Mitochondrial Genetic Disorders: Cell Signaling and Pharmacological Therapies


Abstract
:1. Mitochondrial Energy Metabolism Disorders
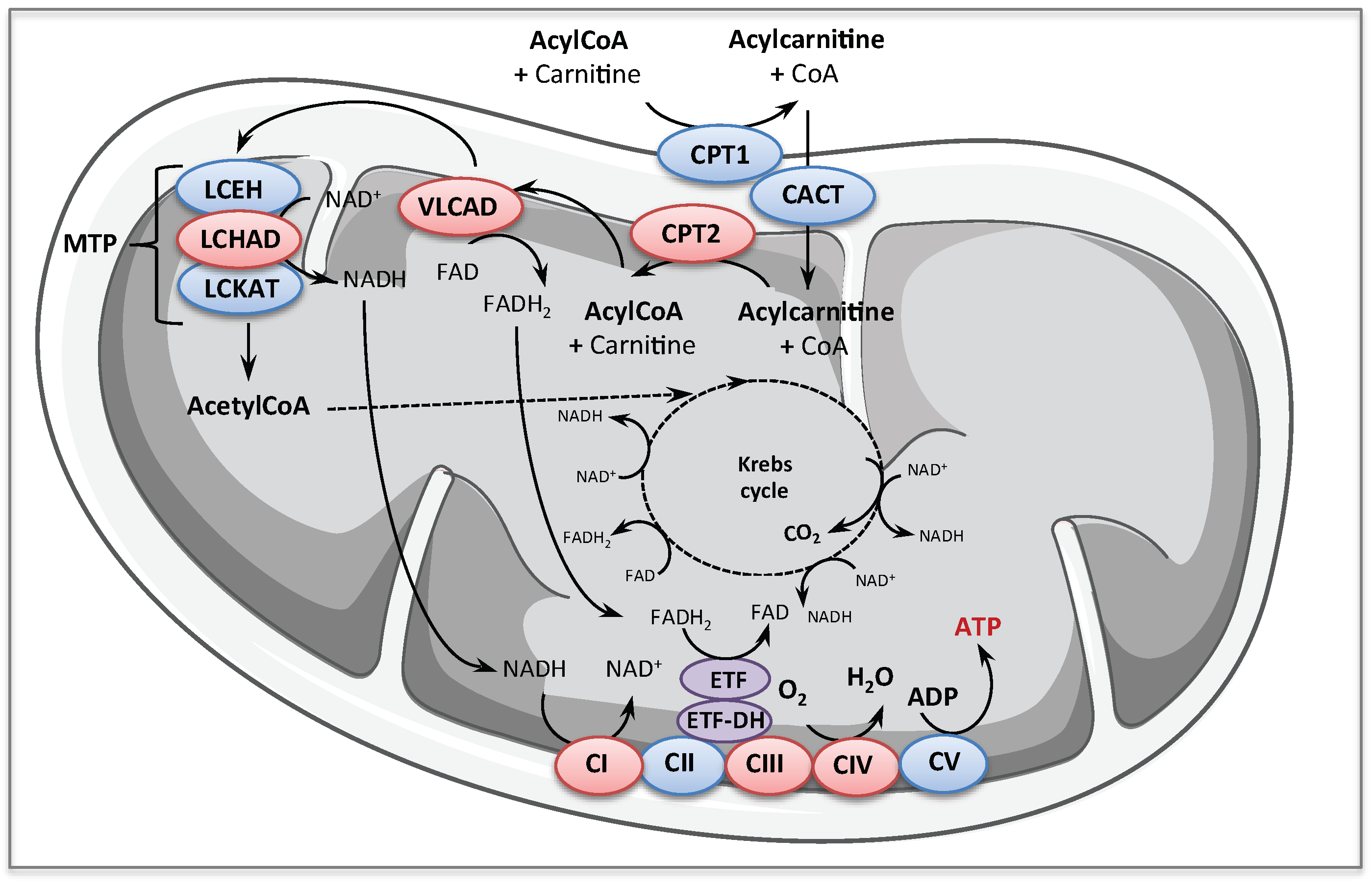
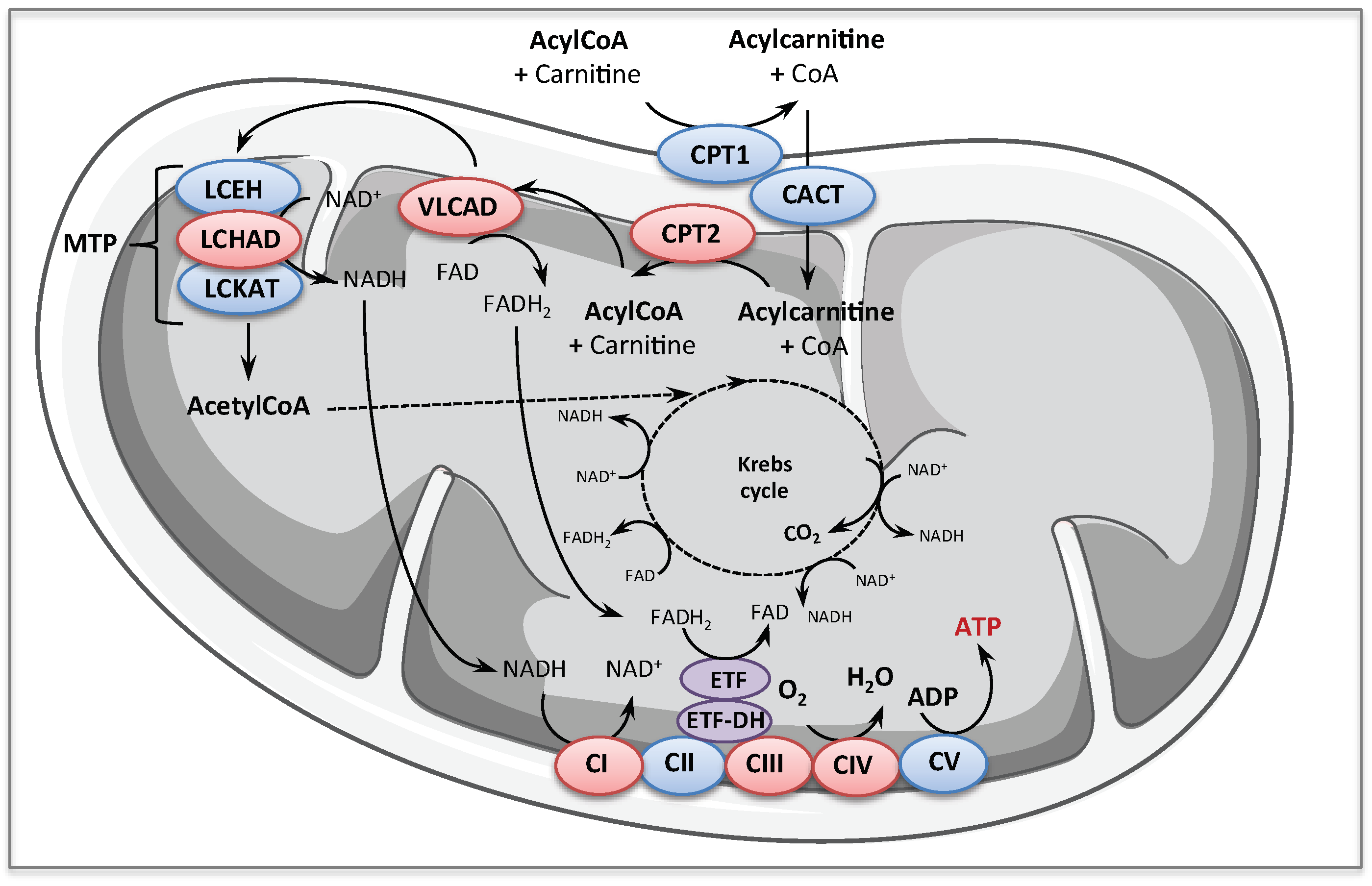
1.1. Fatty Acid Oxidation Disorders
1.2. Respiratory Chain Deficiencies
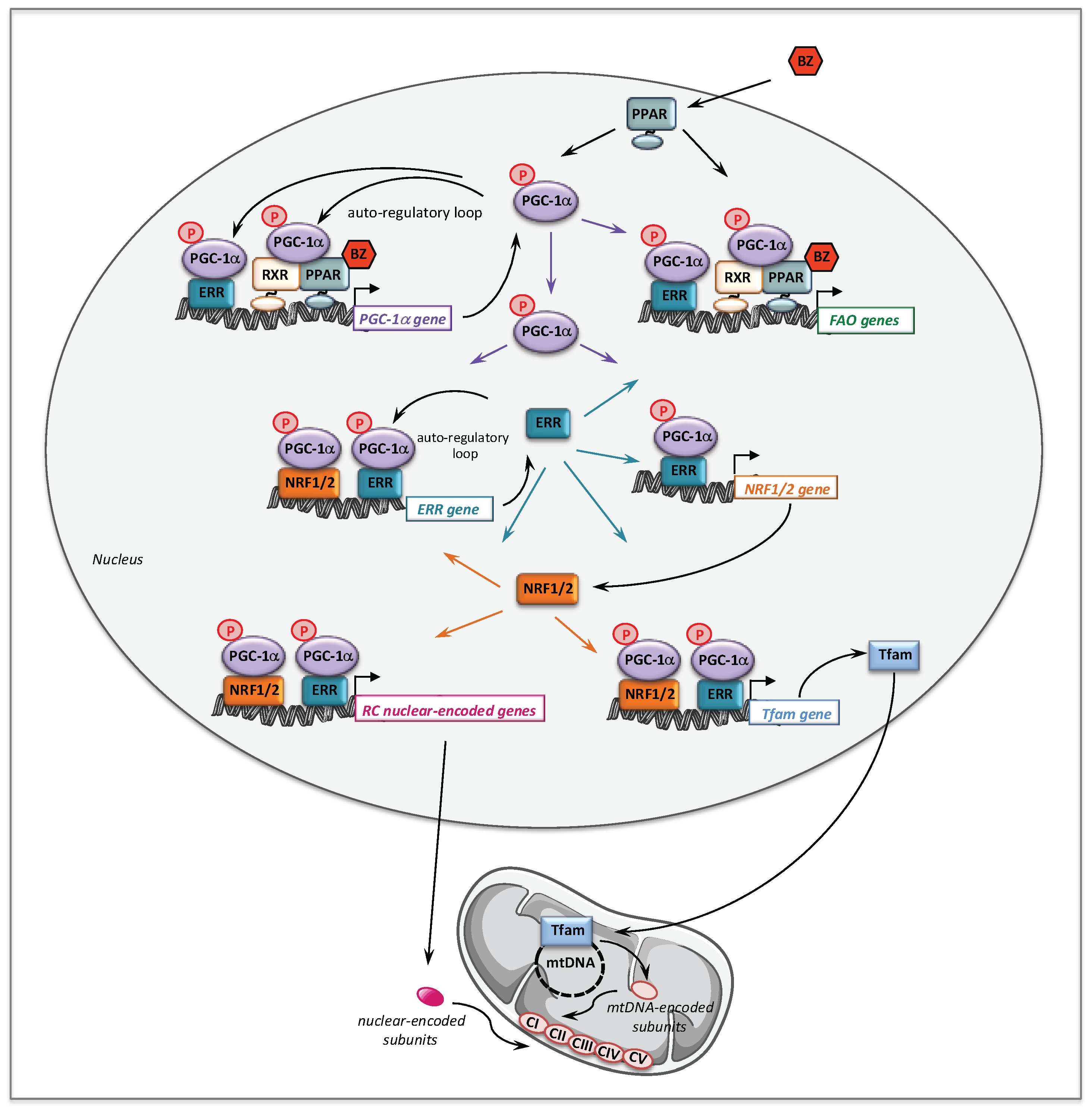
2. Bezafibrate
2.1. Bezafibrate and Fatty Acid Oxidation Disorders
2.2. Bezafibrate and Respiratory Chain Disorders
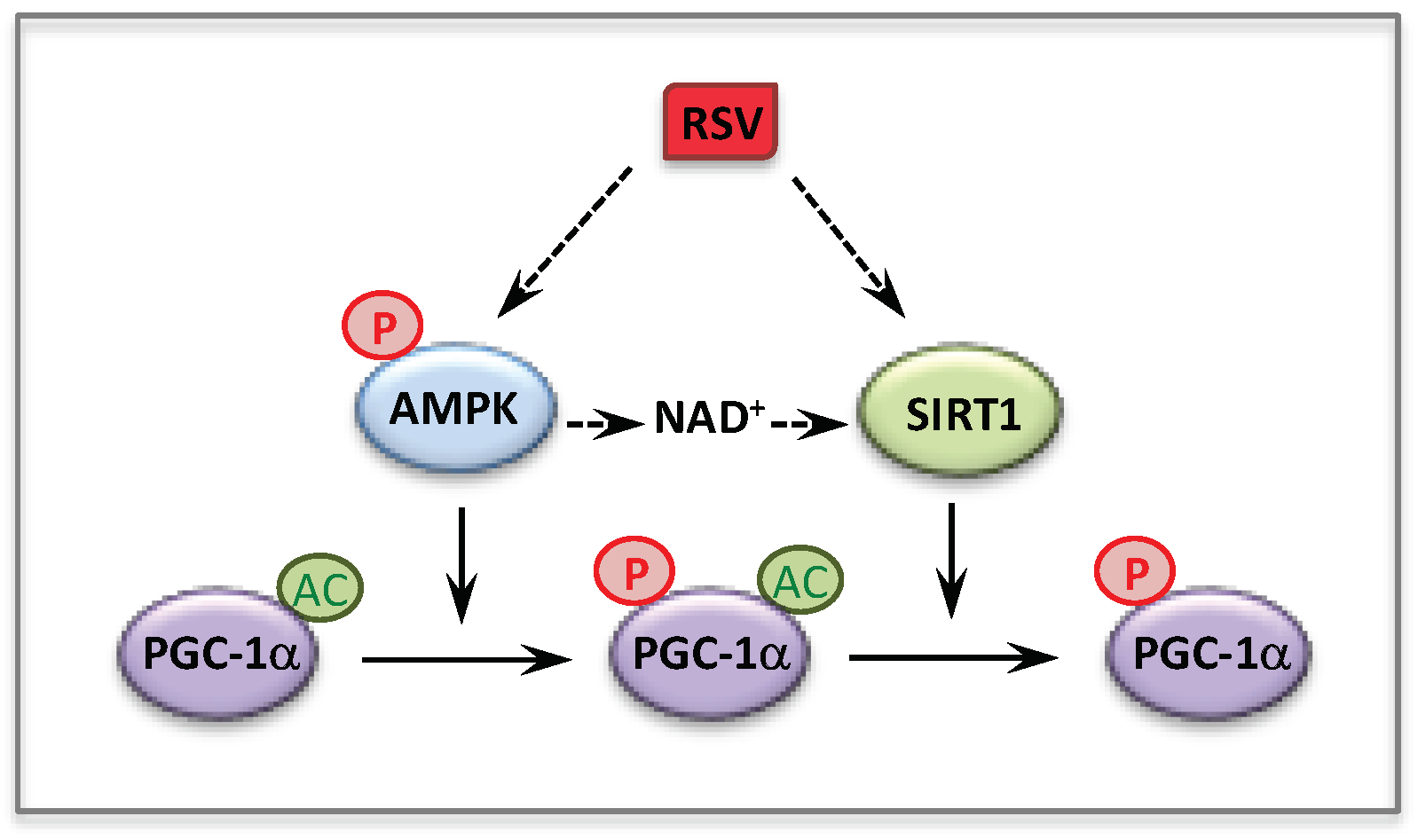
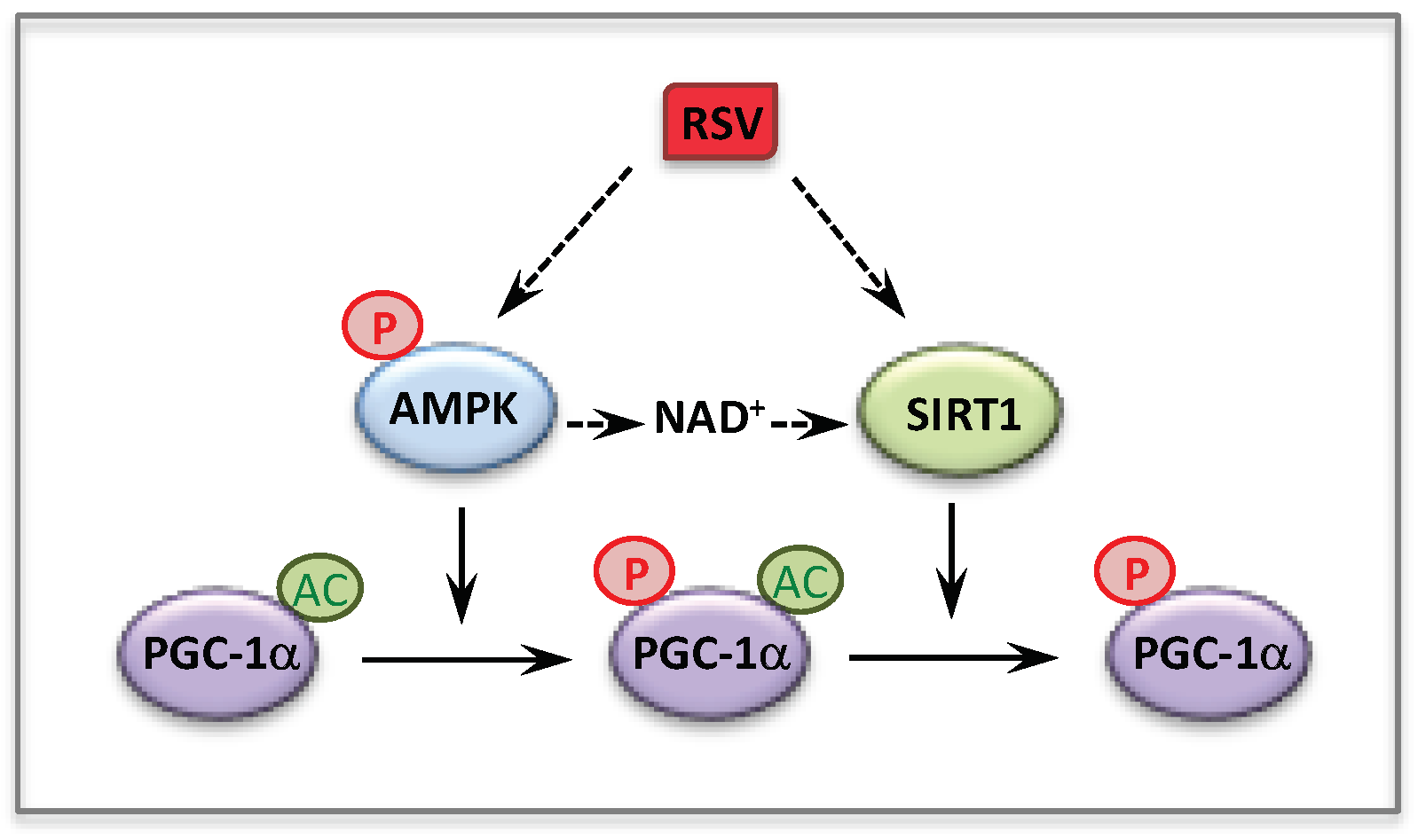
3. Resveratrol
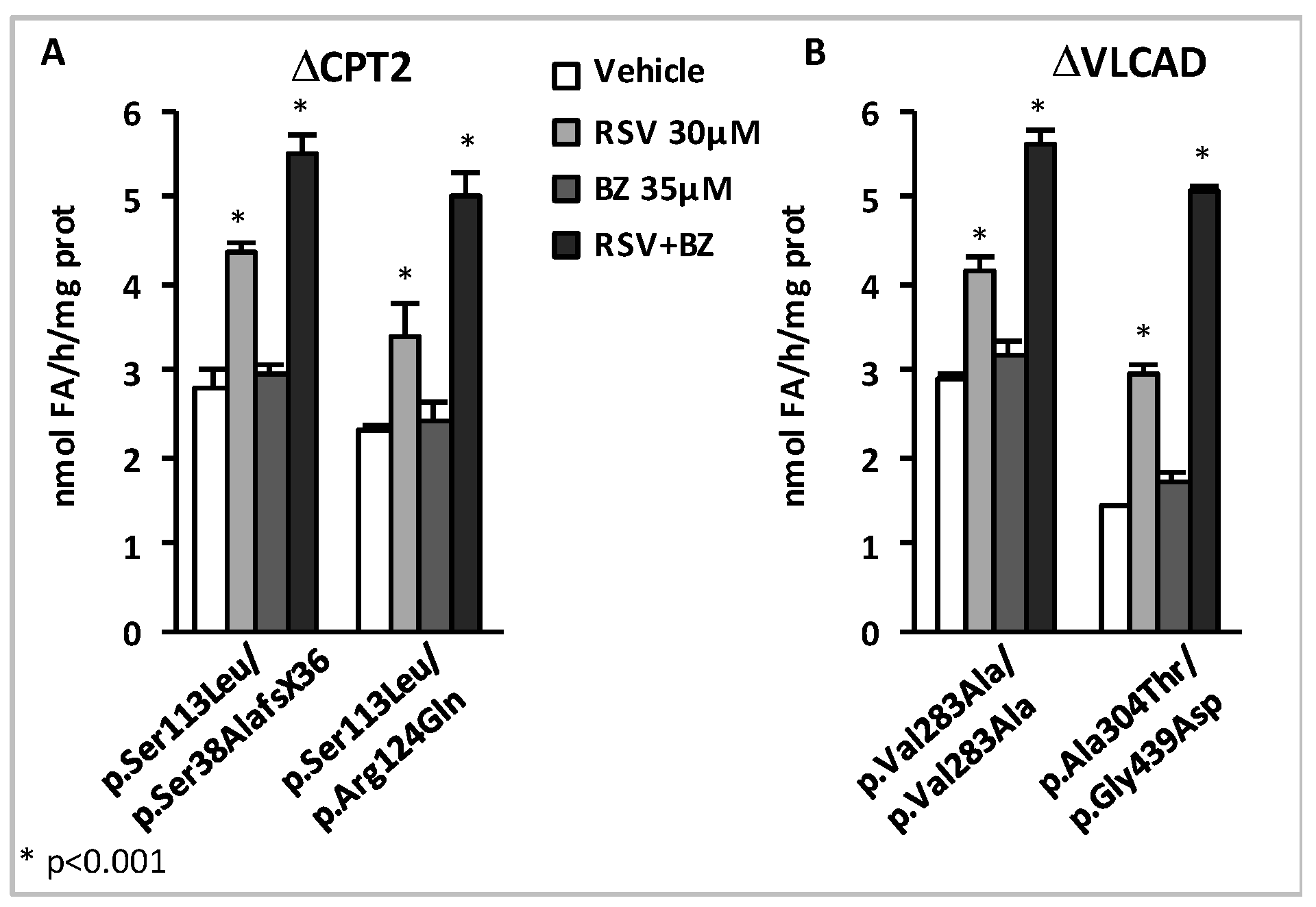
3.1. Resveratrol and Fatty Acid Oxidation Disorders
3.2. Resveratrol and Respiratory Chain Disorders
4. Effects of BZ + RSV Combinations
5. AMPK Activators
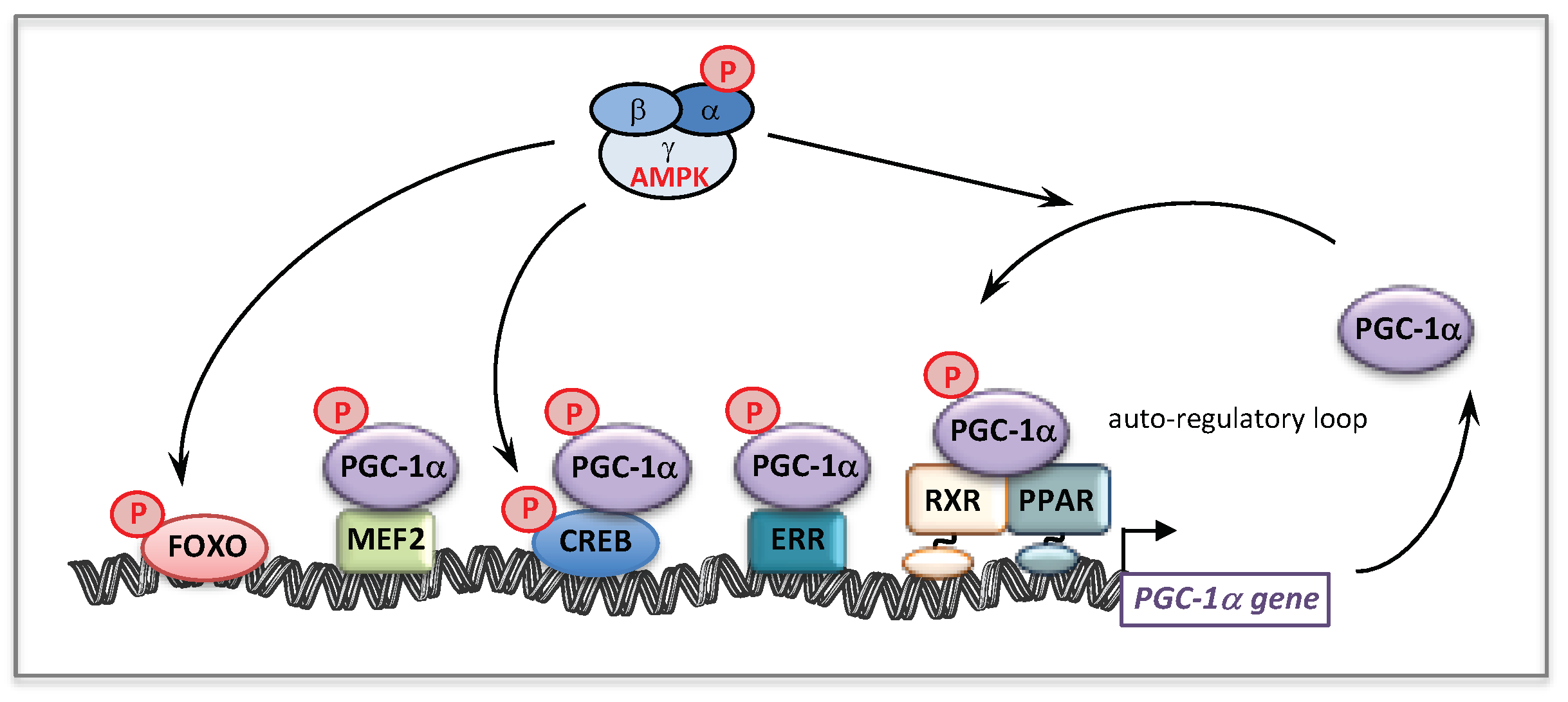
5.1. AMP-Activated Protein Kinase (AMPK)
5.2. AMPK and Mitochondrial Homeostasis
5.3. AMPK Activators and Fatty Acid Oxidation Disorders
5.4. AMPK Activators and Respiratory Chain Disorders
5.5. Others Compounds Tested
6. Antioxidants
6.1. Antioxidants and Fatty Acid Oxidation Disorders
6.2. Antioxidants and Respiratory Chain Disorders
7. NAD+ Precursors
8. Protein Kinase A (PKA) Agonists
9. Other Molecules, Other Pathways
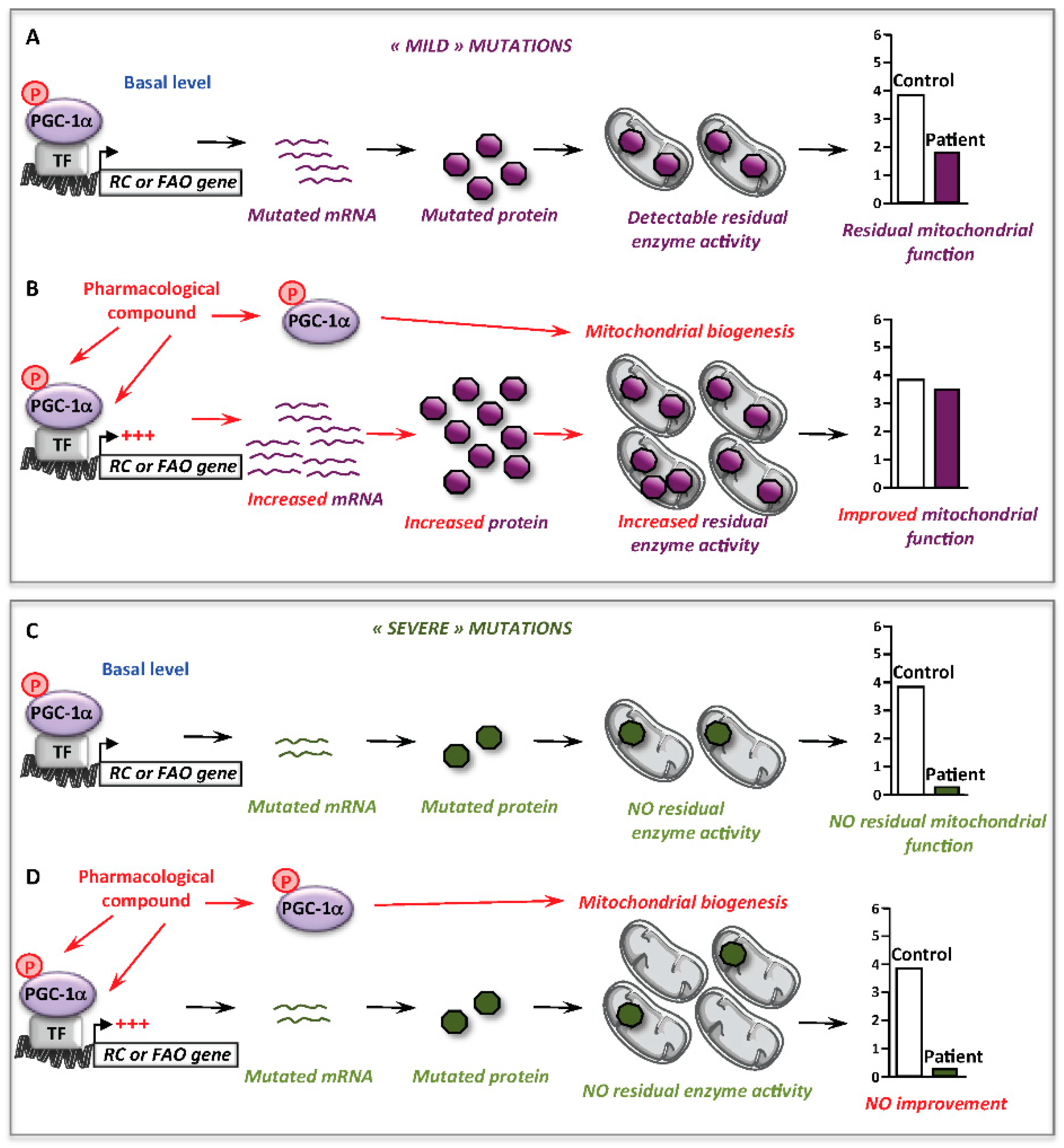
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Patti, M.E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R. Energy metabolism disorders in rare and common diseases. Toward bioenergetic modulation therapy and the training of a new generation of European scientists. Int. J. Biochem. Cell Biol. 2015, 63, 2–9. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef]
- Koopman, W.J.; Beyrath, J.; Fung, C.W.; Koene, S.; Rodenburg, R.J.; Willems, P.H.; Smeitink, J.A. Mitochondrial disorders in children: Toward development of small-molecule treatment strategies. EMBO Mol. Med. 2016, 8, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Viscomi, C. Towards a therapy for mitochondrial disease: An update. Biochem. Soc. Trans. 2018, 46, 1247–1261. [Google Scholar] [CrossRef]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2015. [Google Scholar] [CrossRef]
- Knottnerus, S.J.G.; Bleeker, J.C.; Wust, R.C.I.; Ferdinandusse, S.; IJlst, L.; Wijburg, F.A.; Wanders, R.J.A.; Visser, G.; Houtkooper, R.H. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. 2018, 19, 93–106. [Google Scholar] [CrossRef]
- Baruteau, J.; Sachs, P.; Broue, P.; Brivet, M.; Abdoul, H.; Vianey-Saban, C.; Ogier de Baulny, H. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: A French pediatric study from 187 patients. Complementary data. J. Inherit. Metab. Dis. 2014, 37, 137–139. [Google Scholar] [CrossRef]
- Merritt, J.L., 2nd; Norris, M.; Kanungo, S. Fatty acid oxidation disorders. Ann. Transl. Med. 2018, 6, 473. [Google Scholar] [CrossRef]
- Olpin, S.E. Pathophysiology of fatty acid oxidation disorders and resultant phenotypic variability. J. Inherit. Metab. Dis. 2013, 36, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Kim, Y.M.; Kang, M.; Heo, S.H.; Kim, G.H.; Choi, I.H.; Choi, J.H.; Yoo, H.W.; Lee, B.H. Clinical and genetic characteristics of patients with fatty acid oxidation disorders identified by newborn screening. BMC Pediatr. 2018, 18, 103. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Burrage, L.C.; Gibson, J.B.; Strenk, M.E.; Lose, E.J.; Bick, D.P.; Elsea, S.H.; Sutton, V.R.; Sun, Q.; Graham, B.H.; et al. Recurrent ACADVL molecular findings in individuals with a positive newborn screen for very long chain acyl-coA dehydrogenase (VLCAD) deficiency in the United States. Mol. Genet. Metab. 2015, 116, 139–145. [Google Scholar] [CrossRef]
- Yamada, K.; Taketani, T. Management and diagnosis of mitochondrial fatty acid oxidation disorders: Focus on very-long-chain acyl-CoA dehydrogenase deficiency. J. Hum. Genet. 2019, 64, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, L.; Wang, B.; Liu, S.; Yu, B.; Wang, T. Application of Next-Generation Sequencing Following Tandem Mass Spectrometry to Expand Newborn Screening for Inborn Errors of Metabolism: A Multicenter Study. Front. Genet. 2019, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Rufer, A.C.; Thoma, R.; Benz, J.; Stihle, M.; Gsell, B.; De Roo, E.; Banner, D.W.; Mueller, F.; Chomienne, O.; Hennig, M. The crystal structure of carnitine palmitoyltransferase 2 and implications for diabetes treatment. Structure 2006, 14, 713–723. [Google Scholar] [CrossRef]
- Isackson, P.J.; Bennett, M.J.; Lichter-Konecki, U.; Willis, M.; Nyhan, W.L.; Sutton, V.R.; Tein, I.; Vladutiu, G.D. CPT2 gene mutations resulting in lethal neonatal or severe infantile carnitine palmitoyltransferase II deficiency. Mol. Genet. Metab. 2008, 94, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont, J.P.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Asp. Med. 2004, 25, 495–520. [Google Scholar] [CrossRef]
- Jernberg, J.N.; Bowman, C.E.; Wolfgang, M.J.; Scafidi, S. Developmental regulation and localization of carnitine palmitoyltransferases (CPTs) in rat brain. J. Neurochem. 2017, 142, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the central nervous system. BioMed Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef]
- Knobloch, M.; Pilz, G.A.; Ghesquiere, B.; Kovacs, W.J.; Wegleiter, T.; Moore, D.L.; Hruzova, M.; Zamboni, N.; Carmeliet, P.; Jessberger, S. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep. 2017, 20, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Jones, A.; Deeney, J.T.; Hur, S.K.; Bankaitis, V.A. Inborn Errors of Long-Chain Fatty Acid beta-Oxidation Link Neural Stem Cell Self-Renewal to Autism. Cell Rep. 2016, 14, 991–999. [Google Scholar] [CrossRef] [PubMed]
- McAndrew, R.P.; Wang, Y.; Mohsen, A.W.; He, M.; Vockley, J.; Kim, J.J. Structural basis for substrate fatty acyl chain specificity: Crystal structure of human very-long-chain acyl-CoA dehydrogenase. J. Biol. Chem. 2008, 283, 9435–9443. [Google Scholar] [CrossRef] [PubMed]
- Chegary, M.; Brinke, H.; Ruiter, J.P.; Wijburg, F.A.; Stoll, M.S.; Minkler, P.E.; van Weeghel, M.; Schulz, H.; Hoppel, C.L.; Wanders, R.J.; et al. Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim. Biophys. Acta 2009, 1791, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Hesse, J.; Braun, C.; Behringer, S.; Matysiak, U.; Spiekerkoetter, U.; Tucci, S. The diagnostic challenge in very-long chain acyl-CoA dehydrogenase deficiency (VLCADD). J. Inherit. Metab. Dis. 2018, 41, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Andresen, B.S.; Nation, J.; Boneh, A. VLCAD deficiency: Follow-up and outcome of patients diagnosed through newborn screening in Victoria. Mol. Genet. Metab. 2016, 118, 282–287. [Google Scholar] [CrossRef]
- Pena, L.D.; van Calcar, S.C.; Hansen, J.; Edick, M.J.; Walsh Vockley, C.; Leslie, N.; Cameron, C.; Mohsen, A.W.; Berry, S.A.; Arnold, G.L.; et al. Outcomes and genotype–phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol. Genet. Metab. 2016, 118, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Janeiro, P.; Jotta, R.; Ramos, R.; Florindo, C.; Ventura, F.V.; Vilarinho, L.; Tavares de Almeida, I.; Gaspar, A. Follow-up of fatty acid beta-oxidation disorders in expanded newborn screening era. Eur. J. Pediatr. 2019. [Google Scholar] [CrossRef]
- Bleeker, J.C.; Kok, I.L.; Ferdinandusse, S.; van der Pol, W.L.; Cuppen, I.; Bosch, A.M.; Langeveld, M.; Derks, T.G.J.; Williams, M.; de Vries, M.; et al. Impact of NBS for VLCAD deficiency on genetic, enzymatic and clinical outcomes. J. Inherit. Metab. Dis. 2019. [Google Scholar] [CrossRef]
- Andresen, B.S.; Olpin, S.; Poorthuis, B.J.; Scholte, H.R.; Vianey-Saban, C.; Wanders, R.; Ijlst, L.; Morris, A.; Pourfarzam, M.; Bartlett, K.; et al. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am. J. Hum. Genet. 1999, 64, 479–494. [Google Scholar] [CrossRef]
