Modulation of Receptor Tyrosine Kinase Activity through Alternative Splicing of Ligands and Receptors in the VEGF-A/VEGFR Axis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
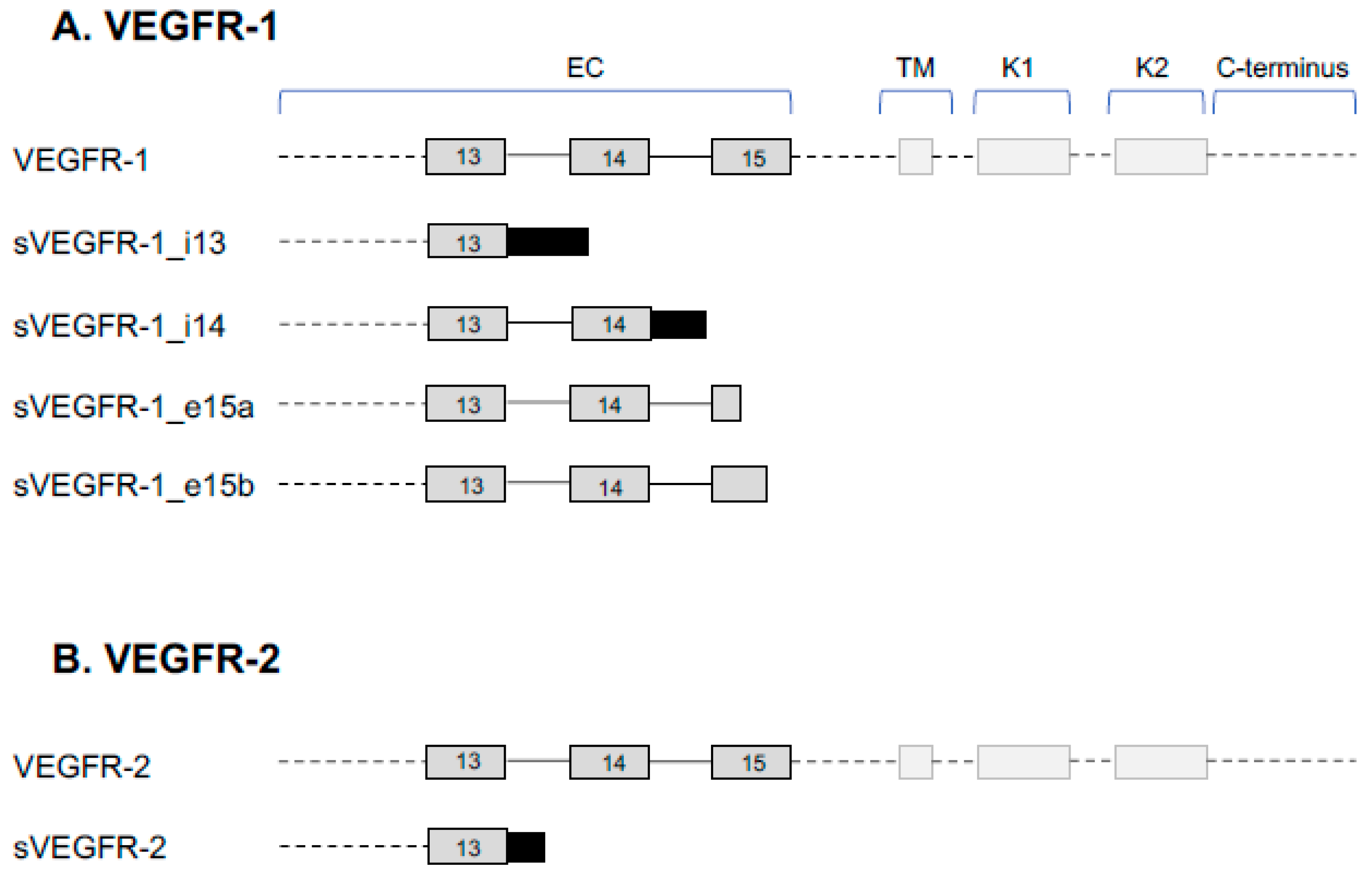
2. VEGFR Splice Variants and Functions
2.1. VEGFR-1 Signaling
2.2. Function of sVEGFR-1
2.3. VEGFR-2 Signaling
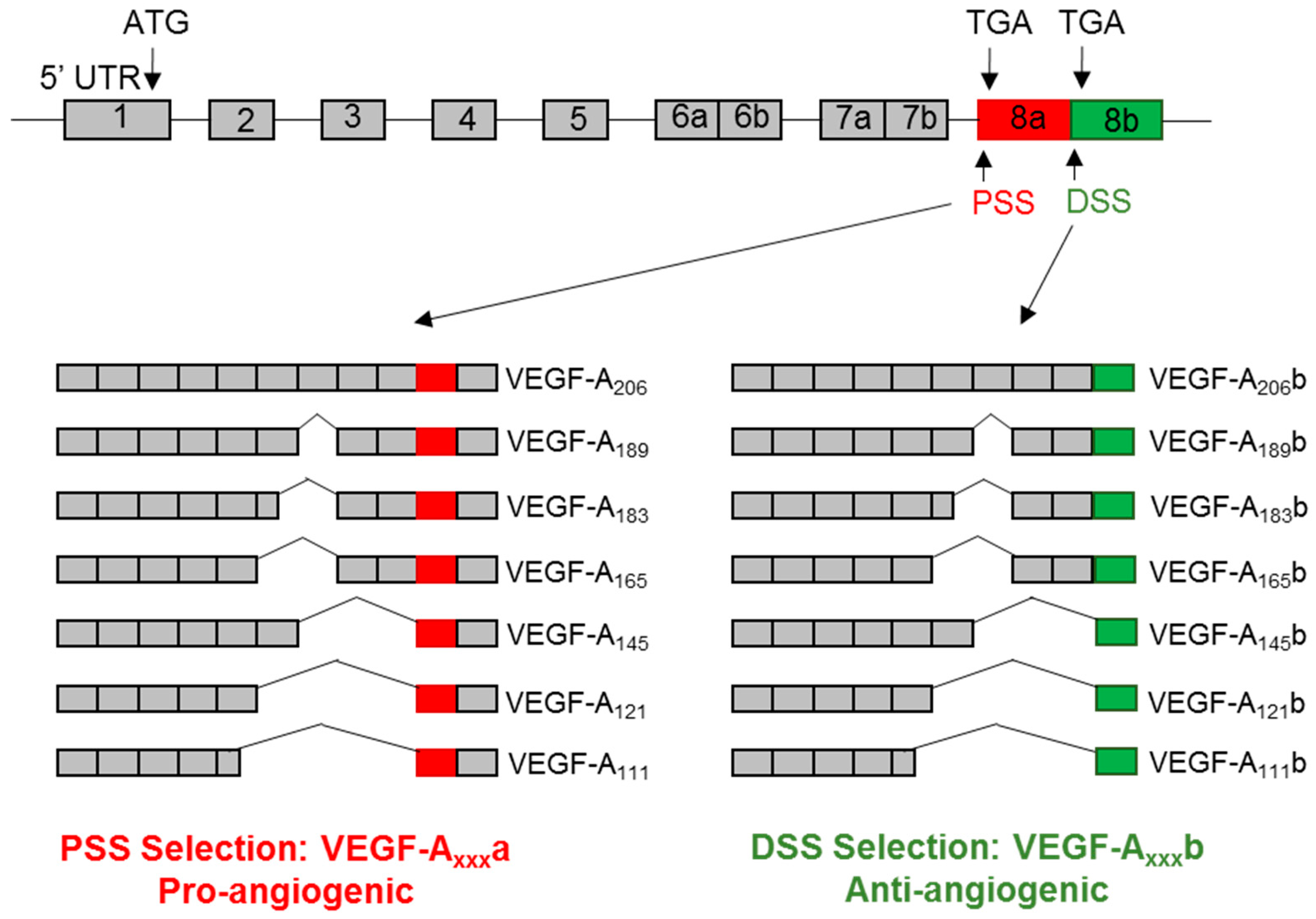
3. VEGF-A Splice Variants
4. VEGFR Signaling
4.1. Role of VEGFR-1 Signlaing and sVEGFR-1 Isoforms
4.2. VEGF-Axxxb Activation of VEGFR-1
4.3. Mehcanisms of VEGFR-2 Signaling
4.4. VEGFR-2 Signaling in Angiogenesis
4.5. VEGFR-2 Signaling in Cell Survival
4.6. VEGFR-2 Signaling in Permeability
4.7. Role of sVEGFR-2
4.8. VEGF-A Isoform Specific Activation of VEGFR-2
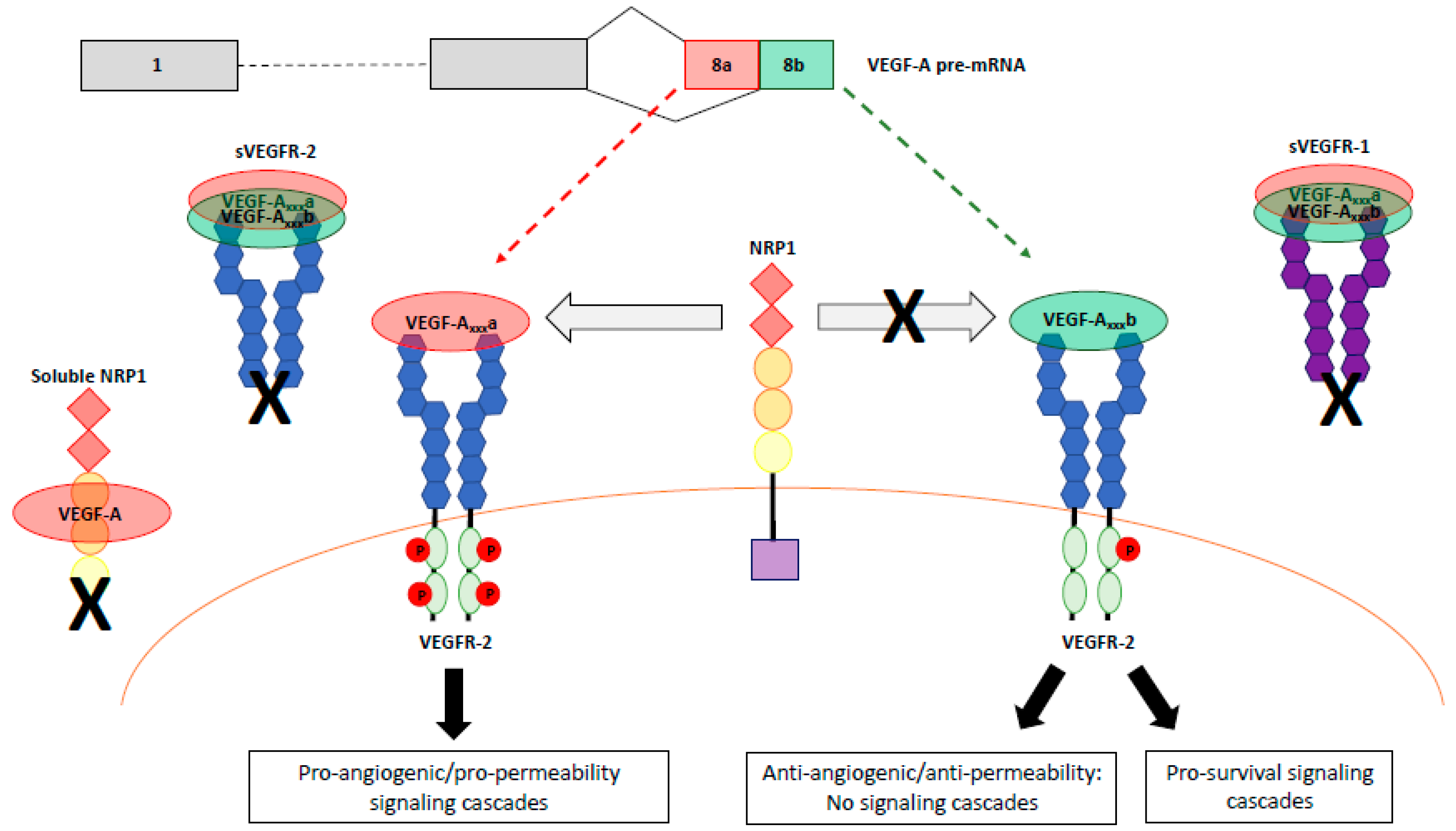
5. VEGFR Signaling Complexes
5.1. VEGFR Heterodimerization
5.2. Roles of Neuropilins NRP1 and NRP2
5.3. NRP1 and NRP2 Splice Variants
6. Regulation of Splicing as a Therapeutic Intervention
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shibuya, M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct. Funct. 2001, 26, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Terman, B.I.; Carrion, M.E.; Kovacs, E.; Rasmussen, B.A.; Eddy, R.L.; Shows, T.B. Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene 1991, 6, 1677–1683. [Google Scholar]
- De Vries, C.; Escobedo, J.A.; Ueno, H.; Houck, K.; Ferrara, N.; Williams, L.T. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992, 21, 989–991. [Google Scholar] [CrossRef]
- Cebe-Suarez, S.; Zehnder-Fjallman, A.; Ballmer-Hofer, K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell. Mol. Life Sci. 2006, 63, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; Condorelli, F.; Park, J.; Ferrara, N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is upregulated by hypoxia. J. Biol. Chem. 1997, 272, 23659–23667. [Google Scholar] [CrossRef]
- Fong, G.H.; Rossant, J.; Gertsenstein, M.; Breitman, M.L. Role of Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef]
- Fong, G.H.; Zhang, L.; Bryce, D.M.; Peng, J. Increased hemangioblast commitment, not vascular disorganization, is the primary defect in flt-1 knock-out mice. Development 1999, 126, 3015–3025. [Google Scholar]
- Hiratsuka, S.; Minowa, O.; Kuno, J.; Noda, T.; Shibuya, M. Flt-1 lacking the tyrosine domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9349–9354. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.L.; Wang, G.; Thomas, K.A. Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem. Biophys. Res. Commun. 1996, 226, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Kearney, J.B.; Johnson, J.H.; Rosenberg, M.P.; Kumar, R.; Bautch, V.L. The vascular endothelial growth factor (VEGF) receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. Am. J. Pathol. 2004, 164, 1531–1535. [Google Scholar] [CrossRef]
- Abou-Faycal, C.; Hatat, A.S.; Gazzeri, S.; Eymin, B. Splice variants of the RTK family: Their role in tumor progression and response to targeted therapy. Int. J. Mol. Sci. 2017, 18, 383. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.P.; Raikwar, N.S.; Kelley, E.A.; Liu, K.Z. Alternate processing of Flt1 transcripts is directed by conserved cis-elements within an intronic region of FLT1 that reciprocally regulates splicing and polyadenylation. Nucleic Acids Res. 2010, 38, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Sun, L.; Tsuruoka, N.; Ishigaki, Y.; Yoshitomi, Y.; Yoshitake, Y.; Yonekura, H. Hypoxia down-regulates sFlt-1 (sVEGFR-1) expression in human microvascular endothelial cells by a mechanism involving mRNA alternative processing. Boichem. J. 2011, 436, 399–407. [Google Scholar] [CrossRef]
- Eubank, T.D.; Roda, J.M.; Liu, H.; O’Neil, T.; Marsh, C.B. Opposing roles for HIF-1α and HIF-2α in the regulation of angiogenesis by mononuclear phagocytes. Blood 2011, 117, 323–332. [Google Scholar] [CrossRef]
- Thomas, R.; Kim, M.H. A HIF-1alpha-dependent autocrine feedback loop promotes survival of serum-deprived prostate cancer cells. Prostate 2009, 69, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liebermann, D.A.; Tront, J.S.; Holtzman, E.J.; Huang, Y.; Hoffman, B.; Geifman-Holtzman, O. Gadd45a stress signaling regulates sFlt-1 expression in preeclampsia. J. Cell. Physiol. 2009, 220, 632–639. [Google Scholar] [CrossRef]
