Niclosamide Triggers Non-Canonical LC3 Lipidation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cell Culture and Transfection
2.3. Immunoblot Assay
2.4. Immunostaining Assay
2.5. Molecule Capture
2.6. Statistical Analysis
3. Results
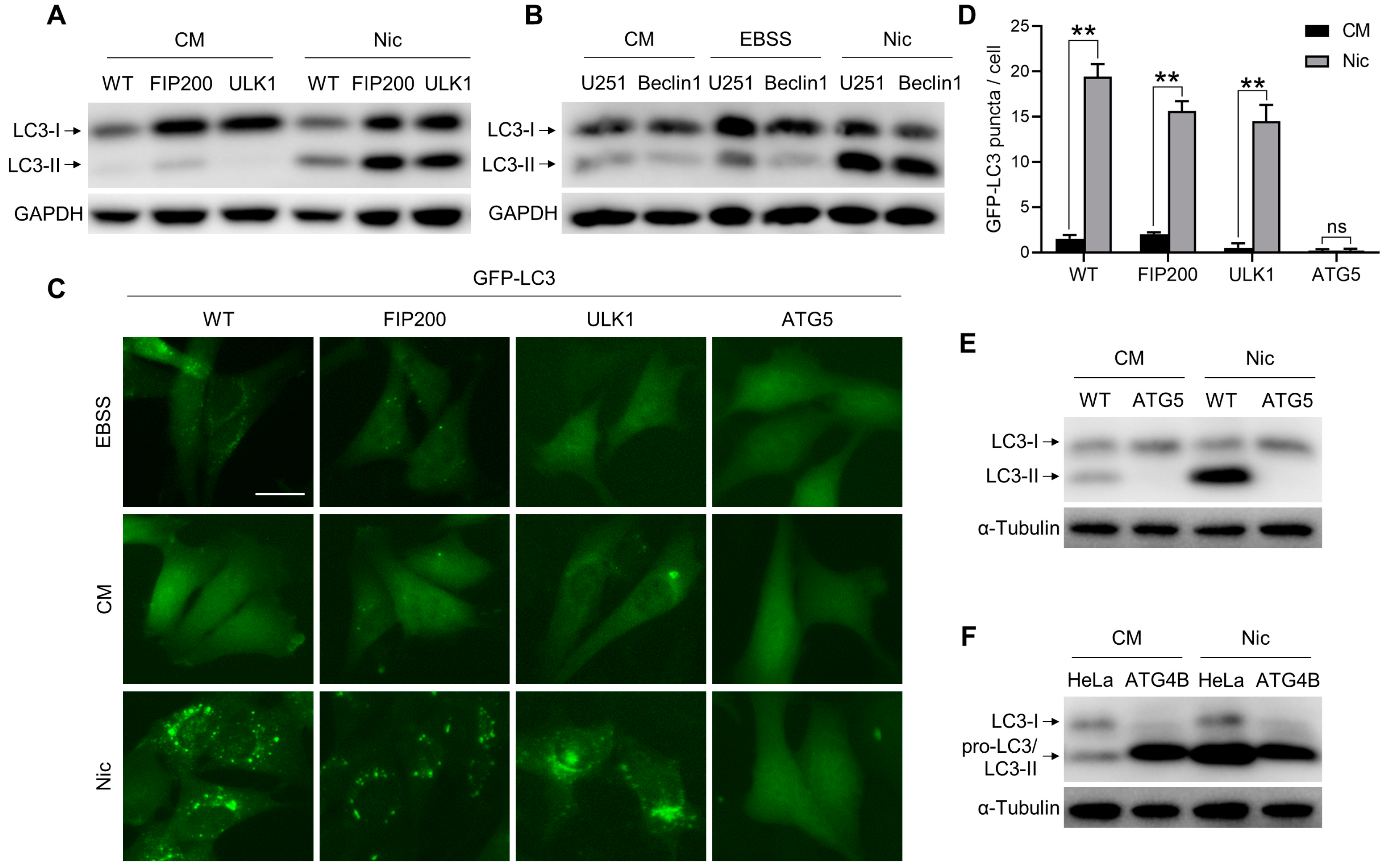
3.1. Nic-Induced NCLL Is Independent of ULK1 Complex and Beclin 1 Complex, But Dependent on Ubiquitin-Like Conjugation Systems