- Diekman, E.F.; Ferdinandusse, S.; van der Pol, L.; Waterham, H.R.; Ruiter, J.P.; Ijlst, L.; Wanders, R.J.; Houten, S.M.; Wijburg, F.A.; Blank, A.C.; et al. Fatty acid oxidation flux predicts the clinical severity of VLCAD deficiency. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 989–994. [Google Scholar] [CrossRef]
- Gobin-Limballe, S.; Djouadi, F.; Aubey, F.; Olpin, S.; Andresen, B.S.; Yamaguchi, S.; Mandel, H.; Fukao, T.; Ruiter, J.P.; Wanders, R.J.; et al. Genetic basis for correction of very-long-chain acyl-coenzyme A dehydrogenase deficiency by bezafibrate in patient fibroblasts: Toward a genotype-based therapy. Am. J. Hum. Genet. 2007, 81, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Fould, B.; Garlatti, V.; Neumann, E.; Fenel, D.; Gaboriaud, C.; Arlaud, G.J. Structural and functional characterization of the recombinant human mitochondrial trifunctional protein. Biochemistry 2010, 49, 8608–8617. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, U.; Khuchua, Z.; Yue, Z.; Bennett, M.J.; Strauss, A.W. General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits similar phenotypes because mutations in either subunit alter TFP complex expression and subunit turnover. Pediatr. Res. 2004, 55, 190–196. [Google Scholar] [CrossRef]
- Olpin, S.E. Fatty acid oxidation defects as a cause of neuromyopathic disease in infants and adults. Clin. Lab. 2005, 51, 289–306. [Google Scholar]
- Purevsuren, J.; Fukao, T.; Hasegawa, Y.; Kobayashi, H.; Li, H.; Mushimoto, Y.; Fukuda, S.; Yamaguchi, S. Clinical and molecular aspects of Japanese patients with mitochondrial trifunctional protein deficiency. Mol. Genet. Metab. 2009, 98, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem. 2018, 62, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genomics Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef]
- Koopman, W.J.; Nijtmans, L.G.; Dieteren, C.E.; Roestenberg, P.; Valsecchi, F.; Smeitink, J.A.; Willems, P.H. Mammalian mitochondrial complex I: Biogenesis, regulation, and reactive oxygen species generation. Antioxid. Redox Signal. 2010, 12, 1431–1470. [Google Scholar] [CrossRef] [PubMed]
- Fassone, E.; Rahman, S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Hoefs, S.J.; Rodenburg, R.J.; Smeitink, J.A.; van den Heuvel, L.P. Molecular base of biochemical complex I deficiency. Mitochondrion 2012, 12, 520–532. [Google Scholar] [CrossRef]
- Valsecchi, F.; Koopman, W.J.; Manjeri, G.R.; Rodenburg, R.J.; Smeitink, J.A.; Willems, P.H. Complex I disorders: Causes, mechanisms, and development of treatment strategies at the cellular level. Dev. Disabil. Res. Rev. 2010, 16, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Ugalde, C.; Janssen, R.J.; van den Heuvel, L.P.; Smeitink, J.A.; Nijtmans, L.G. Differences in assembly or stability of complex I and other mitochondrial OXPHOS complexes in inherited complex I deficiency. Hum. Mol. Genet. 2004, 13, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Koene, S.; Rodenburg, R.J.; van der Knaap, M.S.; Willemsen, M.A.; Sperl, W.; Laugel, V.; Ostergaard, E.; Tarnopolsky, M.; Martin, M.A.; Nesbitt, V.; et al. Natural disease course and genotype–phenotype correlations in Complex I deficiency caused by nuclear gene defects: What we learned from 130 cases. J. Inherit. Metab. Dis. 2012, 35, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, K.; Sofou, K.; Darin, N.; Holme, E.; Kollberg, G.; Asin-Cayuela, J.; Holmberg Dahle, K.M.; Oldfors, A.; Moslemi, A.R.; Tulinius, M. Broad phenotypic variability in patients with complex I deficiency due to mutations in NDUFS1 and NDUFV1. Mitochondrion 2015, 21, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Antonicka, H.; Ogilvie, I.; Taivassalo, T.; Anitori, R.P.; Haller, R.G.; Vissing, J.; Kennaway, N.G.; Shoubridge, E.A. Identification and characterization of a common set of complex I assembly intermediates in mitochondria from patients with complex I deficiency. J. Biol. Chem. 2003, 278, 43081–43088. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Verkaart, S.; Visch, H.J.; van Emst-de Vries, S.; Nijtmans, L.G.; Smeitink, J.A.; Willems, P.H. Human NADH:ubiquinone oxidoreductase deficiency: Radical changes in mitochondrial morphology? Am. J. Physiol. Cell Physiol. 2007, 293, C22–C29. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F. Cytochrome c oxidase deficiency: Patients and animal models. Biochim. Biophys. Acta 2010, 1802, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Bourens, M.; Barrientos, A. Human mitochondrial cytochrome c oxidase assembly factor COX18 acts transiently as a membrane insertase within the subunit 2 maturation module. J. Biol. Chem. 2017, 292, 7774–7783. [Google Scholar] [CrossRef] [PubMed]
- Bohm, M.; Pronicka, E.; Karczmarewicz, E.; Pronicki, M.; Piekutowska-Abramczuk, D.; Sykut-Cegielska, J.; Mierzewska, H.; Hansikova, H.; Vesela, K.; Tesarova, M.; et al. Retrospective, multicentric study of 180 children with cytochrome C oxidase deficiency. Pediatr. Res. 2006, 59, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Kovarova, N.; Pecina, P.; Nuskova, H.; Vrbacky, M.; Zeviani, M.; Mracek, T.; Viscomi, C.; Houstek, J. Tissue- and species-specific differences in cytochrome c oxidase assembly induced by SURF1 defects. Biochim. Biophys. Acta 2016, 1862, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Cerqua, C.; Morbidoni, V.; Desbats, M.A.; Doimo, M.; Frasson, C.; Sacconi, S.; Baldoin, M.C.; Sartori, G.; Basso, G.; Salviati, L.; et al. COX16 is required for assembly of cytochrome c oxidase in human cells and is involved in copper delivery to COX2. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 244–252. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Goto, Y.I. Respiratory Chain Complex Disorganization Impairs Mitochondrial and Cellular Integrity: Phenotypic Variation in Cytochrome c Oxidase Deficiency. Am. J. Pathol. 2017, 187, 110–121. [Google Scholar] [CrossRef]
- Rak, M.; Benit, P.; Chretien, D.; Bouchereau, J.; Schiff, M.; El-Khoury, R.; Tzagoloff, A.; Rustin, P. Mitochondrial cytochrome c oxidase deficiency. Clin. Sci. 2016, 130, 393–407. [Google Scholar] [CrossRef]
- Maj, M.; Sriskandarajah, N.; Hung, V.; Browne, I.; Shah, B.; Weadge, A.; Jamieson, N.L.; Tropak, M.; Cameron, J.M.; Addis, J.B.; et al. Identification of drug candidates which increase cytochrome c oxidase activity in deficient patient fibroblasts. Mitochondrion 2011, 11, 264–272. [Google Scholar] [CrossRef]
- Szeto, H.H.; Birk, A.V. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 2014, 96, 672–683. [Google Scholar] [CrossRef]
- Wagner, B.K.; Kitami, T.; Gilbert, T.J.; Peck, D.; Ramanathan, A.; Schreiber, S.L.; Golub, T.R.; Mootha, V.K. Large-scale chemical dissection of mitochondrial function. Nat. Biotechnol. 2008, 26, 343–351. [Google Scholar] [CrossRef]
- Kanabus, M.; Heales, S.J.; Rahman, S. Development of pharmacological strategies for mitochondrial disorders. Br. J. Pharmacol. 2014, 171, 1798–1817. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Fisman, E.Z. Fibrates are an essential part of modern anti-dyslipidemic arsenal: Spotlight on atherogenic dyslipidemia and residual risk reduction. Cardiovasc. Diabetol. 2012, 11, 125. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef]