- Boeckel, J.N.; Guarani, V.; Koyanangi, M.; Roexe, T.; Lengeling, A.; Schermuly, R.T.; Gellert, P.; Braun, T.; Zeiher, A.; Dimmeler, S. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 3276–3281. [Google Scholar] [CrossRef]
- Ikeda, T.; Yoshitomi, Y.; Shimasaki, T.; Yamaya, H.; Kobata, T.; Ishigaki, Y.; Tomosugi, N.; Yoshitake, Y.; Yonekura, H. Regulation of soluble Flt-1 (VEGFR-1) production by hnRNP D and protein arginine methylation. Mol. Cell. Biochem. 2016, 413, 155–164. [Google Scholar] [CrossRef]
- Raikwar, N.S.; Liu, K.Z.; Thomas, C.P. Protein kinase C regulates FLT1 abundance and stimulates its cleavage in vascular endothelial cells with the release of a soluble PIGF/VEGF antagonist. Exp. Cell Res. 2013, 319, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- Fuh, G.; Li, B.; Crowley, C.; Cunningham, B.; Wells, J.A. Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J. Biol. Chem. 1998, 273, 11197–11204. [Google Scholar] [CrossRef]
- Shinkai, A.; Ito, M.; Anazawa, H.; Yamaguchi, S.; Shitara, K.; Shibuya, M. Mapping of the sites involved in ligand association and dissociation at the extracellular domain of the kinase insert domain-containing receptor for vascular endothelial growth factor. J. Biol. Chem. 1998, 273, 31283–31288. [Google Scholar] [CrossRef]
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.F.; Breitman, M.L.; Schuh, A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Kou, B.; Li, Y.; Zhang, L.; Zhu, G.; Wang, X.; Li, Y.; Xia, J.; Shi, Y. In vivo inhibition of tumor angiogenesis by a soluble VEGFR-2 fragment. Exp. Mol. Pathol. 2004, 76, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Collet, G.; Lamerant-Fayel, N.; Tertil, M.; El Hafny-Rahbi, B.; Stepniewski, J.; Guichard, A.; Foucault-Collet, A.; Klimkiewicz, K.; Petoud, S.; Matejuk, A.; et al. Hypoxia-regulated overexpression of soluble VEGFR2 controls angiogenesis and inhibits tumor growth. Mol. Cancer Ther. 2014, 13, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, R.J.; Hayashi, T.; Cho, W.G.; Kleinman, M.E.; Dridi, S.; Takeda, A.; Baffi, J.Z.; Yamada, K.; Kaneko, H.; Green, M.G.; et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat. Med. 2009, 15, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar] [PubMed]
- Eswarappa, S.M.; Potdar, A.A.; Koch, W.J.; Fan, Y.; Vasu, K.; Linder, D.; Willard, B.; Graham, L.M.; DiCorleto, P.E.; Fox, P.L. Programmed translational readthrough generates antiangiogenic VEGF-Ax. Cell 2014, 157, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Guyot, M.; Pages, G. VEGF splicing and the role of VEGF splice variants: From physiological-pathological conditions to specific pre-mRNA splicing. Methods Mol. Biol. 2015, 1332, 3–23. [Google Scholar] [PubMed]
- Stevens, M.; Oltean, S. Modulation of VEGF-A alternative splicing as a novel treatment in chronic kidney disease. Genes 2018, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Woolard, J.; Bevan, H.S.; Harper, S.J.; Bates, D.O. Molecular diversity of VEGF-A as a regulator of its biological activity. Microcirculation 2009, 16, 572–592. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.I.; Zachary, I.C. Vascular endothelial growth factor regulates stanniocalcin-1 expression via neuropilin-1-dependent regulation of KDR and synergism with fibroblast growth factor-2. Cell Signal. 2008, 20, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Krilleke, D.; DeErkenez, A.; Schubert, W.; Giri, I.; Robinson, G.S.; Ng, Y.S.; Shima, D.T. Molecualr mapping and functional characterization of the VEGF164 heparin-binding domain. J. Biol. Chem. 2007, 282, 28045–28056. [Google Scholar] [CrossRef]
- Lee, T.Y.; Folkman, J.; Javaherian, K. HSPG-binding peptide corresponding to the exon 6a-encoded domain of VEGF inhibits tumor growth by blocking angiogenesis in a murine model. PLoS ONE 2010, 5, e9945. [Google Scholar] [CrossRef]
- Houck, K.; Leung, D.W.; Rowland, A.M.; Winer, J.; Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 1992, 267, 26031–26037. [Google Scholar]
