.; Grismaldo, A.; Sapino, A.; Zaphiropoulos, P.G. Tamoxifen Treatment of Breast Cancer Cells: Impact on Hedgehog/GLI1 Signaling. Int. J. Mol. Sci. 2016, 17, 308. [Google Scholar] [CrossRef] [PubMed]
- Chamoun, Z.; Mann, R.K.; Nellen, D.; von Kessler, D.P.; Bellotto, M.; Beachy, P.A.; Basler, K. Skinny Hedgehog, an Acyltransferase Required for Palmitoylation and Activity of the Hedgehog Signal. Science 2001, 293, 2080–2084. [Google Scholar] [CrossRef]
- Micchelli, C.A.; The, I.; Selva, E.; Mogila, V.; Perrimon, N. Rasp, a putative transmembrane acyltransferase, is required for Hedgehog signaling. Development 2002, 129, 843–851. [Google Scholar] [PubMed]
- Matevossian, A.; Resh, M.D. Hedgehog Acyltransferase as a target in estrogen receptor positive, HER2 amplified, and tamoxifen resistant breast cancer cells. Mol. Cancer 2015, 14, 72. [Google Scholar] [CrossRef]
- Sims-Mourtada, J.; Opdenaker, L.M.; Davis, J.; Arnold, K.M.; Flynn, D. Taxane-induced hedgehog signaling is linked to expansion of breast cancer stem-like populations after chemotherapy. Mol. Carcinog. 2015, 54, 1480–1493. [Google Scholar] [CrossRef]
- Wong, S.Y.; Seol, A.D.; So, P.-L.; Ermilov, A.N.; Bichakjian, C.K.; Epstein, E.H.; Dlugosz, A.A.; Reiter, J.F. Primary cilia can both mediate and suppress Hedgehog pathway–dependent tumorigenesis. Nat. Med. 2009, 15, 1055–1061. [Google Scholar] [CrossRef]
- Laurberg, T.; Tramm, T.; Nielsen, T.; Alsner, J.; Nord, S.; Myhre, S.; Sørlie, T.; Leung, S.; Fan, C.; Perou, C.; et al. Intrinsic subtypes and benefit from postmastectomy radiotherapy in node-positive premenopausal breast cancer patients who received adjuvant chemotherapy—results from two independent randomized trials. Acta Oncol. 2018, 57, 38–43. [Google Scholar] [CrossRef] [PubMed]


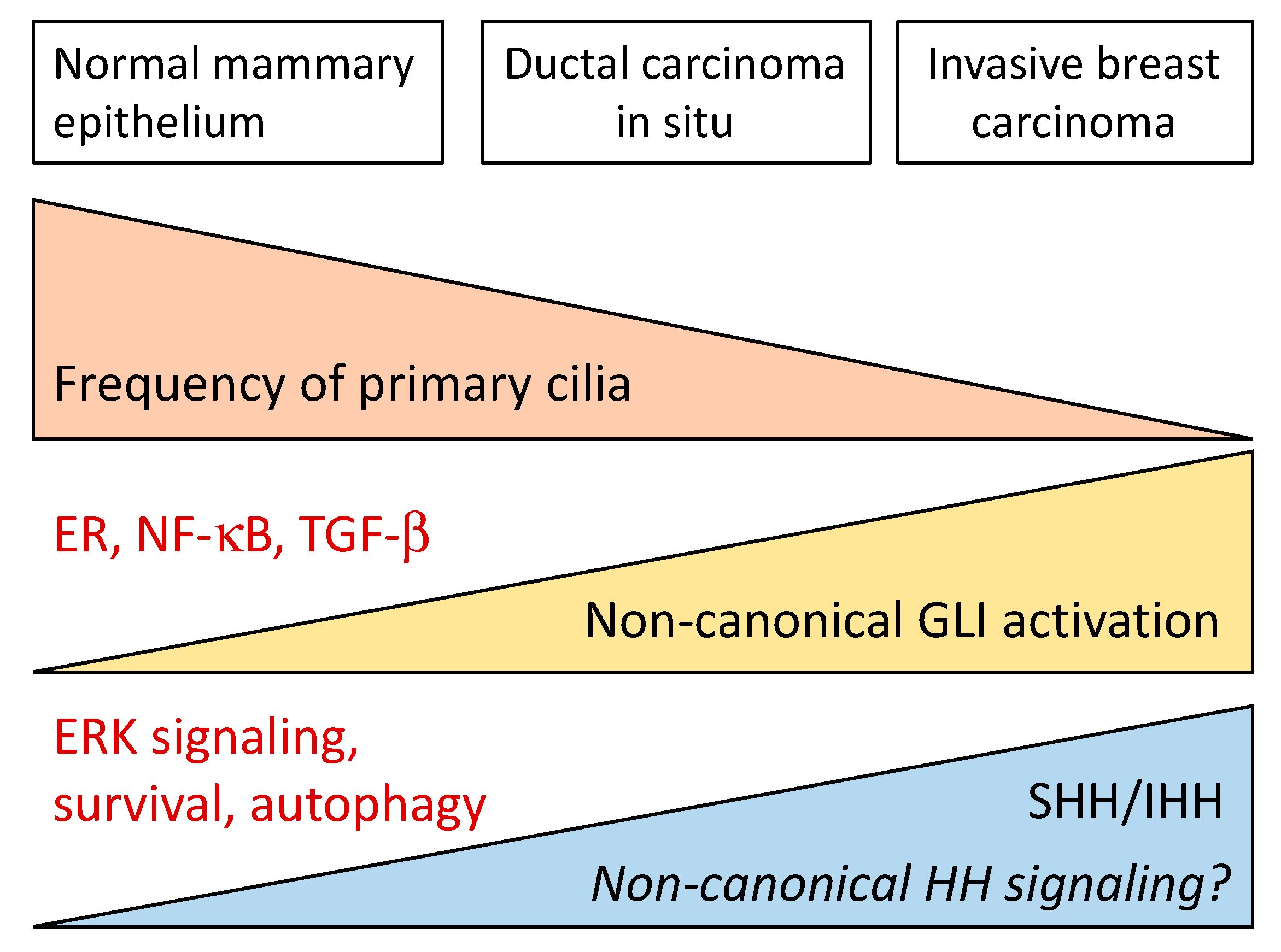{kind=link}
| SHH | DHH | PTCH | SMO | GLI1 | |
|---|---|---|---|---|---|
| ER+ | Overexpressed [4] Positive correlation [4] No correlation [79,88,194,197,198] | Overexpressed [4] Positive correlation [4] | Overexpressed [4] Positive correlation [4] | n.d. | Overexpressed [4] Positive correlation [4,75,194,195,196] High % nuclear staining [102,111] Negative correlation [10,199] No correlation [103,197,198] |
| HER2+ | Overexpressed [197] No correlation [197] No correlation [4,79,88,194,195] | No correlation [4,75,194] | No correlation [75,194,195] | n.d. | Overexpressed [197] Positive correlation [197,198] No correlation [4,75,103,194,195] |
| TNBC | Overexpressed [4,79,195] | Overexpressed [4] | Overexpressed [4] | Overexpressed [4,10] Increased (respect to ER+ and HER2+) [10] | Overexpressed [4,10,102,199] Increased (respect to ER+ and HER2+) [10] High % nuclear staining [199] |
| SHH | DHH | PTCH1 | SMO | GLI1 | ||
|---|---|---|---|---|---|---|
| Expression | Non neoplastic | Low [10,88,203,208] | Low [203] | Low [4,10,67,75,96] | Low or absent [10,67,198] | Low or Absent [4,10,198,199,203] |