- Burri, L.; Thoresen, G.H.; Berge, R.K. The Role of PPARalpha Activation in Liver and Muscle. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARalpha. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Karpe, F.; Ehrenborg, E.E. PPARdelta in humans: Genetic and pharmacological evidence for a significant metabolic function. Curr. Opin. Lipidol. 2009, 20, 333–336. [Google Scholar] [CrossRef]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor beta/delta in skeletal muscle physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Vazquez-Carrera, M.; Montagner, A.; Sng, M.K.; Guillou, H.; Wahli, W. Transcriptional control of physiological and pathological processes by the nuclear receptor PPARbeta/delta. Prog. Lipid Res. 2016, 64, 98–122. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Djouadi, F.; Bonnefont, J.P.; Thuillier, L.; Droin, V.; Khadom, N.; Munnich, A.; Bastin, J. Correction of fatty acid oxidation in carnitine palmitoyl transferase 2-deficient cultured skin fibroblasts by bezafibrate. Pediatr. Res. 2003, 54, 446–451. [Google Scholar] [CrossRef]
- Yao, M.; Yao, D.; Yamaguchi, M.; Chida, J.; Yao, D.; Kido, H. Bezafibrate upregulates carnitine palmitoyltransferase II expression and promotes mitochondrial energy crisis dissipation in fibroblasts of patients with influenza-associated encephalopathy. Mol. Genet. Metab. 2011, 104, 265–272. [Google Scholar] [CrossRef]
- Djouadi, F.; Aubey, F.; Schlemmer, D.; Bastin, J. Peroxisome Proliferator Activated Receptor delta (PPARδ) Agonist But Not PPAR alpha Corrects Carnitine Palmitoyl Transferase 2 Deficiency in Human Muscle Cells. J. Clin. Endocrinol. Metab. 2005, 90, 1791–1797. [Google Scholar] [CrossRef]
- Bugge, A.; Holst, D. PPAR agonists, -Could tissue targeting pave the way? Biochimie 2017, 136, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Aoyama, T.; Burns, A.M.; Gonzalez, F.J. Bezafibrate is a dual ligand for PPARalpha and PPARbeta: Studies using null mice. Biochim. Biophys. Acta 2003, 1632, 80–89. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: The bezafibrate lessons. Cardiovasc. Diabetol. 2005, 4, 14. [Google Scholar] [CrossRef]
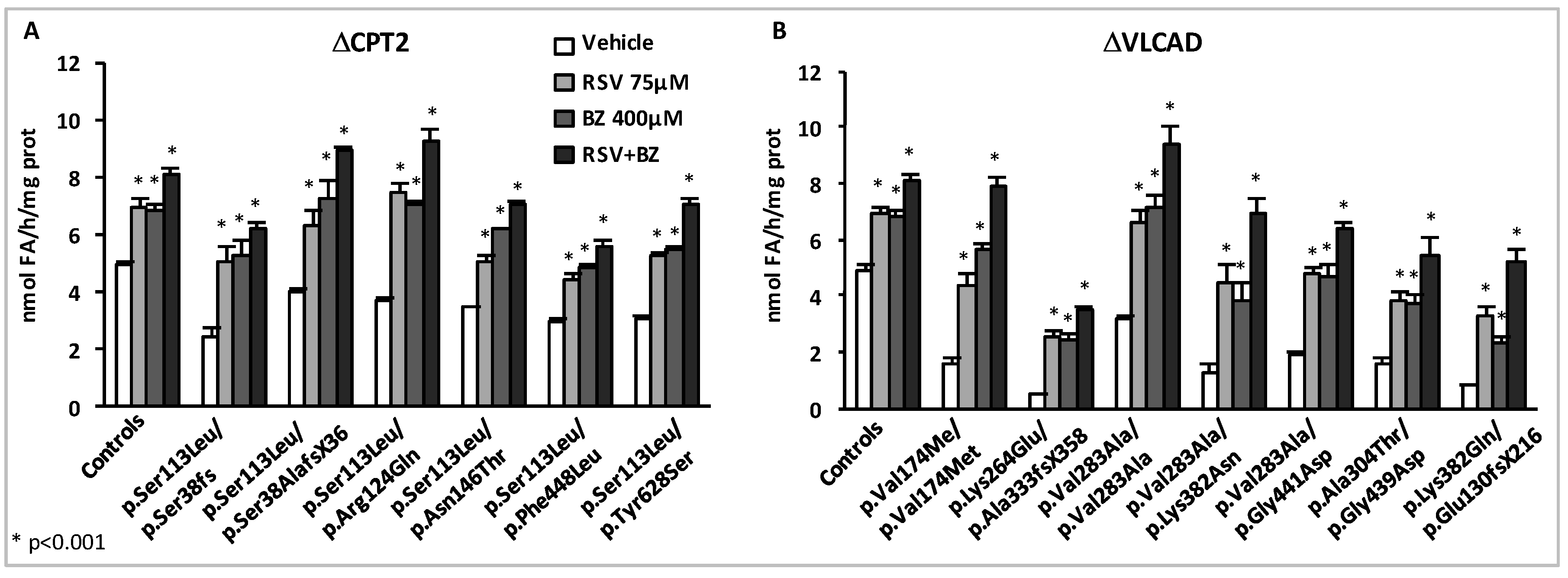
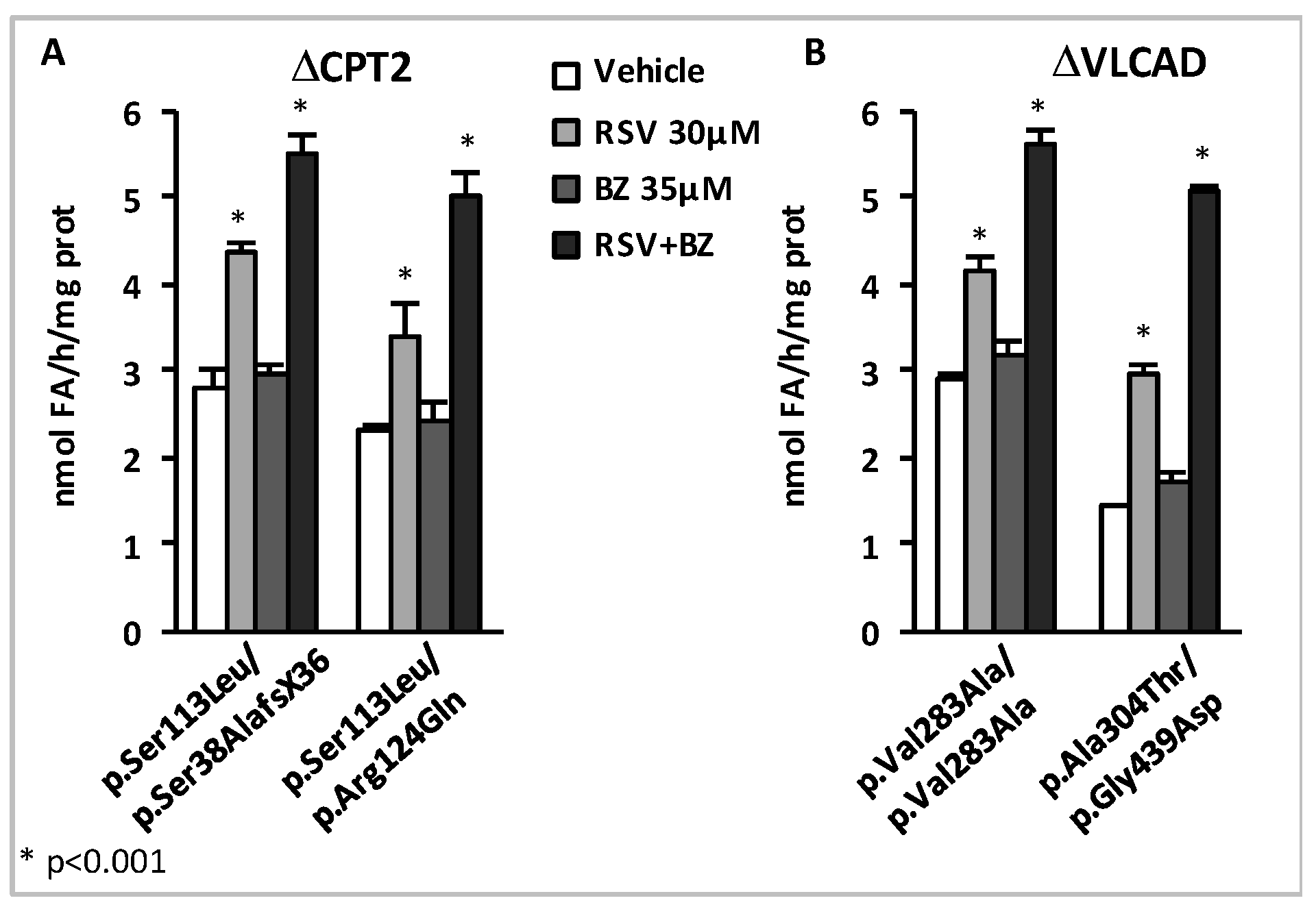
- Yamaguchi, S.; Li, H.; Purevsuren, J.; Yamada, K.; Furui, M.; Takahashi, T.; Mushimoto, Y.; Kobayashi, H.; Hasegawa, Y.; Taketani, T.; et al. Bezafibrate can be a new treatment option for mitochondrial fatty acid oxidation disorders: Evaluation by in vitro probe acylcarnitine assay. Mol. Genet. Metab. 2012, 107, 87–91. [Google Scholar] [CrossRef]
- Yasuno, T.; Osafune, K.; Sakurai, H.; Asaka, I.; Tanaka, A.; Yamaguchi, S.; Yamada, K.; Hitomi, H.; Arai, S.; Kurose, Y.; et al. Functional analysis of iPSC-derived myocytes from a patient with carnitine palmitoyltransferase II deficiency. Biochem. Biophys. Res. Commun. 2014, 448, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Djouadi, F.; Aubey, F.; Schlemmer, D.; Ruiter, J.P.; Wanders, R.J.; Strauss, A.W.; Bastin, J. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum. Mol. Genet. 2005, 14, 2695–2703. [Google Scholar] [CrossRef]
- Gobin-Limballe, S.; McAndrew, R.P.; Djouadi, F.; Kim, J.J.; Bastin, J. Compared effects of missense mutations in Very-Long-Chain Acyl-CoA Dehydrogenase deficiency: Combined analysis by structural, functional and pharmacological approaches. Biochim. Biophys. Acta 2010, 1802, 478–484. [Google Scholar] [CrossRef]
- Li, H.; Fukuda, S.; Hasegawa, Y.; Kobayashi, H.; Purevsuren, J.; Mushimoto, Y.; Yamaguchi, S. Effect of heat stress and bezafibrate on mitochondrial beta-oxidation: Comparison between cultured cells from normal and mitochondrial fatty acid oxidation disorder children using in vitro probe acylcarnitine profiling assay. Brain Dev. 2010, 32, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Djouadi, F.; Habarou, F.; Le Bachelier, C.; Ferdinandusse, S.; Schlemmer, D.; Benoist, J.F.; Boutron, A.; Andresen, B.S.; Visser, G.; de Lonlay, P.; et al. Mitochondrial trifunctional protein deficiency in human cultured fibroblasts: Effects of bezafibrate. J. Inherit. Metab. Dis. 2016, 39, 47–58. [Google Scholar] [CrossRef] [PubMed]
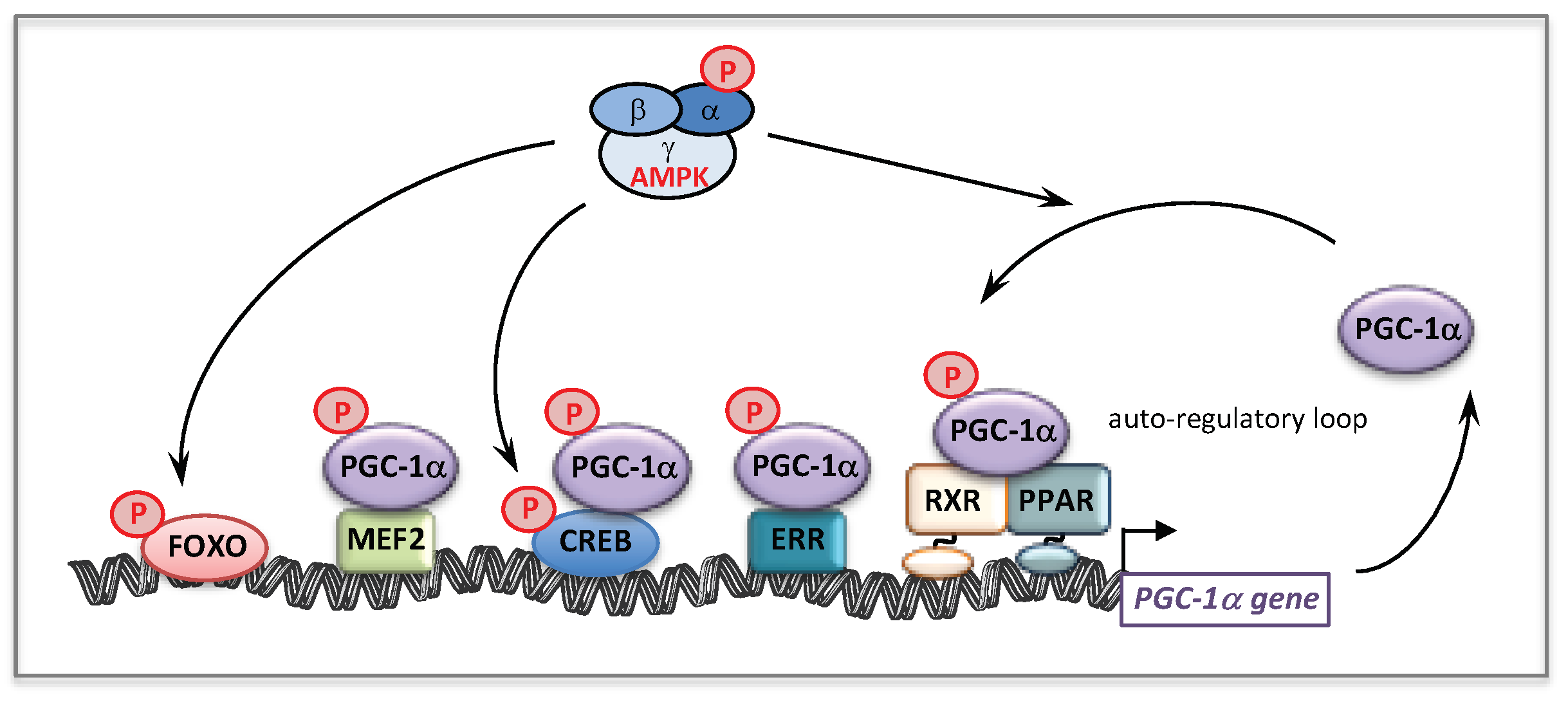
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. TEM 2012, 23, 459–466. [Google Scholar] [CrossRef]
- Hondares, E.; Mora, O.; Yubero, P.; Rodriguez de la Concepcion, M.; Iglesias, R.; Giralt, M.; Villarroya, F. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1alpha gene transcription: An autoregulatory loop controls PGC-1alpha expression in adipocytes via peroxisome proliferator-activated receptor-gamma coactivation. Endocrinology 2006, 147, 2829–2838. [Google Scholar]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef]
- Ramjiawan, A.; Bagchi, R.A.; Albak, L.; Czubryt, M.P. Mechanism of cardiomyocyte PGC-1alpha gene regulation by ERRalpha. Biochem. Cell Biol. Biochim. Biol. Cell. 2013, 91, 148–154. [Google Scholar] [CrossRef]
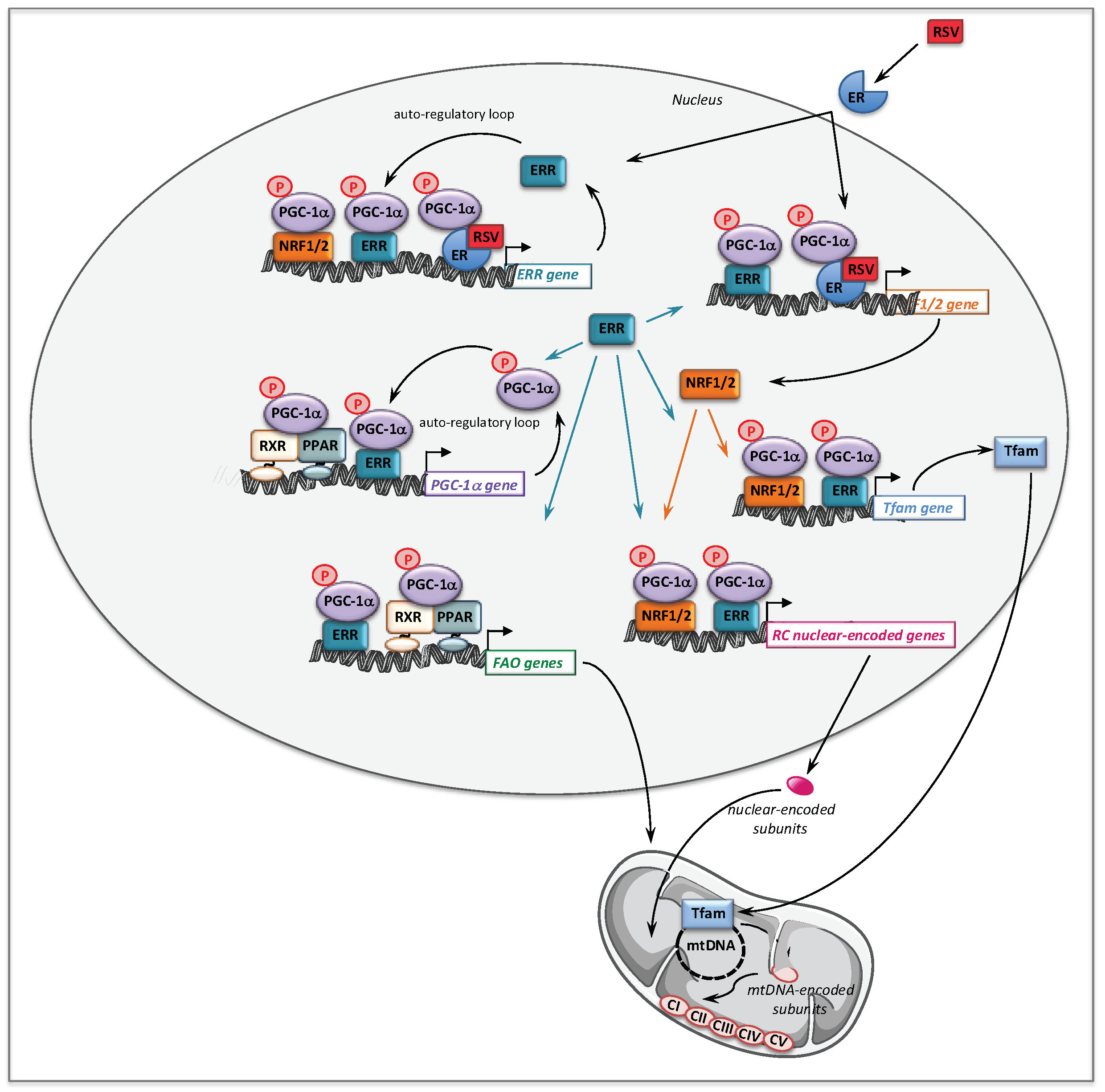
- Eichner, L.J.; Giguere, V. Estrogen related receptors (ERRs): A new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion 2011, 11, 544–552. [Google Scholar] [CrossRef]
- Giguere, V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr. Rev. 2008, 29, 677–696. [Google Scholar] [CrossRef]
- Bonnefont, J.P.; Bastin, J.; Behin, A.; Djouadi, F. Bezafibrate for treatment of an inborn mitochondrial ß-oxidation defect. N. Engl. J. Med. 2009, 360, 838–840. [Google Scholar] [CrossRef]
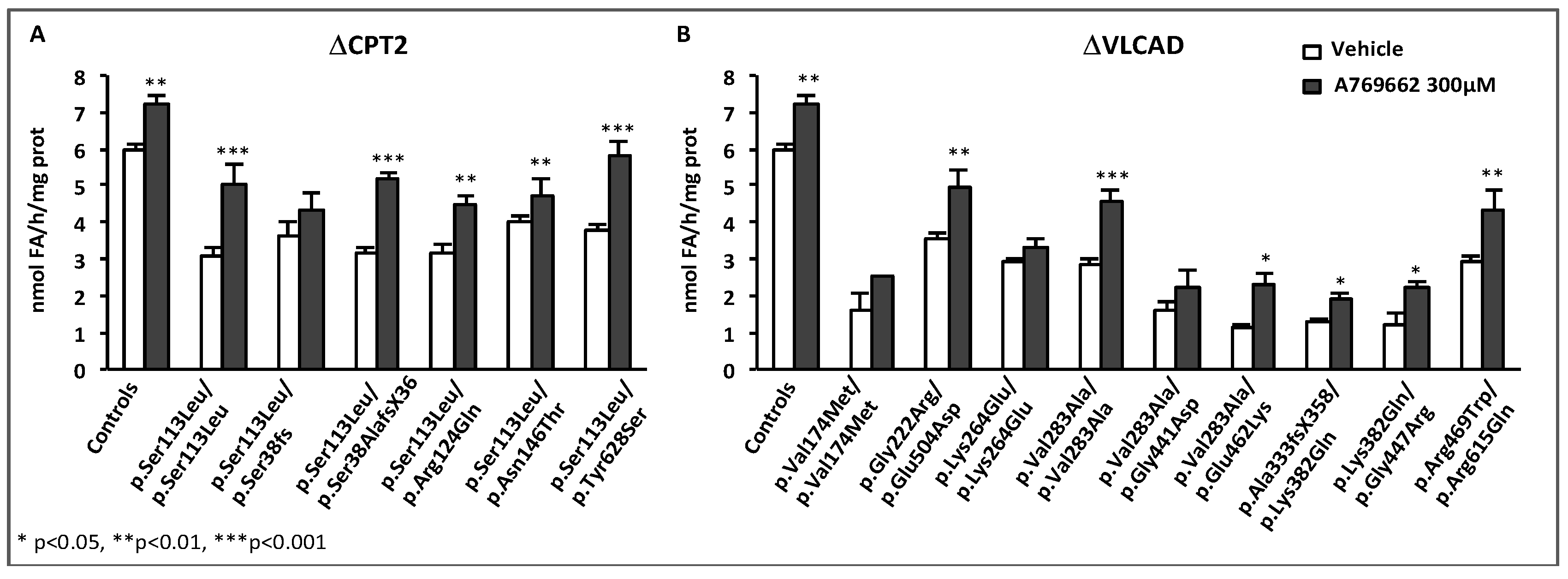
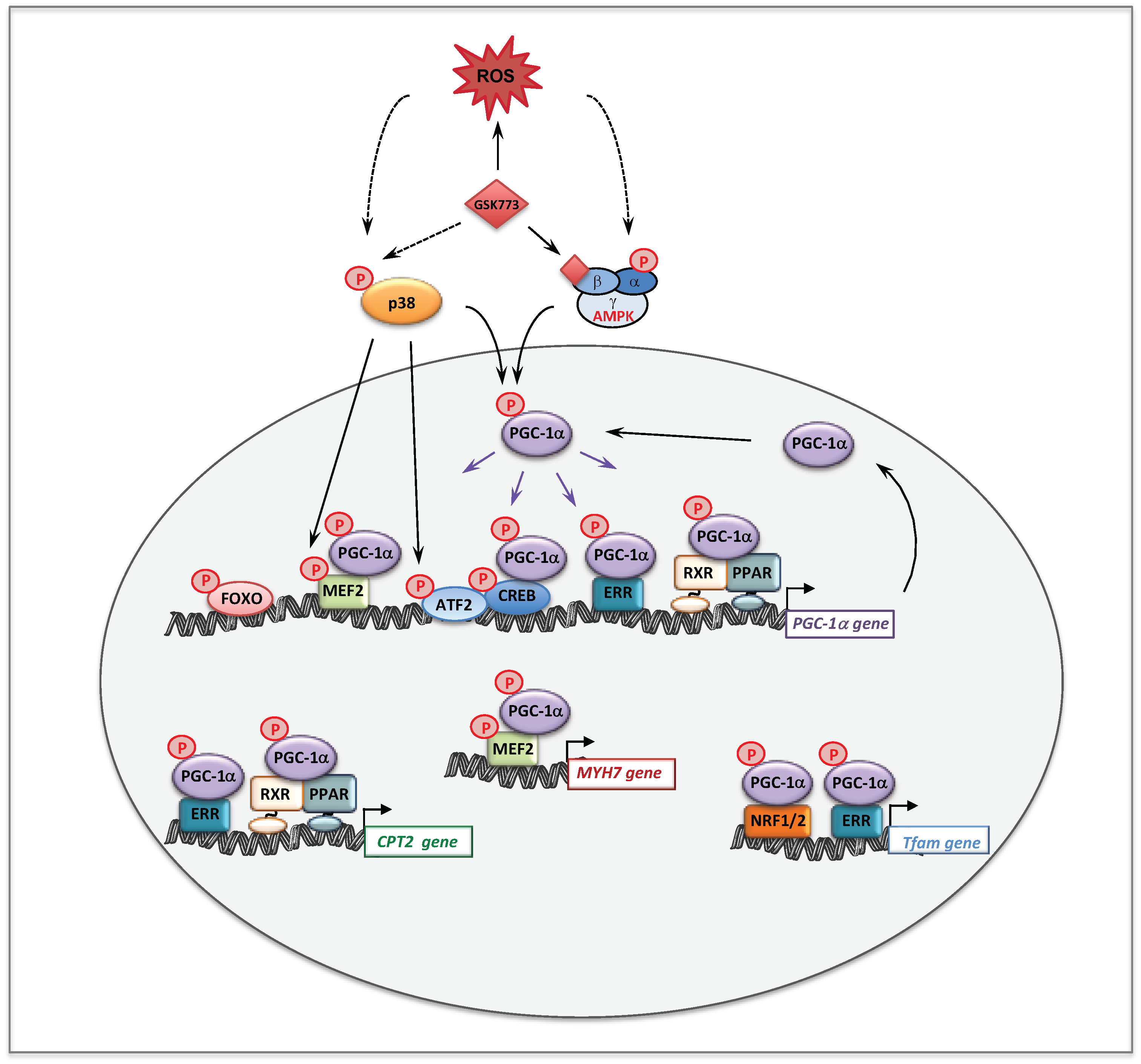
- Boufroura, F.Z.; Le Bachelier, C.; Tomkiewicz-Raulet, C.; Schlemmer, D.; Benoist, J.F.; Grondin, P.; Lamotte, Y.; Mirguet, O.; Mouillet-Richard, S.; Bastin, J.; et al. A new AMPK activator, GSK773, corrects fatty acid oxidation and differentiation defect in CPT2-deficient myotubes. Hum. Mol. Genet. 2018, 27, 3417–3433. [Google Scholar] [CrossRef] [PubMed]
- Aires, V.; Delmas, D.; Le Bachelier, C.; Latruffe, N.; Schlemmer, D.; Benoist, J.F.; Djouadi, F.; Bastin, J. Stilbenes and resveratrol metabolites improve mitochondrial fatty acid oxidation defects in human fibroblasts. Orphanet J. Rare Dis. 2014, 9, 79. [Google Scholar] [CrossRef]
- Bastin, J.; Lopes-Costa, A.; Djouadi, F. Exposure to resveratrol triggers pharmacological correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum. Mol. Genet. 2011, 20, 2048–2057. [Google Scholar] [CrossRef]
- Seminotti, B.; Leipnitz, G.; Karunanidhi, A.; Kochersperger, C.; Roginskaya, V.Y.; Basu, S.; Wang, Y.; Wipf, P.; Van Houten, B.; Mohsen, A.W.; et al. Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zolkipli, Z.; Pedersen, C.B.; Lamhonwah, A.M.; Gregersen, N.; Tein, I. Vulnerability to oxidative stress in vitro in pathophysiology of mitochondrial short-chain acyl-CoA dehydrogenase deficiency: Response to antioxidants. PLoS ONE 2011, 6, e17534. [Google Scholar] [CrossRef]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 2299–2301. [Google Scholar]
- Wang, Y.X.; Zhang, C.L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef]
- Bastin, J.; Aubey, F.; Rotig, A.; Munnich, A.; Djouadi, F. Activation of peroxisome proliferator-activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients’ cells lacking its components. J. Clin. Endocrinol. Metab. 2008, 93, 1433–1441. [Google Scholar] [CrossRef]
- Hofer, A.; Noe, N.; Tischner, C.; Kladt, N.; Lellek, V.; Schauss, A.; Wenz, T. Defining the action spectrum of potential PGC-1alpha activators on a mitochondrial and cellular level in vivo. Hum. Mol. Genet. 2014, 23, 2400–2415. [Google Scholar] [CrossRef] [PubMed]
- Casarin, A.; Giorgi, G.; Pertegato, V.; Siviero, R.; Cerqua, C.; Doimo, M.; Basso, G.; Sacconi, S.; Cassina, M.; Rizzuto, R.; et al. Copper and bezafibrate cooperate to rescue cytochrome c oxidase deficiency in cells of patients with SCO2 mutations. Orphanet J. Rare Dis. 2012, 7, 21. [Google Scholar] [CrossRef]