- Nowak, D.G.; Woolard, J.; Amin, E.M.; Konopatskaya, O.; Saleem, M.A.; Churchill, A.J.; Ladomery, M.R.; Harper, S.J.; Bates, D.O. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 2008, 121, 3487–3495. [Google Scholar] [CrossRef]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef]
- Merdzhanova, G.; Gout, S.; Keramidas, M.; Edmond, V.; Coll, J.L.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. The transcription factor E2F1 and the SR protein SC35 control the ratio of pro-angiogenic verus antiangiogenic isoforms of vascular endothelial growth factor-A to inhibit noevascularization in vivo. Oncogene 2010, 29, 5392–5403. [Google Scholar] [CrossRef]
- Inoue, T.; Kibata, K.; Suzuki, M.; Nakamura, S.; Motoda, R.; Orita, K. Identification of a vascular endothelial growth factor (VEGF) antagonist, sFlt-1, from a human hematopoietic cell line NALM-16. FEBS Lett. 2000, 469, 14–18. [Google Scholar] [CrossRef]
- Kearney, J.B.; Kappas, N.C.; Ellerstrom, C.; DiPaola, F.W.; Bautch, V.L. The VEGF receptor flt-1 (VEGFR-1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood 2004, 103, 4527–4535. [Google Scholar] [CrossRef] [PubMed]
- Kappas, N.C.; Zeng, G.; Chappell, J.C.; Kearney, J.B.; Hazarika, S.; Kallianos, K.G.; Patterson, C.; Annex, B.H.; Bautch, V.L. The VEGF receptor Flt-1 spatially modulates Flk-1 signaling and blood vessel branching. J. Cell Biol. 2008, 181, 847–858. [Google Scholar] [CrossRef]
- Chappell, J.C.; Taylor, S.M.; Ferrara, N.; Bautch, V.L. Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev. Cell 2009, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Orecchia, A.; Lacal, P.M.; Schietroma, C.; Morea, V.; Zambruno, G.; Failla, C.M. Vascualr endothelial growth factor receptor-1 is deposited in the extracellular matrix by endothelial cells and is a ligand for the alpha 5 beta integrin. J. Cell Sci. 2003, 116, 3479–3489. [Google Scholar] [CrossRef] [PubMed]
- Failla, C.M.; Carbo, M.; Morea, V. Positive and negative regulation of angiogenesis by soluble vascular endothelial growth factor receptor-1. Int. J. Mol. Sci. 2018, 19, 1306. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tjwa, M.; Van Hove, I.; Enholm, B.; Neven, E.; Paavonen, K.; Jeltsch, M.; Juan, T.D.; Sievers, R.E.; Chorianopoulos, E.; et al. Reevaluation of the role of VEGF-B suggests a restricted role in the revascularization of the ischemic myocardium. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Bando, H.; Mori, T.; Takahashi, K.; Matsumoto, H.; Yasutome, M.; Weich, H.; Toi, M. Overexpression of soluble vascular endothelial growth factor receptor 1 in colorectal cancer: Association with progression and prognosis. Cancer Sci 2007, 98, 405–410. [Google Scholar] [CrossRef]
- Lamszus, K.; Ulbricht, U.; Matschke, J.; Brockmann, M.A.; Fillbrandt, R.; Westphal, M. Levels of soluble vascular endothelial growth factor (VEGF) receptor 1 in astrocytic tumors and its relation to malignancy, vascularity, and VEGF-A. Clin. Cancer Res. 2003, 9, 1399–1405. [Google Scholar]
- Toi, M.; Bando, H.; Ogawa, T.; Muta, M.; Hornig, C.; Weich, H.A. Significance of vascular endothelial growth factor (VEGF)/soluble VEGF receptor-1 relationship in breast cancer. Int. J. Cancer 2002, 98, 14–18. [Google Scholar] [CrossRef]
- Nagaoka, S.; Yoshida, T.; Akiyoshi, J.; Akiba, J.; Hisamoto, T.; Yoshida, Y.; Abe, M.; Koga, H.; Toirimura, T.; Ueno, T.; et al. The ratio of placenta growth factor to soluble vascular endothelial growth factor receptor-1 predicts the prognosis of hepatocellular carcinoma. Oncol. Rep. 2010, 23, 1647–1654. [Google Scholar]
- Ilhan, N.; Ilhan, N.; Deveci, F. Functional significance of vascular endothelial growth factor and its receptor (receptor-1) in various lung cancer types. Clin. Biochem. 2004, 37, 840–845. [Google Scholar] [CrossRef]
- Ruffini, F.; Failla, C.M.; Orecchia, A.; Bani, M.R.; Dorio, A.S.; Fortes, C.; Zambruno, G.; Graziani, G.; Giavazzi, R.; D’Atri, S.; et al. Expression of soluble vascular endothelial growth factor receptor-1 in cutaneous melanoma: Role in tumour progression. Br. J. Dermatol. 2011, 164, 1061–1070. [Google Scholar] [CrossRef]
- Wierzbowska, A.; Robak, T.; Wrzesien-Kus, A.; Krawczynska, A.; Lech-Maranda, E.; Urbanska-Rys, H. Circulating VEGF and its soluble receptors sVEGFR-1 and sVEGFR-2 in patients with acute leukemia. Eur. Cytokine Netw. 2003, 14, 149–153. [Google Scholar] [PubMed]