| DCIS | Increased * [10,88,111] | Increased * [4] | Low or absent [10,67,96] Increased * [4,75,198] | Increased * [10,67,198] | Increased * [4,10,75,88,111,198,199] | |
| IDC | Increased * [10,79,88,111] | Increased * [4] | Low or absent [10,67,96] Increased * [4,75,79,198] | Increased * [10,67,198] | Increased * [10,88,111,198,199] | |
| Correlations | ER | Positive [195] None [3,9,12,14,15] | Positive [75,194,195]) | Positive [96] None [198] | None [198] | Positive [75,194,195,196] None [103,197,198] Negative [10,199] |
| HER2 | None [3,8,12,14,15] | None [7,8,15] | None [75,194,195] | n.d. | Positive [4,9,14] None [8,15,17] | |
| Ki-67 | Positive [4,79,195] | Positive [4] | Positive [4] | n.d. | Positive [4] None [199] | |
| Age | Positive [4] None [88] | Positive [4] | n.d. | Positive [4] None [4]) | Positive [4] None [199] | |
| Histological type | Positive [88] | n.d. | n.d. | n.d. | None [199] | |
| Histological grade | Positive [4,10,79,195,197] | Positive [4] | Positive [67] | Positive [10,67] | Positive [4,197] None [199] | |
| Tumor size | Positive [4,79,198] None [88] | Positive [4] | Positive [198] Negative [96] | Positive [198] | Positive [4] None [197,198] | |
| Early disease onset | Positive [4] | Positive [4] | n.d. | Positive [4] | Positive [4] | |
| Stage | Positive [4,88,195] | Positive [4] | n.d. | Positive [10] | Positive [10,103] None [199] | |
| Lymph node involvement | Positive [4,200] None [88] | Positive [4] | Negative [96] | Positive [198] | Positive [4,10,103] None [197] | |
| Distant metastasis | Positive [4,79,195] | Positive [4] | n.d. | None [4]) | Positive [8,16] | |
| Pre-menopause | Positive [4] | Positive [4] | n.d. | n.d. | Positive [4] | |
| Post-menopause | Positive [195] | n.d. | Positive [4] | n.d. | n.d. | |
| Poor prognosis | Positive [8,9,12,13,19,20,21] | Positive [4] | Positive [4] | Positive [4] | Positive [4,98,103,199] | |
| Recurrence | Positive [198,200,202] | Positive [198] | Positive [198] | Positive [198] | Positive [198,199] None [103] | |
| Independent predictor | ER, age, DM, OS [4,195] | ER, age, DM, mortality, OS [4,195] | Age, mortality [4] | n.d. | ER, age, DM, OS [4] | ER, mortality [4] |
| Treatment | Target | Location | Setting | Clinical Status | Status | Number of Patients | Trial Identifier | Reference |
|---|---|---|---|---|---|---|---|---|
| Vismodegib + RO4929097s | Smoothened Antagonist + Gamma Secretase Inhibitor | United States | Advanced BC (metastatic or unresectable) | Phase I | T | 13 | NCT01071564 | No results posted |
| Neoadjuvant Vismodegib + Paclitaxel + Epirubicin + Cyclophosphamide | Smoothened Antagonist + antimicrotubule agent +Topoisomerase inhibitor + antineoplastic agent | Spain | TNBC | Phase II | R | 40 | NCT02694224 | No results posted |
| Sonidegib (LDE225) + Paclitaxel | Smoothened Antagonist + antimicrotubule agent | Switzerland | Solid tumors | Phase I | C | 2 BC out of 18 | NCT01954355 | [230] |
| Sonidegib (LDE225) + Docetaxel | Smoothened Antagonist + antimicrotubule agent | Spain | Advanced TNBC | Phase I | C | 12 | NCT02027376 | [231] |