- Ioannou, N.; Hargreaves, I.P.; Allen, G.; Duberley, K.; Land, J.M.; Heales, S.J. Bezafibrate induced increase in mitochondrial electron transport chain complex IV activity in human astrocytoma cells: Implications for mitochondrial cytopathies and neurodegenerative diseases. Biofactors 2010, 36, 468–473. [Google Scholar] [CrossRef]
- Wenz, T.; Wang, X.; Marini, M.; Moraes, C.T. A metabolic shift induced by a PPAR panagonist markedly reduces the effects of pathogenic mitochondrial tRNA mutations. J. Cell. Mol. Med. 2011, 15, 2317–2325. [Google Scholar] [CrossRef]
- Golubitzky, A.; Dan, P.; Weissman, S.; Link, G.; Wikstrom, J.D.; Saada, A. Screening for active small molecules in mitochondrial complex I deficient patient’s fibroblasts, reveals AICAR as the most beneficial compound. PLoS ONE 2011, 6, e26883. [Google Scholar] [CrossRef]
- Soiferman, D.; Ayalon, O.; Weissman, S.; Saada, A. The effect of small molecules on nuclear-encoded translation diseases. Biochimie 2014, 100, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, L.; Smeitink, J.A.; van Emst-de Vries, S.E.; Vogels, C.; Pellegrini, M.; Jonckheere, A.I.; Rodenburg, R.J.; Buydens, L.M.; Beyrath, J.; Willems, P.H.; et al. Quantifying small molecule phenotypic effects using mitochondrial morpho-functional fingerprinting and machine learning. Sci. Rep. 2015, 5, 8035. [Google Scholar] [CrossRef] [PubMed]
- Distelmaier, F.; Visch, H.J.; Smeitink, J.A.; Mayatepek, E.; Koopman, W.J.; Willems, P.H. The antioxidant Trolox restores mitochondrial membrane potential and Ca2+ -stimulated ATP production in human complex I deficiency. J. Mol. Med. 2009, 87, 515–522. [Google Scholar] [CrossRef]
- Koopman, W.J.; Verkaart, S.; van Emst-de Vries, S.E.; Grefte, S.; Smeitink, J.A.; Nijtmans, L.G.; Willems, P.H. Mitigation of NADH: Ubiquinone oxidoreductase deficiency by chronic Trolox treatment. Biochim. Biophys. Acta 2008, 1777, 853–859. [Google Scholar] [CrossRef]
- Leipnitz, G.; Mohsen, A.W.; Karunanidhi, A.; Seminotti, B.; Roginskaya, V.Y.; Markantone, D.M.; Grings, M.; Mihalik, S.J.; Wipf, P.; Van Houten, B.; et al. Evaluation of mitochondrial bioenergetics, dynamics, endoplasmic reticulum-mitochondria crosstalk, and reactive oxygen species in fibroblasts from patients with complex I deficiency. Sci. Rep. 2018, 8, 1165. [Google Scholar] [CrossRef]
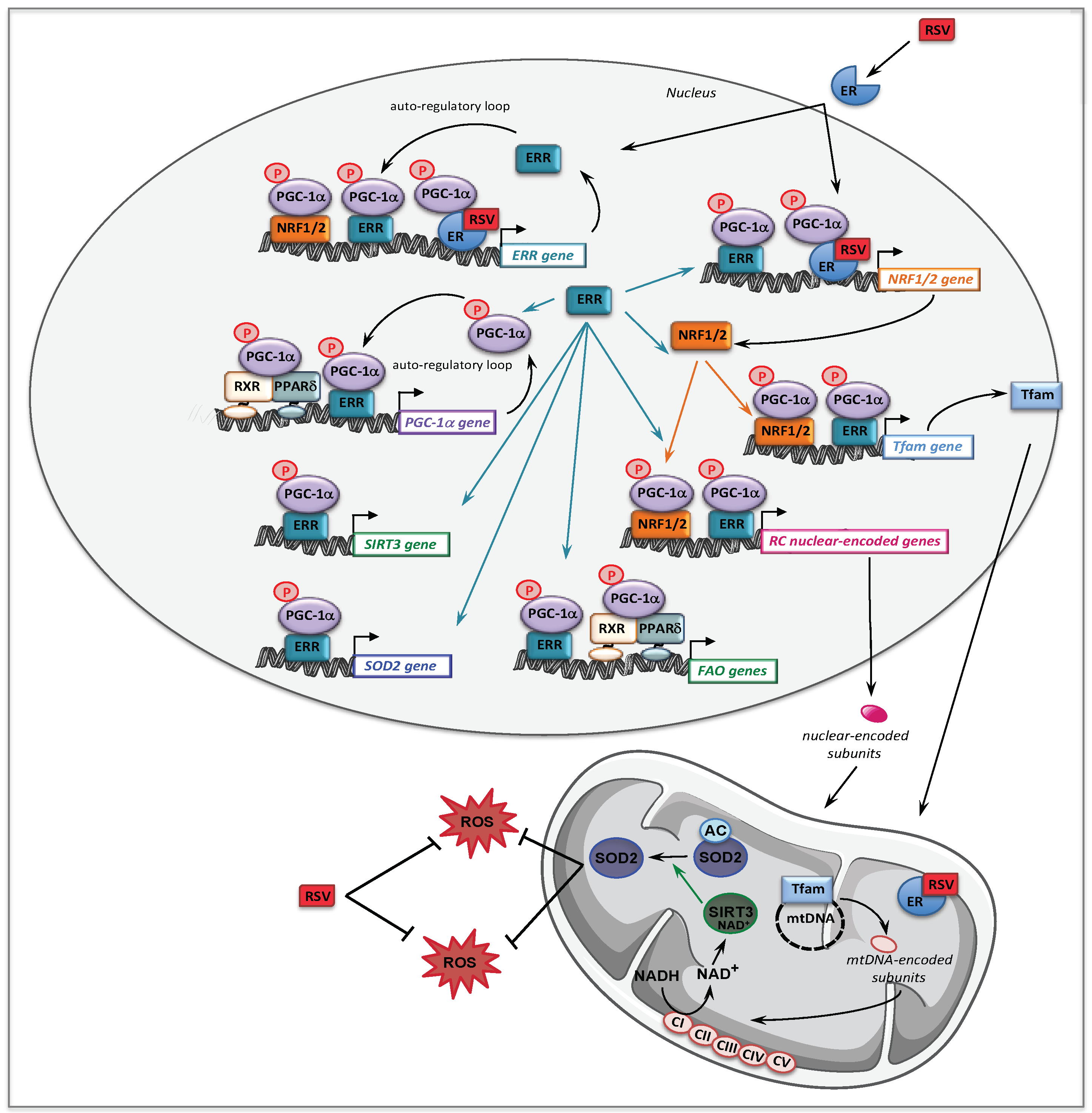
- Lopes Costa, A.; Le Bachelier, C.; Mathieu, L.; Rotig, A.; Boneh, A.; De Lonlay, P.; Tarnopolsky, M.A.; Thorburn, D.R.; Bastin, J.; Djouadi, F. Beneficial effects of resveratrol on respiratory chain defects in patients’ fibroblasts involve estrogen receptor and estrogen-related receptor alpha signaling. Hum. Mol. Genet. 2014, 23, 2106–2119. [Google Scholar] [CrossRef]
- Mathieu, L.; Costa, A.L.; Le Bachelier, C.; Slama, A.; Lebre, A.S.; Taylor, R.W.; Bastin, J.; Djouadi, F. Resveratrol attenuates oxidative stress in mitochondrial Complex I deficiency: Involvement of SIRT3. Free Radic. Biol. Med. 2016, 96, 190–198. [Google Scholar] [CrossRef]
- Felici, R.; Lapucci, A.; Cavone, L.; Pratesi, S.; Berlinguer-Palmini, R.; Chiarugi, A. Pharmacological NAD-Boosting Strategies Improve Mitochondrial Homeostasis in Human Complex I-Mutant Fibroblasts. Mol. Pharmacol. 2015, 87, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Scacco, S.; Piccoli, C.; Bellomo, F.; Petruzzella, V.; Trentadue, R.; Minuto, M.; Ripoli, M.; Capitanio, N.; Zeviani, M.; et al. Dysfunctions of cellular oxidative metabolism in patients with mutations in the NDUFS1 and NDUFS4 genes of complex I. J. Biol. Chem. 2006, 281, 10374–10380. [Google Scholar] [CrossRef] [PubMed]
- Pirinen, E.; Canto, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef]
- Polyak, E.; Ostrovsky, J.; Peng, M.; Dingley, S.D.; Tsukikawa, M.; Kwon, Y.J.; McCormack, S.E.; Bennett, M.; Xiao, R.; Seiler, C.; et al. N-acetylcysteine and vitamin E rescue animal longevity and cellular oxidative stress in pre-clinical models of mitochondrial complex I disease. Mol. Genet. Metab. 2018, 123, 449–462. [Google Scholar] [CrossRef]
- De Paepe, B.; Vandemeulebroecke, K.; Smet, J.; Vanlander, A.; Seneca, S.; Lissens, W.; Van Hove, J.L.; Deschepper, E.; Briones, P.; Van Coster, R. Effect of resveratrol on cultured skin fibroblasts from patients with oxidative phosphorylation defects. Phytother. Res. PTR 2014, 28, 312–316. [Google Scholar] [CrossRef]
- Burelle, Y.; Bemeur, C.; Rivard, M.E.; Thompson Legault, J.; Boucher, G.; Morin, C.; Coderre, L.; Des Rosiers, C. Mitochondrial vulnerability and increased susceptibility to nutrient-induced cytotoxicity in fibroblasts from leigh syndrome French canadian patients. PLoS ONE 2015, 10, e0120767. [Google Scholar] [CrossRef]
- Abdulhag, U.N.; Soiferman, D.; Schueler-Furman, O.; Miller, C.; Shaag, A.; Elpeleg, O.; Edvardson, S.; Saada, A. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. EJHG 2015, 23, 159–164. [Google Scholar] [CrossRef]
- Douiev, L.; Saada, A. The pathomechanism of cytochrome c oxidase deficiency includes nuclear DNA damage. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Douiev, L.; Soiferman, D.; Alban, C.; Saada, A. The Effects of Ascorbate, N-Acetylcysteine, and Resveratrol on Fibroblasts from Patients with Mitochondrial Disorders. J. Clin. Med. 2016, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Salazar, E.; Brosel, S.; Yang, H.; Schon, E.A.; Manfredi, G. Modulation of mitochondrial protein phosphorylation by soluble adenylyl cyclase ameliorates cytochrome oxidase defects. EMBO Mol. Med. 2009, 1, 392–406. [Google Scholar] [CrossRef]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, C.; Bottani, E.; Civiletto, G.; Cerutti, R.; Moggio, M.; Fagiolari, G.; Schon, E.A.; Lamperti, C.; Zeviani, M. In Vivo Correction of COX Deficiency by Activation of the AMPK/PGC-1alpha Axis. Cell Metab. 2011, 14, 80–90. [Google Scholar] [CrossRef]
- Yatsuga, S.; Suomalainen, A. Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum. Mol. Genet. 2012, 21, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.M.; Hida, A.; Garcia, S.; Prolla, T.A.; Moraes, C.T. Long-term bezafibrate treatment improves skin and spleen phenotypes of the mtDNA mutator mouse. PLoS ONE 2012, 7, e44335. [Google Scholar] [CrossRef] [PubMed]
- Djouadi, F.; Bastin, J. Species differences in the effects of bezafibrate as a potential treatment of mitochondrial disorders. Cell Metab. 2011, 14, 715–716. [Google Scholar] [CrossRef] [PubMed]
- Romanino, K.; Mazelin, L.; Albert, V.; Conjard-Duplany, A.; Lin, S.; Bentzinger, C.F.; Handschin, C.; Puigserver, P.; Zorzato, F.; Schaeffer, L.; et al. Myopathy caused by mammalian target of rapamycin complex 1 (mTORC1) inactivation is not reversed by restoring mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 20808–20813. [Google Scholar] [CrossRef] [PubMed]
- Schafer, C.; Moore, V.; Dasgupta, N.; Javadov, S.; James, J.F.; Glukhov, A.I.; Strauss, A.W.; Khuchua, Z. The Effects of PPAR Stimulation on Cardiac Metabolic Pathways in Barth Syndrome Mice. Front. Pharmacol. 2018, 9, 318. [Google Scholar] [CrossRef]
- Huang, Y.; Powers, C.; Madala, S.K.; Greis, K.D.; Haffey, W.D.; Towbin, J.A.; Purevjav, E.; Javadov, S.; Strauss, A.W.; Khuchua, Z. Cardiac metabolic pathways affected in the mouse model of barth syndrome. PLoS ONE 2015, 10, e0128561. [Google Scholar] [CrossRef]
- Jang, S.; Lewis, T.S.; Powers, C.; Khuchua, Z.; Baines, C.P.; Wipf, P.; Javadov, S. Elucidating Mitochondrial Electron Transport Chain Supercomplexes in the Heart During Ischemia-Reperfusion. Antioxid. Redox Signal. 2017, 27, 57–69. [Google Scholar] [CrossRef]