- Harris, A.L.; Reusch, P.; Barleon, B.; Hang, C.; Dobbs, N.; Marme, D. Soluble Tie2 and Flt1 extracellular domains in serum of patients with renal cancer and response to antiangiogenic therapy. Clin. Cancer Res. 2001, 7, 1992–1997. [Google Scholar]
- Kulapaditharom, B.; Boonkitticharoen, V.; Sritara, C. Plasma vascular endothelial growth factor dysregulation in defining aggressiveness of head and neck squamous cell carcinoma. J. Oncol. 2012, 2012, 687934. [Google Scholar] [CrossRef] [PubMed]
- Bando, H.; Weich, H.A.; Brokelmann, M.; Horiguchi, S.; Funata, N.; Ogawa, T.; Toi, M. Association between intratumoral free and total VEGF, soluble VEGFR-1, VEGFR-2 and prognosis in breast cancer. Br. J. Cancer 2005, 92, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Tolany, S.M.; Boucher, Y.; Duda, D.G.; Martin, J.D.; Seano, G.; Ancukiewicz, M.; Barry, W.T.; Goel, S.; Lahdenrata, J.; Isakoff, S.J.; et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc. Natl. Acad. Sci. USA 2015, 112, 14325–14330. [Google Scholar] [CrossRef] [PubMed]
- Willett, C.G.; Duda, D.G.; di Tomaso, E.; Boucher, Y.; Ancukiewicz, M.; Sahani, D.V.; Lahdenranta, J.; Chung, D.C.; Fischman, A.J.; Lauwers, G.Y.; et al. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: A multidisciplinary phase II study. J. Clin. Oncol. 2009, 27, 3020–3026. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.Y.; Merchen, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Selke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef]
- McKeeman, G.C.; Ardill, J.E.; Caldwell, C.M.; Hunter, A.J.; McClure, N. Soluble vascular endothelial growth factor receptor-1 (sFlt-1) is increased throughout gestation in patients who have preeclampsia. Am. J. Obstet. Gynecol. 2004, 191, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Krysiak, O.; Bretschneider, A.; Zhong, E.; Webb, J.; Hopp, H.; Verlohren, S.; Fuhr, N.; Lanowska, M.; Nonnenmacher, A.; Vetter, R.; et al. Soluble vascular endothelial growth factor receptor-1 (sFLT-1) mediates downregulation of FLT-1 and prevents activated neutrophils from women with preeclampsia from additional migration by VEGF. Circ. Res. 2005, 97, 1253–1261. [Google Scholar] [CrossRef]
- Palmer, K.R.; Tong, S.; Kaitu’u-Lino, T.J. Plcental-specific sFLT-1: Role in pre-eclamptic pathophysiology and its translational possibilities for clinical prediction and diagnosis. Mol. Hum. Reprod. 2017, 23, 69–78. [Google Scholar] [PubMed]
- Di Marco, G.S.; Kentrup, D.; Reuter, S.; Mayer, A.B.; Golle, L.; Tiemann, K.; Fobker, M.; Engelbertz, C.; Breithardt, G.; Brande, E.; et al. Soluble Flt-1 links microvascular disease with heart failure in CKD. Basic Res. Cardiol. 2015, 110, 30. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Reuter, S.; Hillebrand, U.; Amler, S.; Konig, M.; Larger, E.; Oberleithner, H.; Brand, E.; Pavenstadt, H.; Brand, M. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J. Am. Soc. Npehrol. 2009, 20, 2235–2245. [Google Scholar] [CrossRef]
- Ku, C.H.; White, K.E.; Dei Cas, A.; Hayward, A.; Webster, Z.; Bilous, R.; Marshall, S.; Viberti, G.; Gnudi, L. Inducible overexpression of sFlt-1 in podocytes ameliorates glomerulopathy in diabetic mice. Diabetes 2008, 57, 2824–2833. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Sison, K.; Li, C.; Tian, R.; Wnuk, M.; Sung, H.K.; Jeansson, M.; Zhang, C.; Tucholska, M.; Jones, N.; et al. Soluble FLT1 binds lipid microdomains in podocytes to control cell morphology and glomerular barrier function. Cell 2012, 151, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Ambati, B.K.; Nozaki, M.; Singh, N.; Takeda, A.; Jani, P.D.; Suthar, T.; Albuquerque, R.J.; Richter, E.; Sakurai, E.; Newcomb, M.T.; et al. Corneal avascualrity is due to soluble VEGF receptor-1. Nature 2006, 443, 993–997. [Google Scholar] [CrossRef]