| Sonidegib (LDE225) + BKM120 | Smoothened Antagonist + PI3K Inhibitor | Australia, Europe and United States | Solid tumors including TNBC and ER/PR+/Her2- metastatic BC | Phase Ib | C | 120 | NCT01576666 | No results posted |
| Erismodegib (LDE225) | Smoothened Antagonist | United states | TNBC | Phase II | W (Poor accrual) | 68 | NCT01757327 | No results posted |
| Itraconazole | Anti-angiogenesis and HH pathway inhibitor | United States | Metastatic BC | Pilot trial | C | 13 | NCT00798135 | [237] |
| Vismodegib + Rabeprazole or Itraconazole or Fluconazole | Smoothened Antagonist + proton pump inhibitor or antifungal drug | United States | Healthy volunteer | Phase I | C | 92 | NCT01772290 | [238] |
| Arsenic Trioxide | HH/Gli pathway inhibitor | United States | Advanced BC | Phase II | W (no subjects recruited) | 0 | NCT00075413 | No results posted |
| Curcumin | NF-kB DNA binding Inhibitor | United States | BC | Phase II | C | 3 | NCT01740323 | No results posted |
| Curcumin (DS) + Docetaxel | Dietary phytonutrient + antimicrotubule agent | France | Her2- advanced or metastatic BC | Phase II | T | 42 | NCT00852332 | No results posted |
| Curcumin (iv) + Paclitaxel | Antiproliferative, anti-invasive, and antiangiogenic + antimicrotubule agent | Armenia | Advanced and Metastatic BC | Phase II | R | 75 | NCT03072992 | No results posted |
| Curcumin (DS) | Anti-inflammatory effect | United States | obese women at high risk for breast cancer | Pilot trial | ANR | 30 | NCT01975363 | No results posted |
| Atorvastatin | Cholesterol synthesis inhibitor | United States | Pre-menopausal women with a strong family history of breast and/or ovarian cancer | Phase II | C | 100 | NCT00914017 | [239] |
| Vitamin D3 | Smoothened inhibitor | Canada | Primary BC | Phase II | C | 83 | NCT01948128 | No results posted |
| Vitamin D3 + neoadjuvant Progesterone | Apoptotic agent + antiproliferative, cytotoxic | India | Large Operable and Locally Advanced BC | Phase III | ANR | 800 | NCT01608451 | No results posted |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riobo-Del Galdo, N.A.; Lara Montero, Á.; Wertheimer, E.V. Role of Hedgehog Signaling in Breast Cancer: Pathogenesis and Therapeutics. Cells 2019, 8, 375. https://doi.org/10.3390/cells8040375
Riobo-Del Galdo NA, Lara Montero Á, Wertheimer EV. Role of Hedgehog Signaling in Breast Cancer: Pathogenesis and Therapeutics. Cells. 2019; 8(4):375. https://doi.org/10.3390/cells8040375
Chicago/Turabian StyleRiobo-Del Galdo, Natalia A., Ángela Lara Montero, and Eva V. Wertheimer. 2019. "Role of Hedgehog Signaling in Breast Cancer: Pathogenesis and Therapeutics" Cells 8, no. 4: 375. https://doi.org/10.3390/cells8040375
APA StyleRiobo-Del Galdo, N. A., Lara Montero, Á., & Wertheimer, E. V. (2019). Role of Hedgehog Signaling in Breast Cancer: Pathogenesis and Therapeutics. Cells, 8(4), 375. https://doi.org/10.3390/cells8040375