- Bonnefont, J.P.; Bastin, J.; Laforet, P.; Aubey, F.; Mogenet, A.; Romano, S.; Ricquier, D.; Gobin-Limballe, S.; Vassault, A.; Behin, A.; et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin. Pharmacol. Ther. 2010, 88, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Orngreen, M.C.; Madsen, K.L.; Preisler, N.; Andersen, G.; Vissing, J.; Laforet, P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: A randomized clinical trial. Neurology 2014, 82, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Shiraishi, H.; Oki, E.; Ishige, M.; Fukao, T.; Hamada, Y.; Sakai, N.; Ochi, F.; Watanabe, A.; Kawakami, S.; et al. Open-label clinical trial of bezafibrate treatment in patients with fatty acid oxidation disorders in Japan. Mol. Genet. Metab. Rep. 2018, 15, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, M. Of the phenolic substances of white hellbore (Veratrum Grandiflorum LOES. fil.). J. Fac. Sci. Hokkaido Imp. Univ. 1940, 3, 1–16. [Google Scholar]
- Nonomura, S.; Kanagawa, H.; Makimoto, A. [Chemical Constituents of Polygonaceous Plants. I. Studies on the Components of Ko-J O-Kon. (Polygonum Cuspidatum Sieb. Et Zucc.)]. Yakugaku zasshi: J. Pharm. Soc. Jpn. 1963, 83, 988–990. [Google Scholar] [CrossRef]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Bastin, J.; Djouadi, F. Resveratrol and Myopathy. Nutrients 2016, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Milbrandt, J. Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7217–7222. [Google Scholar] [CrossRef]
- Dolinsky, V.W.; Jones, K.E.; Sidhu, R.S.; Haykowsky, M.; Czubryt, M.P.; Gordon, T.; Dyck, J.R. Improvements in skeletal muscle strength and cardiac function induced by resveratrol during exercise training contribute to enhanced exercise performance in rats. J. Physiol. 2012, 590, 2783–2799. [Google Scholar] [CrossRef]
- Menzies, K.J.; Singh, K.; Saleem, A.; Hood, D.A. Sirtuin 1-mediated effects of exercise and resveratrol on mitochondrial biogenesis. J. Biol. Chem. 2013, 288, 6968–6979. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef]
- Mizuguchi, Y.; Hatakeyama, H.; Sueoka, K.; Tanaka, M.; Goto, Y.I. Low dose resveratrol ameliorates mitochondrial respiratory dysfunction and enhances cellular reprogramming. Mitochondrion 2017, 34, 43–48. [Google Scholar] [CrossRef]
- Morvan, D.; Demidem, A. NMR metabolomics of fibroblasts with inherited mitochondrial Complex I mutation reveals treatment-reversible lipid and amino acid metabolism alterations. Metab. Off. J. Metab. Soc. 2018, 14, 55. [Google Scholar] [CrossRef]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef]
- Bitterman, J.L.; Chung, J.H. Metabolic effects of resveratrol: Addressing the controversies. Cell. Mol. Life Sci. CMLS 2015, 72, 1473–1488. [Google Scholar] [CrossRef]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [PubMed]
- Higashida, K.; Kim, S.H.; Jung, S.R.; Asaka, M.; Holloszy, J.O.; Han, D.H. Effects of resveratrol and SIRT1 on PGC-1alpha activity and mitochondrial biogenesis: A reevaluation. PLoS Biol. 2013, 11, e1001603. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McDonagh, T.; Heltweg, B.; Hixon, J.; Westman, E.A.; Caldwell, S.D.; Napper, A.; Curtis, R.; DiStefano, P.S.; Fields, S.; et al. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 280, 17038–17045. [Google Scholar] [CrossRef]
- Pacholec, M.; Bleasdale, J.E.; Chrunyk, B.; Cunningham, D.; Flynn, D.; Garofalo, R.S.; Griffith, D.; Griffor, M.; Loulakis, P.; Pabst, B.; et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [Google Scholar] [CrossRef]
- Bowers, J.L.; Tyulmenkov, V.V.; Jernigan, S.C.; Klinge, C.M. Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology 2000, 141, 3657–3667. [Google Scholar] [CrossRef]
- Chen, J.Q.; Cammarata, P.R.; Baines, C.P.; Yager, J.D. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim. Biophys. Acta 2009, 1793, 1540–1570. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Canto, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, W.S.; Kim, K.H.; Yoon, M.J.; Cho, H.J.; Shen, Y.; Ye, J.M.; Lee, C.H.; Oh, W.K.; Kim, C.T.; et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 2006, 55, 2256–2264. [Google Scholar] [CrossRef] [PubMed]
- Bastin, J.; Djouadi, F. Combination of Bezafibrate and of Resveratrol or Resveratrol Derivatives for the Treatment and Prevention of Diseases Involving a Mitochondrial Energy Dysfunction. Patent US20160317483A1, 3 November 2016. [Google Scholar]
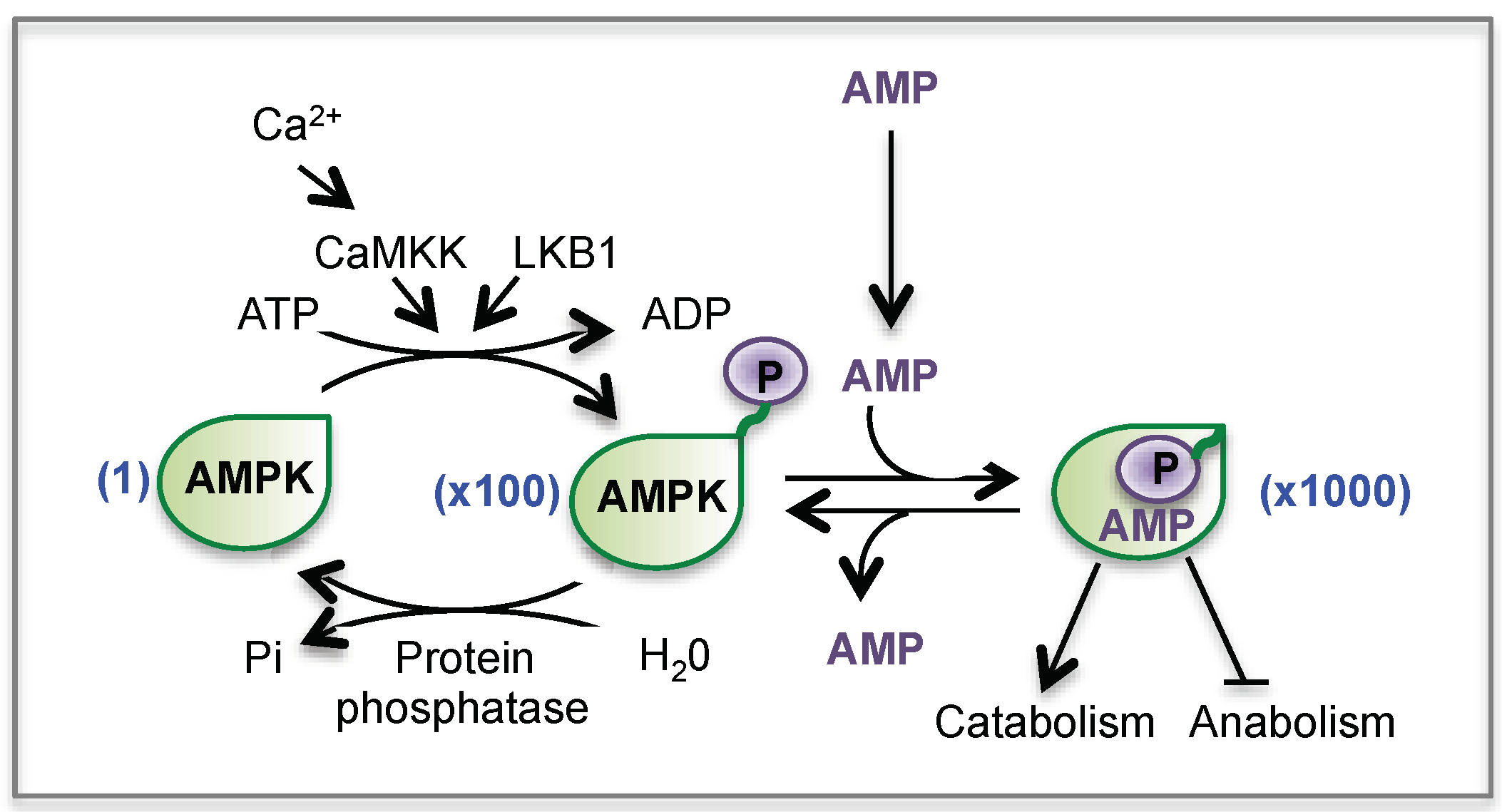
- Carling, D.; Mayer, F.V.; Sanders, M.J.; Gamblin, S.J. AMP-activated protein kinase: nature’s energy sensor. Nat. Chem. Biol. 2011, 7, 512–518. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Hardie, D.G. Sensing of energy and nutrients by AMP-activated protein kinase. Am. J. Clin. Nutr. 2011, 93, 891S–896S. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell. Mol. Life Sci. CMLS 2010, 67, 3407–3423. [Google Scholar] [CrossRef]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.M.; Herway, S.T.; Fillmore, N.; Kim, H.; Brown, J.D.; Barrow, J.R.; Winder, W.W. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J. Appl. Physiol. (1985) 2008, 104, 429–438. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK activation: A therapeutic target for type 2 diabetes? Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 241–253. [Google Scholar] [CrossRef]
- Olivier, S.; Foretz, M.; Viollet, B. Promise and challenges for direct small molecule AMPK activators. Biochem. Pharmacol. 2018, 153, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Guigas, B.; Sakamoto, K.; Taleux, N.; Reyna, S.M.; Musi, N.; Viollet, B.; Hue, L. Beyond AICA riboside: In search of new specific AMP-activated protein kinase activators. IUBMB Life 2009, 61, 18–26. [Google Scholar] [CrossRef]
- Van Den Neste, E.; Van den Berghe, G.; Bontemps, F. AICA-riboside (acadesine), an activator of AMP-activated protein kinase with potential for application in hematologic malignancies. Expert Opin. Investig. Drugs 2010, 19, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Mullane, K. Acadesine: The prototype adenosine regulating agent for reducing myocardial ischaemic injury. Cardiovasc. Res. 1993, 27, 43–47. [Google Scholar] [CrossRef]
- Guigas, B.; Taleux, N.; Foretz, M.; Detaille, D.; Andreelli, F.; Viollet, B.; Hue, L. AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. Biochem. J. 2007, 404, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef]
- Hoppeler, H. Molecular networks in skeletal muscle plasticity. J. Exp. Biol. 2016, 219, 205–213. [Google Scholar] [CrossRef]
- Mounier, R.; Theret, M.; Lantier, L.; Foretz, M.; Viollet, B. Expanding roles for AMPK in skeletal muscle plasticity. Trends Endocrinol. Metab. TEM 2015, 26, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Candau, R.B.; Csibi, A.; Pagano, A.F.; Raibon, A.; Bernardi, H. The role of AMP-activated protein kinase in the coordination of skeletal muscle turnover and energy homeostasis. Am. J. Physiol. Cell Physiol. 2012, 303, C475–C485. [Google Scholar] [CrossRef] [PubMed]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. AMPK and PPARdelta agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Peralta, S.; Garcia, S.; Yin, H.Y.; Arguello, T.; Diaz, F.; Moraes, C.T. Sustained AMPK activation improves muscle function in a mitochondrial myopathy mouse model by promoting muscle fiber regeneration. Hum. Mol. Genet. 2016, 25, 3178–3191. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
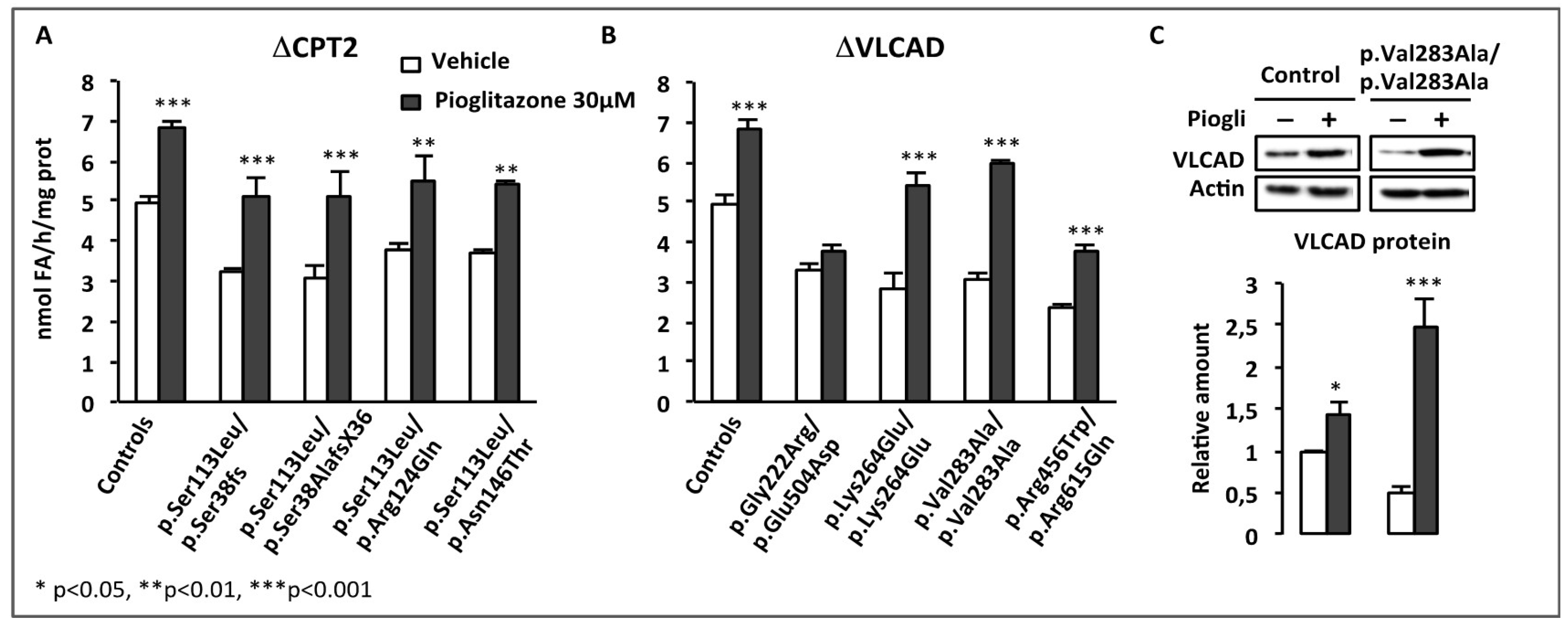
- Coletta, D.K.; Sriwijitkamol, A.; Wajcberg, E.; Tantiwong, P.; Li, M.; Prentki, M.; Madiraju, M.; Jenkinson, C.P.; Cersosimo, E.; Musi, N.; et al. Pioglitazone stimulates AMP-activated protein kinase signalling and increases the expression of genes involved in adiponectin signalling, mitochondrial function and fat oxidation in human skeletal muscle in vivo: A randomised trial. Diabetologia 2009, 52, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Fryer, L.G.; Parbu-Patel, A.; Carling, D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 2002, 277, 25226–25232. [Google Scholar] [CrossRef]