- Uehara, H.; Mamalis, C.; McFadden, M.; Taggart, M.; Stagg, B.; Passi, S.; Earle, P.; Chakravarthy, U.; Hogg, R.E.; Ambati, B.K. The reduction of serum soluble Flt-1 in patients with neovascular age-related macular degeneration. Am. J. Ophthalmol. 2015, 159, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, N.I.; Yano, K.; Okada, H.; Fischer, C.; Howell, M.; Spokes, K.C.; Ngo, L.; Angus, D.C.; Aird, W.C. A prospective, observational study of soluble FLT-1 and vascular endothelial growth factor in sepsis. Shock 2008, 29, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Dumnicka, P.; Kusnierz-Cabala, B.; Sporek, M.; Mazur-Laskowska, M.; Gil, K.; Kuzniewski, M.; Ceranowicz, Z.; Warzacha, Z.; Dembinski, A.; Bonior, J.; et al. Serum concentrations of angiopoietin-2 and soluble fms-like tyrosine kinase 1 (sFlt-1) are associated with coagulopathy among patients with acute pancreatitis. Int. J. Mol. Sci. 2017, 18, 753. [Google Scholar] [CrossRef]
- Goldman, C.K.; Kendll, R.L.; Cabrera, G.; Soroceanu, L.; Heike, Y.; Gillespie, G.Y.; Siegal, G.P.; Mao, X.; Bett, A.J.; Huckle, W.R.; et al. Paracrine expression of a native soluble vascular endothelial growth factor receptor inhibits tumor growth, metastasis, and mortality rate. Proc. Natl. Acad. Sci. USA 1998, 95, 8795–8800. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Defresne, F.; Lair, F.; Vandermeulen, G.; Rath, G.; Dessy, C.; Preat, V.; Feron, O. Delivery of soluble VEGF receptor 1 (sFlt1) by gene electrotransfer as a new antiangiogenic cancer therapy. Mol. Pharm. 2011, 8, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Ueno, H.; Nakanishi, Y.; Sakamoto, T.; Inoue, K.; Shimizu, K.; Oohashi, H.; Hara, N. Suppression of tumor angiogenesis and growth by gene transfer of a soluble form of vascular endothelial growth factor receptor into a remote organ. Cancer Res. 2000, 60, 2169–2177. [Google Scholar] [PubMed]
- Shiose, S.; Sakamoto, T.; Yoshikawa, H.; Hata, Y.; Kawano, Y.; Ishibashi, T.; Inomata, H.; Takayama, K.; Ueno, H. Gene transfer of a soluble receptor of VEGF inhibits the growth of experimental eyelid malignant melanoma. Investig. Ophthalmol. Vis. Sci 2000, 41, 2395–2403. [Google Scholar]
- Ganta, V.C.; Choi, M.; Kutateladze, A.; Annex, B.H. VEGF165b modulates endothelial VEGFR1-STAT3 signaling pathway and angiogenesis in human and experimental peripheral arterial disease. Circ. Res. 2017, 120, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Ruch, C.; Skiniotis, G.; Steinmetz, M.O.; Walz, T.; Ballmer-Hoffer, K. Structure of a VEGF-VEGF receptor complex determined by electron microscopy. Nat. Struct. Mol. Biol. 2007, 14, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Sarabipour, S.; Ballmer-Hofer, K.; Hristova, K. VEGFR-2 conformational switch in response to ligand binding. Elife 2016, 5, e13876. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Kisko, K.; Schleier, T.; Missimer, J.; Ballmer-Hofer, K. Functional and structural characterization of the kinase insert domain and the carboxy terminal domain in VEGF receptor 2 activation. FASEB J. 2014, 28, 4914–4923. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef]
- Dougher, M.; Terman, B.I. Autophosphorylation of KDR in the kinase domain is required for maximal VEGF-stimulated kinase activity and receptor internalization. Oncogene 1999, 18, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Bohman, S.; Dixelius, J.; Berge, T.; Dimberg, A.; Magnusson, P.; Wang, L.; Wikner, C.; Qi, J.H.; Wernstedt, C.; et al. VEGF receptor-2 Y951 signlaing and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 2005, 24, 2342–2353. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Sanyal, S.; Mukhopadhyay, D. Tyrosine residues 951 and 1059 of vascular endothelial growth factor receptor-2 (KDR) are essential for vascular permeability factor/vascular endothelial growth factor-induced endothelium migration and proliferation, respectively. J. Biol. Chem. 2001, 276, 32714–32719. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamaguchi, S.; Chida, K.; Shibuya, M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 2001, 20, 2768–2778. [Google Scholar] [CrossRef]
- Lanahan, A.A.; Lech, D.; Dubrac, A.; Zhang, Z.W.; Eichmann, A.; Simons, M. PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 2014, 130, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Haj, F.G.; Markova, B.; Klaman, L.D.; Bohmer, F.D.; Neel, B.G. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatase-1B. J. Biol. Chem. 2003, 278, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Jackobsson, L.; Bentley, K.; Gerhardt, H. VEGFRs and Notch: A dynamic collaboration in vascular patterning. Biochem. Soc. Trans. 2009, 37, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Meadows, K.N.; Bryant, P.; Pumiglia, K. Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J. Biol. Chem. 2001, 276, 49289–49298. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Ueno, H.; Shibuya, M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 1999, 18, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Ohgimoto, K.; Kataoka, Y.; Yoshida, N.; Shibuya, M. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Abu-ghazaleh, R.; Kabir, J.; Jia, H.; Lobo, M.; Zachary, I. Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. Biochem. J. 2001, 360, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascualr endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Frank, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Harper, S.J. Regulation of vascular permeability by vascular endothelial growth factors. Vasc. Pharmacol. 2002, 39, 225–237. [Google Scholar] [CrossRef]
- Garrido-urbani, S.; Bradfield, P.F.; Lee, B.P.; Imhof, B.A. Vascular and epithelial junctions: A barrier for leucocyte migration. Biochem. Soc. Trans. 2008, 36, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells in Akt-dependent phosphorylation. Nature 1999, 339, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Ebos, J.M.; Bocci, G.; Man, S.; Thorpe, P.E.; Hicklin, D.J.; Zhou, D.; Jia, X.; Kerbel, R.S. A naturally occurring soluble form of vascular endothelial growth factor receptor 2 detected in mouse and human plasma. Mol. Cancer Res. 2003, 2, 315–326. [Google Scholar]
- Kawamura, H.; Li, X.; Harper, S.J.; Bates, D.O.; Claesson_Welsh, L. Vascualr endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 2008, 68, 4683–4692. [Google Scholar] [CrossRef]
- Catena, R.; Larzabal, L.; Larrayoz, M.; Molina, E.; Hermida, J.; Agorreta, J.; Montes, R.; Ruben, P.; Montuenga, L.M.; Calvo, A. VEGF121b and VEGF165b are weakly angiogenic isoforms of VEGF-A. Mol. Cancer 2010, 9, 320. [Google Scholar] [CrossRef]
- Kikuchi, R.; Nakamura, K.; MacLauchlan, S.; Ngo, D.T.; Shimizu, I.; Fuster, J.J.; Katanasaka, Y.; Yoshida, S.; Qiu, Y.; Yamaguchi, T.P.; et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med. 2014, 20, 1464–1471. [Google Scholar] [CrossRef]
- Ngo, D.T.; Farb, M.G.; Kikuchi, R.; Karki, S.; Tiwari, S.; Bigornia, S.J.; Bates, D.O.; LaValley, M.P.; Hamburg, N.M.; Vita, J.A.; et al. Antiangiogenic actions of vascular endothelial growth factor-A165b, an inhibitory isoform of vascular endothelial growth factor-A, in human obesity. Circulation 2014, 130, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Neal, C.R.; Salmon, A.H.J.; Bayes, D.O.; Harper, S.J.; Oltean, S.O. VEGF-A165b protects against proteinuria in a mouse model with progressive depletion of all endogenous VEGF-A splice variants from the kidney. J. Physiol. 2017, 595, 6281–6298. [Google Scholar] [CrossRef] [PubMed]
- Oltean, O.; Qiu, Y.; Ferguson, J.K.; Stevens, M.; Neal, C.; Russell, A.; Kaura, A.; Arkill, K.P.; Harris, K.; Symonds, C.; et al. Vascular endothelial growth factor-A165b is protective and restores endothelial glycocalyx in diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1889–1904. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Neal, C.R.; Salmon, A.H.J.; Bates, D.O.; Harper, S.J.; Oltean, O. Vascular endothelial growth factor-A165b restores normal glomerular water permeability in a diphtheria-toxin mouse model of glomerular injury. Nephron 2018, 139, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Oltean, O.; Neal, C.R.; Mavrou, A.; Patel, P.; Ahad, T.; Alsop, C.; Lee, T.; Sison, K.; Qiu, Y.; Harper, S.J.; et al. VEGF165b overexpression restores normal glomerular water permeability in VEGF164-overexpressing adult mice. Am. J. Physiol. Renal. Physiol. 2012, 303, F1026–F1036. [Google Scholar] [CrossRef] [PubMed]
- Varey, A.H.; Rennel, E.S.; Qiu, Y.; Bevan, H.S.; Perrin, R.M.; Raffy, S.; Dixon, A.R.; Paraskeva, C.; Zaccheo, O.; Hassan, A.B.; et al. VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: Balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br. J. Cancer 2008, 98, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef]