- LeBrasseur, N.K.; Kelly, M.; Tsao, T.S.; Farmer, S.R.; Saha, A.K.; Ruderman, N.B.; Tomas, E. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E175–E181. [Google Scholar] [CrossRef]
- Cha, B.S.; Ciaraldi, T.P.; Park, K.S.; Carter, L.; Mudaliar, S.R.; Henry, R.R. Impaired fatty acid metabolism in type 2 diabetic skeletal muscle cells is reversed by PPARgamma agonists. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E151–E159. [Google Scholar] [CrossRef]
- Sakamoto, J.; Kimura, H.; Moriyama, S.; Odaka, H.; Momose, Y.; Sugiyama, Y.; Sawada, H. Activation of human peroxisome proliferator-activated receptor (PPAR) subtypes by pioglitazone. Biochem. Biophys. Res. Commun. 2000, 278, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Rabol, R.; Boushel, R.; Almdal, T.; Hansen, C.N.; Ploug, T.; Haugaard, S.B.; Prats, C.; Madsbad, S.; Dela, F. Opposite effects of pioglitazone and rosiglitazone on mitochondrial respiration in skeletal muscle of patients with type 2 diabetes. Diabetes Obes. Metab. 2010, 12, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Devchand, P.R.; Liu, T.; Altman, R.B.; FitzGerald, G.A.; Schadt, E.E. The Pioglitazone Trek via Human PPAR Gamma: From Discovery to a Medicine at the FDA and Beyond. Front. Pharmacol. 2018, 9, 1093. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.K.; Cornelius, N.; Gregersen, N. Genetic and cellular modifiers of oxidative stress: What can we learn from fatty acid oxidation defects? Mol. Genet. Metab. 2013, 110, S31–S39. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.K.; Cornelius, N.; Gregersen, N. Redox signalling and mitochondrial stress responses; lessons from inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Wajner, M.; Amaral, A.U. Mitochondrial dysfunction in fatty acid oxidation disorders: Insights from human and animal studies. Biosci. Rep. 2015, 36, e00281. [Google Scholar] [CrossRef]
- Tonin, A.M.; Grings, M.; Busanello, E.N.; Moura, A.P.; Ferreira, G.C.; Viegas, C.M.; Fernandes, C.G.; Schuck, P.F.; Wajner, M. Long-chain 3-hydroxy fatty acids accumulating in LCHAD and MTP deficiencies induce oxidative stress in rat brain. Neurochem. Int. 2010, 56, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Simon, K.R.; Tonin, A.M.; Busanello, E.N.; Moura, A.P.; Ferreira, G.C.; Wajner, M.; Streck, E.L.; Schuck, P.F. Toxicity of octanoate and decanoate in rat peripheral tissues: Evidence of bioenergetic dysfunction and oxidative damage induction in liver and skeletal muscle. Mol. Cell. Biochem. 2012, 361, 329–335. [Google Scholar] [CrossRef]
- Schmidt, S.P.; Corydon, T.J.; Pedersen, C.B.; Bross, P.; Gregersen, N. Misfolding of short-chain acyl-CoA dehydrogenase leads to mitochondrial fission and oxidative stress. Mol. Genet. Metab. 2010, 100, 155–162. [Google Scholar] [CrossRef]
- Hagenbuchner, J.; Scholl-Buergi, S.; Karall, D.; Ausserlechner, M.J. Very long-/and long Chain-3-Hydroxy Acyl CoA Dehydrogenase Deficiency correlates with deregulation of the mitochondrial fusion/fission machinery. Sci. Rep. 2018, 8, 3254. [Google Scholar] [CrossRef] [PubMed]
- Seifert, E.L.; Estey, C.; Xuan, J.Y.; Harper, M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 2010, 285, 5748–5758. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Sestili, P. Reactive oxygen species in skeletal muscle signaling. J. Signal Transduct. 2012, 2012, 982794. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef]
- Roestenberg, P.; Manjeri, G.R.; Valsecchi, F.; Smeitink, J.A.; Willems, P.H.; Koopman, W.J. Pharmacological targeting of mitochondrial complex I deficiency: The cellular level and beyond. Mitochondrion 2012, 12, 57–65. [Google Scholar] [CrossRef]
- Verkaart, S.; Koopman, W.J.; van Emst-de Vries, S.E.; Nijtmans, L.G.; van den Heuvel, L.W.; Smeitink, J.A.; Willems, P.H. Superoxide production is inversely related to complex I activity in inherited complex I deficiency. Biochim. Biophys. Acta 2007, 1772, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Wojtala, A.; Karkucinska-Wieckowska, A.; Sardao, V.A.; Szczepanowska, J.; Kowalski, P.; Pronicki, M.; Duszynski, J.; Wieckowski, M.R. Modulation of mitochondrial dysfunction-related oxidative stress in fibroblasts of patients with Leigh syndrome by inhibition of prooxidative p66Shc pathway. Mitochondrion 2017, 37, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, M.; Vijayvergiya, C.; Gajewski, C.D.; DeVivo, D.C.; Lenaz, G.; Wiedmann, M.; Manfredi, G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Mol. Genet. 2004, 13, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Voets, A.M.; Lindsey, P.J.; Vanherle, S.J.; Timmer, E.D.; Esseling, J.J.; Koopman, W.J.; Willems, P.H.; Schoonderwoerd, G.C.; De Groote, D.; Poll-The, B.T.; et al. Patient-derived fibroblasts indicate oxidative stress status and may justify antioxidant therapy in OXPHOS disorders. Biochim. Biophys. Acta 2012, 1817, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Lopez, L.C.; Gilkerson, R.W.; Dorado, B.; Coku, J.; Naini, A.B.; Lagier-Tourenne, C.; Schuelke, M.; Salviati, L.; Carrozzo, R.; et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3733–3743. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.; Rivera, H.; Sanchez-Arago, M.; Blazquez, A.; Merinero, B.; Ugalde, C.; Arenas, J.; Cuezva, J.M.; Martin, M.A. Mitochondrial bioenergetics and dynamics interplay in complex I-deficient fibroblasts. Biochim. Biophys. Acta 2010, 1802, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Verkaart, S.; Koopman, W.J.; Cheek, J.; van Emst-de Vries, S.E.; van den Heuvel, L.W.; Smeitink, J.A.; Willems, P.H. Mitochondrial and cytosolic thiol redox state are not detectably altered in isolated human NADH:ubiquinone oxidoreductase deficiency. Biochim. Biophys. Acta 2007, 1772, 1041–1051. [Google Scholar] [CrossRef]
- Hargreaves, I.P. Coenzyme Q10 as a therapy for mitochondrial disease. Int. J. Biochem. Cell Biol. 2014, 49, 105–111. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Soiferman, D.; Moore, D.G.; Burte, F.; Saada, A. Evaluating the therapeutic potential of idebenone and related quinone analogues in Leber hereditary optic neuropathy. Mitochondrion 2017, 36, 36–42. [Google Scholar] [CrossRef]
- Bodmer, M.; Vankan, P.; Dreier, M.; Kutz, K.W.; Drewe, J. Pharmacokinetics and metabolism of idebenone in healthy male subjects. Eur. J. Clin. Pharmacol. 2009, 65, 493–501. [Google Scholar] [CrossRef]
- Giorgio, V.; Schiavone, M.; Galber, C.; Carini, M.; Da Ros, T.; Petronilli, V.; Argenton, F.; Carelli, V.; Acosta Lopez, M.J.; Salviati, L.; et al. The idebenone metabolite QS10 restores electron transfer in complex I and coenzyme Q defects. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 901–908. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef]