- Rennel, E.S.; Hamdollah-Zadeh, M.A.; Wheatley, E.R.; Magnussen, A.; Schuler, Y.; Kelly, S.P.; Finucane, C.; Ellison, D.; Cebe-Suarez, S.; Ballmer-Hofer, K.; et al. Recombinant human VEGF165b protein is an effective anti-cancer agent in mice. Eur. J. Cancer 2008, 44, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Boudria, A.; Abou Faycal, C.; Jia, T.; Gout, S.; Keramidas, M.; Didier, C.; Lemaitre, N.; Manet, S.; Coll, J.L.; Toffart, A.C.; et al. VEGF165b. a splice variant of VEGF-A, promotes lung tumor progression and escape from anti-angiogenic therapies through a B1 integrin/VEGFR autocrine loop. Oncogene 2018. [Google Scholar] [CrossRef]
- Keyt, B.A.; Berleau, L.T.; Nguyen, H.V.; Chen, H.; Heinsohn, H.; Vandlen, R.; Ferrara, N. The carboxyl-terminal domain (111-165) of vascular endothelial growth factor is critical for its mitogenic potency. J. Biol. Chem. 1996, 271, 7788–7795. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Chathery, Y.; Wu, Y.; Rathore, N.; Tong, R.K.; Peale, F.; Bagri, A.; Tessier-Lavigne, M.; Koch, A.W.; Watts, R.J. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J. Biol. Chem. 2007, 282, 24049–24056. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, G.W.; Bruns, A.F.; Wheatcroft, S.B.; Ponnambalam, S. VEGF-A isoform-specific regulation of calcium ion flux, transcriptional activation and endothelial cell migration. Biol. Open 2015, 4, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, G.W.; Smith, G.A.; Abdul-Zani, I.; Yuldasheva, N.; Mughal, N.A.; Homer-Vanniasinkam, S.; Kearney, M.T.; Zachary, I.C.; Tomlinson, D.C.; Harrison, M.A.; et al. VEGF-A isoforms program differential VEGFR2 signal transduction, trafficking and proteolysis. Biol. Open 2016, 5, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Fuster, M.M.; Lawrence, R.; Esko, J.D. Heparin sulfate regulates VEGF165- and VEGF121-mediated vascular hyperpermeability. J. Biol. Chem. 2011, 286, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.M.; Waltenberger, J.; Yachechko, R.; Mirzapoiazova, T.; Sham, J.S.; Lee, C.G.; Elias, J.A.; Verin, A.D. Neuropilin-1 regulates vascular endothelial growth factor-mediated endothelial permeability. Circ. Res. 2005, 96, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Herve, M.A.; Buteau-Lozano, H.; Mourah, S.; Calvo, F.; Perrot-Applanat, M. VEGF189 stimulates endothelial cells proliferation and migration in vu=itro and up-regulates the expression of Flk-1/KDR mRNA. Exp. Cell Res. 2005, 309, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Rundqvist, H.; Branco, C.; Johnson, R.S. Autocrine VEGF isoforms differentially regulate endothelial cell behavior. Front. Cell Dev. Biol. 2016, 4, 99. [Google Scholar] [CrossRef]
- Mac Gabhann, F.; Popel, A.S. Dimerization of VEGF receptors and implications for signal transduction: A computational study. Biophys. Chem. 2007, 128, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Andersson, C.; Roomans, G.M.; Ito, N.; Claesson-Welsh, L. Signaling properties of VEGF receptor-1 and -2 homo- and heterodimers. Int. J. Biochem. Cell Biol. 2001, 33, 315–324. [Google Scholar] [CrossRef]
- Alam, A.; Herault, J.P.; Barron, P.; Favier, B.; Fons, P.; Delesque-Touchard, N.; Senegas, I.; Laboudie, P.; Bonnin, J.; Cassan, C.; et al. Heterodimerization with vascular endothelial growth factor receptor-2 (VEGFR-2) is necessary for VEGFR-3 activity. Biochem. Biophys. Res. Commun. 2004, 324, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, H.; Kitsukawa, T.; Kawakami, A.; Takagi, S.; Shimizu, M.; Hirata, T. Roles of a neuronal cell-surface molecule, neuropilin, in nerve fiber fasciculation and guidance. Cell Tissue Res. 1997, 290, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Vander Kooi, C.W.; Jusino, M.A.; Perman, B.; Neau, D.B.; Bellamy, H.D.; Leahy, D.J. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. USA 2007, 104, 6152–6157. [Google Scholar] [CrossRef]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Wang, L.; Zeng, H.; Wang, P.; Soker, S.; Mukhopadhyay, D. Neuropilin-1-mediated vascular permeability factor/vascular endothelial growth factor-dependent endothelial cell migration. J. Biol. Chem. 2003, 278, 48848–48860. [Google Scholar] [CrossRef] [PubMed]