- Parodi-Rullan, R.M.; Chapa-Dubocq, X.; Rullan, P.J.; Jang, S.; Javadov, S. High Sensitivity of SIRT3 Deficient Hearts to Ischemia-Reperfusion Is Associated with Mitochondrial Abnormalities. Front. Pharmacol. 2017, 8, 275. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dolle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Imai, S. The NAD World: A new systemic regulatory network for metabolism and aging—Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem. Biophys. 2009, 53, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Pittelli, M.; Felici, R.; Pitozzi, V.; Giovannelli, L.; Bigagli, E.; Cialdai, F.; Romano, G.; Moroni, F.; Chiarugi, A. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol. 2011, 80, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Chang, P. New PARP targets for cancer therapy. Nat. Rev. Cancer 2014, 14, 502–509. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. Targeting sirtuin 1 to improve metabolism: All you need is NAD(+)? Pharmacol. Rev. 2012, 64, 166–187. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Auwerx, J. Modulating NAD(+) metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Auwerx, J. NAD(+) metabolism: A therapeutic target for age-related metabolic disease. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 397–408. [Google Scholar] [CrossRef]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. TEM 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, J.C.; Houtkooper, R.H. Sirtuin activation as a therapeutic approach against inborn errors of metabolism. J. Inherit. Metab. Dis. 2016, 39, 565–572. [Google Scholar] [CrossRef]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef]
- Felici, R.; Cavone, L.; Lapucci, A.; Guasti, D.; Bani, D.; Chiarugi, A. PARP inhibition delays progression of mitochondrial encephalopathy in mice. Neurother. J. Am. Soc. Exp. Neurother. 2014, 11, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsstrom, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef] [PubMed]
- van de Weijer, T.; Phielix, E.; Bilet, L.; Williams, E.G.; Ropelle, E.R.; Bierwagen, A.; Livingstone, R.; Nowotny, P.; Sparks, L.M.; Paglialunga, S.; et al. Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes 2015, 64, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Gattoni, G.; Papa, F.; Santeramo, A.; Pacelli, C.; Cocco, T.; Micelli, L.; Sardaro, N.; Larizza, M.; Scivetti, M.; et al. The beta-adrenoceptor agonist isoproterenol promotes the activity of respiratory chain complex I and lowers cellular reactive oxygen species in fibroblasts and heart myoblasts. Eur. J. Pharmacol. 2011, 652, 15–22. [Google Scholar] [CrossRef]
- Peterson, Y.K.; Cameron, R.B.; Wills, L.P.; Trager, R.E.; Lindsey, C.C.; Beeson, C.C.; Schnellmann, R.G. beta2-Adrenoceptor agonists in the regulation of mitochondrial biogenesis. Bioorg. Med. Chem. Lett. 2013, 23, 5376–5381. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef]
- Papa, S.; Rasmo, D.D.; Technikova-Dobrova, Z.; Panelli, D.; Signorile, A.; Scacco, S.; Petruzzella, V.; Papa, F.; Palmisano, G.; Gnoni, A.; et al. Respiratory chain complex I, a main regulatory target of the cAMP/PKA pathway is defective in different human diseases. FEBS Lett. 2012, 586, 568–577. [Google Scholar] [CrossRef]
- Papa, S.; Scacco, S.; De Rasmo, D.; Signorile, A.; Papa, F.; Panelli, D.; Nicastro, A.; Scaringi, R.; Santeramo, A.; Roca, E.; et al. cAMP-dependent protein kinase regulates post-translational processing and expression of complex I subunits in mammalian cells. Biochim. Biophys. Acta 2010, 1797, 649–658. [Google Scholar] [CrossRef]
- Wills, L.P.; Trager, R.E.; Beeson, G.C.; Lindsey, C.C.; Peterson, Y.K.; Beeson, C.C.; Schnellmann, R.G. The beta2-adrenoceptor agonist formoterol stimulates mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 2012, 342, 106–118. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, L.; Qi, Y.; Xu, H. Mitochondrial cAMP signaling. Cell. Mol. Life Sci. CMLS 2016, 73, 4577–4590. [Google Scholar] [CrossRef]
- Gerhart-Hines, Z.; Dominy, J.E., Jr.; Blattler, S.M.; Jedrychowski, M.P.; Banks, A.S.; Lim, J.H.; Chim, H.; Gygi, S.P.; Puigserver, P. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol. Cell 2011, 44, 851–863. [Google Scholar] [CrossRef]
- Vockley, J.; Charrow, J.; Ganesh, J.; Eswara, M.; Diaz, G.A.; McCracken, E.; Conway, R.; Enns, G.M.; Starr, J.; Wang, R.; et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol. Genet. Metab. 2016, 119, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Marsden, D.; McCracken, E.; DeWard, S.; Barone, A.; Hsu, K.; Kakkis, E. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment--A retrospective chart review. Mol. Genet. Metab. 2015, 116, 53–60. [Google Scholar] [CrossRef]
- Olsen, R.K.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.C.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain J. Neurol. 2007, 130, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.K.J.; Konarikova, E.; Giancaspero, T.A.; Mosegaard, S.; Boczonadi, V.; Matakovic, L.; Veauville-Merllie, A.; Terrile, C.; Schwarzmayr, T.; Haack, T.B.; et al. Riboflavin-Responsive and -Non-responsive Mutations in FAD Synthase Cause Multiple Acyl-CoA Dehydrogenase and Combined Respiratory-Chain Deficiency. Am. J. Hum. Genet. 2016, 98, 1130–1145. [Google Scholar] [CrossRef]
- Xu, J.; Li, D.; Lv, J.; Xu, X.; Wen, B.; Lin, P.; Liu, F.; Ji, K.; Shan, J.; Li, H.; et al. ETFDH Mutations and Flavin Adenine Dinucleotide Homeostasis Disturbance Are Essential for Developing Riboflavin-Responsive Multiple Acyl-Coenzyme A Dehydrogenation Deficiency. Ann. Neurol. 2018, 84, 659–673. [Google Scholar] [CrossRef]
- Tubbs, E.; Rieusset, J. Metabolic signaling functions of ER-mitochondria contact sites: Role in metabolic diseases. J. Mol. Endocrinol. 2017, 58, R87–R106. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Sagua, R.; Parra, V.; Lopez-Crisosto, C.; Diaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Arduino, D.M.; Perocchi, F. Pharmacological modulation of mitochondrial calcium homeostasis. J. Physiol. 2018, 596, 2717–2733. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2015, 3, 83. [Google Scholar] [CrossRef]
- Schrader, M.; Godinho, L.F.; Costello, J.L.; Islinger, M. The different facets of organelle interplay-an overview of organelle interactions. Front. Cell Dev. Biol. 2015, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Shai, N.; Yifrach, E.; van Roermund, C.W.T.; Cohen, N.; Bibi, C.; IJlst, L.; Cavellini, L.; Meurisse, J.; Schuster, R.; Zada, L.; et al. Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 2018, 9, 1761. [Google Scholar] [CrossRef]
- Birault, V.; Solari, R.; Hanrahan, J.; Thomas, D.Y. Correctors of the basic trafficking defect of the mutant F508del-CFTR that causes cystic fibrosis. Curr. Opin. Chem. Biol. 2013, 17, 353–360. [Google Scholar] [CrossRef]
- Davis, P.B. Another Beginning for Cystic Fibrosis Therapy. N. Engl. J. Med. 2015, 373, 274–276. [Google Scholar] [CrossRef]
- Quon, B.S.; Rowe, S.M. New and emerging targeted therapies for cystic fibrosis. BMJ 2016, 352, i859. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutated Gene | Amino Acid Change | Amino Acid Change | Effects | References |
|---|---|---|---|---|
| p.Pro50His | p.Asp608His | + | [97] | |
| p.Arg51Gly | p.Glu174Lys | + | [84] | |
| p.Ser113Leu | p.Ser113Leu | + | [78,80,97,98], this review | |
| p.Ser113Leu | p.Ser38fs | + | [78,99,100], this review | |
| p.Ser113Leu | p.Ser38AlafsX35 | + | [97,98] | |
| p.Ser113Leu | p.Ser38AlafsX36 | + | [100], this review | |
| CPT2 | p.Ser113Leu | p.Arg124Gln | + | [78,99,100], this review |
| p.Ser113Leu | p.Asn146Thr | + | [78,100], this review | |
| p.Ser113Leu | p.Phe188Ser | + | [80] | |
| p.Ser113Leu | p.Trp201X | + | [97,98] | |
| p.Ser113Leu | p.Gly377Asp | + | [97,98] | |
| p.Ser113Leu | p.Phe448Leu | + | this review | |
| p.Ser113Leu | p.Tyr628Ser | + | [80,100] | |
| p.Asp328Gly | p.Asp328Gly | - | [78,100] | |
| p.Gln13X | p.Arg286Gly | - | [39,87] | |
| p.Gly34fsX60 | p.Gly34fsX60 | - | [39] | |
| p.Pro65Leu+Lys247Gln | p.Pro65Leu+Lys247Gln | - | [39] | |
| p.Pro89Ser | p.Ala536fsX550 | ± | [39] | |
| p.Pro91Gln | p.Gly193Arg | + | [39] | |
| p.Asn122Asp | p.Asn122Asp | - | [39,86,87] | |
| p.Ser148Gly | p.Arg511Trp | ± | [39] | |
| p.Val174Met | p.Val174Met | + | [39,86,87,100], this review | |
| p.Val174Met | p.Glu609Lys | + | [101] | |