- Favier, B.; Alam, A.; Barron, P.; Bonnin, J.; Laboudie, P.; Fons, P.; Mandron, M.; Herault, J.P.; Neufeld, G.; Savi, P.; et al. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood 2006, 108, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, H.; Li, X.; Goishi, K.; van Meeteren, L.A.; Jakobsson, L.; Cebe-Suarez, S.; Shimizu, A.; Edholm, D.; Ballmer-Hofer, K.; Kjellen, L.; et al. Neuropilin-1 in regulation of VEGF-induced activation of p38MAPK and endothelial cell organization. Blood 2008, 112, 3638–3649. [Google Scholar] [CrossRef] [PubMed]
- Gluzman-Poltorak, Z.; Cohen, T.; Shibuya, M.; Neufeld, G. Vascualr endothelial growth factor receptor-1 and neuropilin-2 form complexes. J. Biol. Chem. 2001, 276, 18688–18694. [Google Scholar] [CrossRef]
- Fujisawa, H.; Kitsukawa, T. Receptors for collapsing/semaphorins. Curr. Opin. Neurobiol. 1998, 8, 587–592. [Google Scholar] [CrossRef]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Wollard, J. Molecular pharmacology of VEGF-A isoforms: Binding and signaling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Sarabipour, S.; Mac Gabhann, F. VEGF-A121a binding to neuropilins- A concept revisited. Cell Adhes. Migr. 2018, 12, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, M.; Gagnon, M.L.; Klagsburn, M. Genomic organization of human nueopilin-1 and neuropilin-2 genes: Identification and distribution of splice variants and soluble forms. Genomics 2000, 70, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.L.; Bienlenberg, D.R.; Gechtman, Z.; Miao, H.Q.; Takashima, S.; Soker, S.; Klagsbrun, M. Identification of a natural soluble neuopilin-1 that binds vascular endothelial growth factor: In vivo expression and antitumor activity. Proc. Natl. Acad. Sci. USA 2000, 97, 2573–2578. [Google Scholar] [CrossRef] [PubMed]
- Cackowski, F.C.; Xu, L.; Hu, B.; Cheng, S.Y. Identification of two novel alternatively spliced Neuropilin-1 isoforms. Genomics 2010, 84, 82–94. [Google Scholar] [CrossRef]
- Tao, Q.; Spring, S.C.; Terman, B.I. Characterization of a new alternatively spliced neuropilin-1 isoform. Angiogenesis 2003, 6, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Germmill, R.M.; Nasarra, P.; Nair-Menon, J.; Cappuzzo, F.; Landi, L.; D’Incecco, A.; Uramoto, H.; Yoshida, T.; Haura, E.B.; Armeson, K.; et al. The neuropilin 2 isoform NRP2b uniquely supports TGFbeta-mediated progression in lung cancer. Sci. Signal. 2017, 10, eaag0528. [Google Scholar] [CrossRef] [PubMed]
- Gammons, M.V.; Dick, A.D.; Harper, S.J.; Bates, D.O. SRPK1 inhibition modulates VEGF splicing to reduce pathological neovascularization in a rat model of retinipathy of prematurity. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5797–5806. [Google Scholar] [CrossRef] [PubMed]
- Batson, J.; Toop, H.D.; Redondo, C.; Babaei-Jadidi, R.; Chaikuad, A.; Wearmouth, S.F.; Gibbons, B.; Allen, C.; Tallant, C.; Zhang, J.; et al. Development of potent, selective SRPK1 inhibitors as potential topical therapeutics for neovascular eye disease. ACS Chem. Biol. 2017, 12, 825–832. [Google Scholar] [CrossRef]
- Stevens, M.; Neal, C.R.; Craciun, E.C.; Dronca, M.; Harper, S.J.; Oltean, S. The natural drug DIAVIT is protective in a type II mouse model of diabetic nephropathy. PLoS ONE 2019, 14, e0212910. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stevens, M.; Oltean, S. Modulation of Receptor Tyrosine Kinase Activity through Alternative Splicing of Ligands and Receptors in the VEGF-A/VEGFR Axis. Cells 2019, 8, 288. https://doi.org/10.3390/cells8040288
Stevens M, Oltean S. Modulation of Receptor Tyrosine Kinase Activity through Alternative Splicing of Ligands and Receptors in the VEGF-A/VEGFR Axis. Cells. 2019; 8(4):288. https://doi.org/10.3390/cells8040288
Chicago/Turabian StyleStevens, Megan, and Sebastian Oltean. 2019. "Modulation of Receptor Tyrosine Kinase Activity through Alternative Splicing of Ligands and Receptors in the VEGF-A/VEGFR Axis" Cells 8, no. 4: 288. https://doi.org/10.3390/cells8040288
APA StyleStevens, M., & Oltean, S. (2019). Modulation of Receptor Tyrosine Kinase Activity through Alternative Splicing of Ligands and Receptors in the VEGF-A/VEGFR Axis. Cells, 8(4), 288. https://doi.org/10.3390/cells8040288