| p.Gly185Ser | p.Asn252_His293del42 | - | [39,87,100] | |
| p.Gly185Ser | p.Gly294Glu | - | [39] | |
| p.Gly222Arg | p.Gly222Arg | - | [39,87] | |
| p.Gly222Arg | p.Glu504Asp | + | [39,99,100], this review | |
| p.Arg229X | p.Arg613Trp | - | [39] | |
| p.Asn252_His293del42 | p.Gly441Asp | - | [39,87,100] | |
| p.Thr260Met | p.Ala640fsX679 | - | [39,87] | |
| p.Lys264Glu | p.Lys264Glu | +; ± | [39,100], this review | |
| p.Lys264Glu | p.Ala333fsX358 | ± | [39,84], this review | |
| VLCAD | p.Lys264Glu | p.Met437Val | + | [39,100] |
| p.Val283Ala | p.Val283Ala | + | [39,86,99,100], this review | |
| p.Val283Ala | p.Lys382Asn | ± | this review | |
| p.Val283Ala | p.Gly441Asp | + | [39,100], this review | |
| p.Val283Ala | p.Glu462Lys | ± | [39,100], this review | |
| p.Val283Ala | p.? | + | [101] | |
| p.Lys299Met | p.Leu502Gln | ± | [39] | |
| p.Ala304Thr | p.Gly439Asp | ± | [39,87], this review | |
| p.Ala333fsX358 | p.Lys382Gln | + | [39,100], this review | |
| p.Arg366His | p.Arg453X | ± | [39,87] | |
| p.Lys382Gln | p.Gly447Arg | +; ± | [39,84,99,100], this review | |
| p.Lys382Gln | p.Glu130fsX216 | ± | [39,87], this review | |
| p.Asp405His | p.Arg450His | ± | [39,87] | |
| p.Ala416Thr | p.Arg450His | ± | [39,100], | |
| p.Ala416Thr | p.Lys600fsX679 | ± | [39,87] | |
| p.Arg453Gln | p.Arg453Gln | - | [39,87] | |
| p.Arg456His | p.Arg615Gln | + | [39,100], this review | |
| p.Arg469Trp | p.Arg469Trp | - | [39] | |
| p.Lys540Pro | p.? | + | [101] | |
| p.Thr37SerfsX6 | p.? | + | [89] | |
| p.Lys267SerfsX7 | p.Lys267SerfsX7 | - | [89] | |
| p.Lys353IlefsX19 | p.Lys353IlefsX19 | - | [89] | |
| p.Glu446Lys | p.Gly703Arg | - | [89] | |
| p.Pro467_Ile495del | Pro467_Ile495del | + | [89] | |
| p.Glu510Gln | p.Glu510Gln | - | [89] | |
| HADHA | p.Glu510Gln | p.Thr37SerfsX6 | + | [89] |
| p.Glu510Gln | p.Ile305Asn | + | [89] | |
| p.Glu510Gln | p.Gly328Arg and p.Gln358Lys | + | [89] | |
| p.Glu510Gln | p.Leu571Pro | - | [89] | |
| p.Glu510Gln | p.Arg676His | - | [89] | |
| p.Glu510Gln | p.? | - | [102] | |
| p.Lys664ValfsX2 | p.Lys664ValfsX2 | - | [89] | |
| p.Val705Asp | p.Val705Asp | - | [89] | |
| p.Asn114Ser | p.? | + | [89] | |
| p.Arg247Cys | p.Asp273IlefsX20 | - | [89] | |
| HADHB | p.Ser383Leu | p.Ser383Leu | - | [89] |
| p.Asn389Asp | p.Asn389Asp | - | [89] | |
| p.Arg444Lys | p.Arg444Lys | - | [89] | |
| p.Val455Gly | p.Val455Gly | - | [89] | |
| p.Arg83Cys | p.Arg83Cys | ± | [102] | |
| SCAD | p.Arg83Cys | p.Gly185Ser | ± | [102] |
| p.Gly185Ser | p.Gly185Ser | ± | [102] | |
| p.Arg380Trp | p.Arg380Trp | ± | [102] | |
| MCAD | p.Lys329Glu | p.? | ± | [102] |
| Patients | Mutated Gene | Amino Acid Change | Amino Acid Change | Effects | References |
|---|---|---|---|---|---|
| p.Arg59X | p.Thr423Met | + | [112,113,114] | ||
| p.Tyr204Cys | p.Cys206Gly | + | [105,115,116] | ||
| p.Glu214Lys | ex 8 del | + | [116] | ||
| NDUFV1 | p.Ala432Pro | p.Gly388X | + | [116] | |
| p.Glu377Lys | p.Glu377Lys | - | [116] | ||
| p.Arg386His | p.Pro252Arg | - | [116] | ||
| p.Arg386Cys | p.Ser251fsX44 | - | [116] | ||
| p.Gln381Arg | p.Arg386His | - | [117] | ||
| p.Arg386Cys | p.Arg386Cys | + | [117] | ||
| NDUFV2 | ex 2 del | ex 2 del | + | [105,116] | |
| p.Ala183Thr | p.Tyr70fs*6 | + | [117] | ||
| p.Gly19_Val40del | p.Gly19_Val40del | + | [117] | ||
| NDUFS1 | Del222 | p.Asp252Gly | - | [105] | |
| del entire gene | p.Met707Val | - | [116] | ||
| P.Arg241Trp | p.Arg557X | - | [116] | ||
| p.Asp380Val | ? | - | [117] | ||
| p.Val228Ala | p.Asp252Gly | ± | [117] | ||
| CI-deficient | p.Gln522Lys | p.Gln522Lys | + | [118,119,120] | |
| p.Arg557X | p.Asp618Asn | - | [112,113,114] | ||
| NDUFS2 | p.Arg228Gln | p.Arg228Gln | + | [110,112,113,114] | |
| p.Met292Thr | p.Met443Lys | + | [117] | ||
| p.Met292Thr | p.Arg118Gln | - | [117] | ||
| p.Ser413Pro | p.Ser413Pro | - | [112,117] | ||
| NDUFS3 | p.Thr145Ile | p.Arg199Trp | + | [105,116] | |
| NDUFS4 | ex 3-4 del | ex 3-4 del | - | [105] | |
| p.Trp97X | p.Trp97X | + | [112,117,119] | ||
| p.Arg106X | p.Arg106X | + | [112,113] | ||
| NDUFS6 | ex 3 and 4 del | ex 3 and 4 del | - | [116] | |
| p.Leu23Trpfs*35 | p.Leu23Trpfs*35 | ± | [117] | ||
| NDUFS7 | Ala6_Arg213del | Ala6_Arg213del | - | [117] | |
| p.Val122Met | p.Val122Met | + | [112,113,114] | ||
| p.Arg145His | p.Arg145His | + | [117] | ||
| NDUFS8 | p.Arg54Trp | p.Gly20Arg | - | [121] | |
| p.Arg94Cys | p.Arg94Cys | ± | [112,113,114] | ||
| C8orf38 | p.Gln99Arg | p.Gln99Arg | - | [116] | |
| C20orfF7 | p.Gly250Val | p.Gly250Val | ± | [110] | |
| NDUFAF4 | p.Thr194Cys | p.Thr194Cys | ± | [110] | |
| FOXRED1 | p.Arg352Trp | p.Arg352Trp | ± | [110] | |
| NDUFA12L | p.Met1Leu | p.Met1Leu | ± | [110] | |
| ACAD9 | p.Arg518His | p.Arg518His | + | [115] | |
| CII-deficient | NFU1 | p.Gly208Cys | p.Gly208Cys | - | [122] |
| FP | p.Gly555Glu | p.Gly555Glu | - | [122] | |
| CIII-deficient | BCS1 | p.Pro99Leu | p.Pro99Leu | + | [105] |
| COX10 | p.Asn204Lys | p.Asn204Lys | + | [105,116] | |
| SURF1 | p.Leu105X | p.Leu105X | - | [116,122] | |
| p.Gly180Glu | ? | - | [116] | ||
| CIV-deficient | p.Pro183fsX189 | p.Pro183fsX189 | - | [105] | |
| p.Ser282Cysfs | p.Ser282Cysfs | - | [122] | ||
| LRPPRC | P.Ala354Val | P.Ala354Val | - | [123] | |
| COX6B1 | p.Arg20Cys | p.Arg20Cys | ± | [124,125,126] | |
| COX4I1 | p.Lys101Asn | p.Lys101Asn | + | [125] | |
| SCO2 | p.Gln53X | p.Glu140Lys | + | [127] | |
| EFT | p.Arg333trp | p.Arg333trp | ± | [111,126] | |
| Mutiple RC defects | GFM1 | p.Leu398Pro | p.Leu398Pro | ± | [111] |
| MRPSS22 | p.Arg170His | p.Arg170His | ± | [111,126] | |
| TRMU | p.Tyr77His | p.Tyr77His | ± | [111] |
| Compounds | Disorders | References | |
|---|---|---|---|
| BZ (PPARα/δ) | FAO | [39,78,80,84,85,86,87,88,89,97,102] | |
| RC | [105,107,109,110,111,124,129,130,131,134] | ||
| GW7647 (PPARα) | FAO | [80] | |
| PPAR agonists | RC | [105] | |
| GW0742 (PPARδ) | FAO | [80] | |
| RC | [105] | ||
| Pioglitazone (PPARγ) | FAO | this review | |
| Stilbenes | trans-RSV | FAO | [99,100,101] |
| RC | [110,111,115,116,117,122,124,126,149,150] | ||
| cis-RSV | FAO | [99] | |
| piceid | FAO | [99] | |
| RSV metabolites | RSV-3-O-glucuronide | [99] | |
| RSV-4-O-glucuronide | FAO | [99] | |
| RSV-3-O-sulfate | [99] | ||
| di-hydro-RSV | [99] | ||
| AMPK activators | AICA-riboside | FAO | [100], this review |
| RC | [110,111,124,129,192] | ||
| A769662 | FAO | [100], this review | |
| GSK773 | FAO | [98] | |
| Antioxidants | NAC | FAO | [101,102] |
| RC | [111,115,124,126] | ||
| Trolox | FAO | [101] | |
| RC | [112,113,114,115] | ||
| MitoQ | FAO | [101] | |
| RC | [115] | ||
| Quinone analogues | RC | [220,222] | |
| JP4-039 | FAO | [101] | |
| RC | [115] | ||
| XJB-5-131 | FAO | [101] | |
| Vitamin C or Ascorbate | FAO | [102] | |
| RC | [111,126] | ||
| NAD+ | NAD+ precursors | RC | [118,125,242,244,245] |
| PARP-1 inhibitor | RC | [118,243,244] | |
| PKA agonists | Dibutyryl-cAMP | RC | [119] |
| 8-Br-cAMP | RC | [127] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djouadi, F.; Bastin, J. Mitochondrial Genetic Disorders: Cell Signaling and Pharmacological Therapies. Cells 2019, 8, 289. https://doi.org/10.3390/cells8040289
Djouadi F, Bastin J. Mitochondrial Genetic Disorders: Cell Signaling and Pharmacological Therapies. Cells. 2019; 8(4):289. https://doi.org/10.3390/cells8040289
Chicago/Turabian StyleDjouadi, Fatima, and Jean Bastin. 2019. "Mitochondrial Genetic Disorders: Cell Signaling and Pharmacological Therapies" Cells 8, no. 4: 289. https://doi.org/10.3390/cells8040289
APA StyleDjouadi, F., & Bastin, J. (2019). Mitochondrial Genetic Disorders: Cell Signaling and Pharmacological Therapies. Cells, 8(4), 289. https://doi.org/10.3390/cells8040289