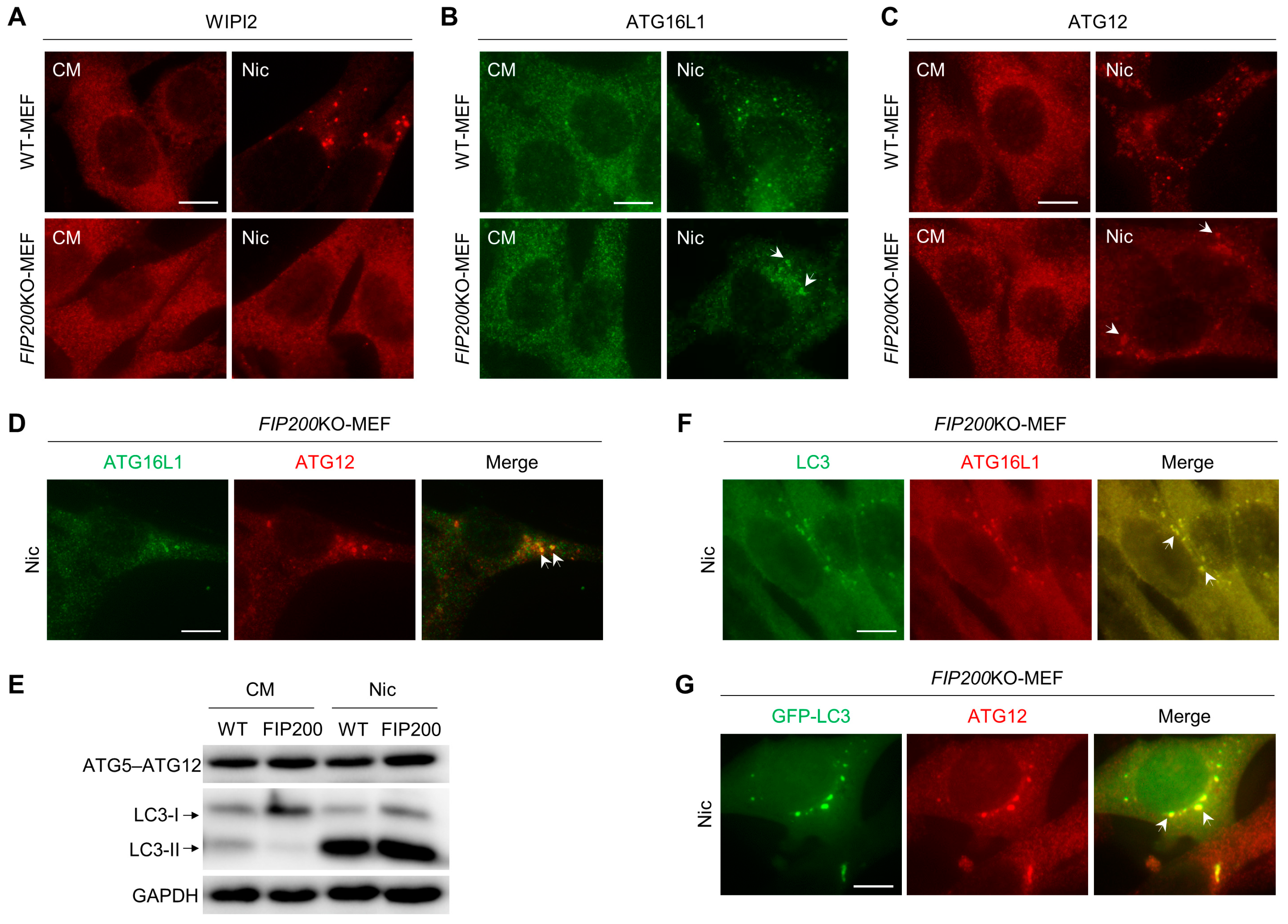
3.2. Nic Recruits the ATG12-ATG5-ATG16L1 Complex via a WIPI2-Independent Pathway
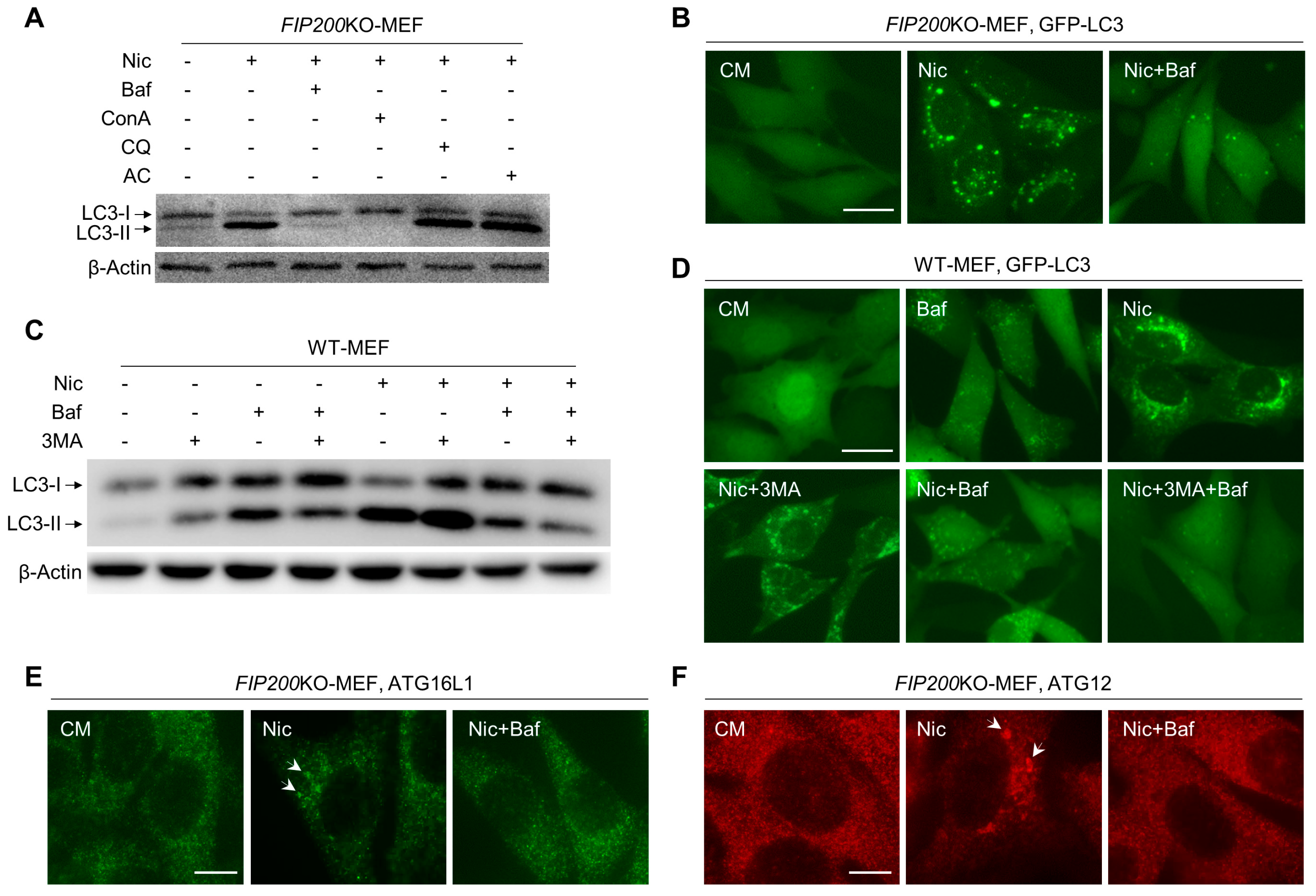
3.3. Baf Is Able to Inhibit Nic-Induced NCLL
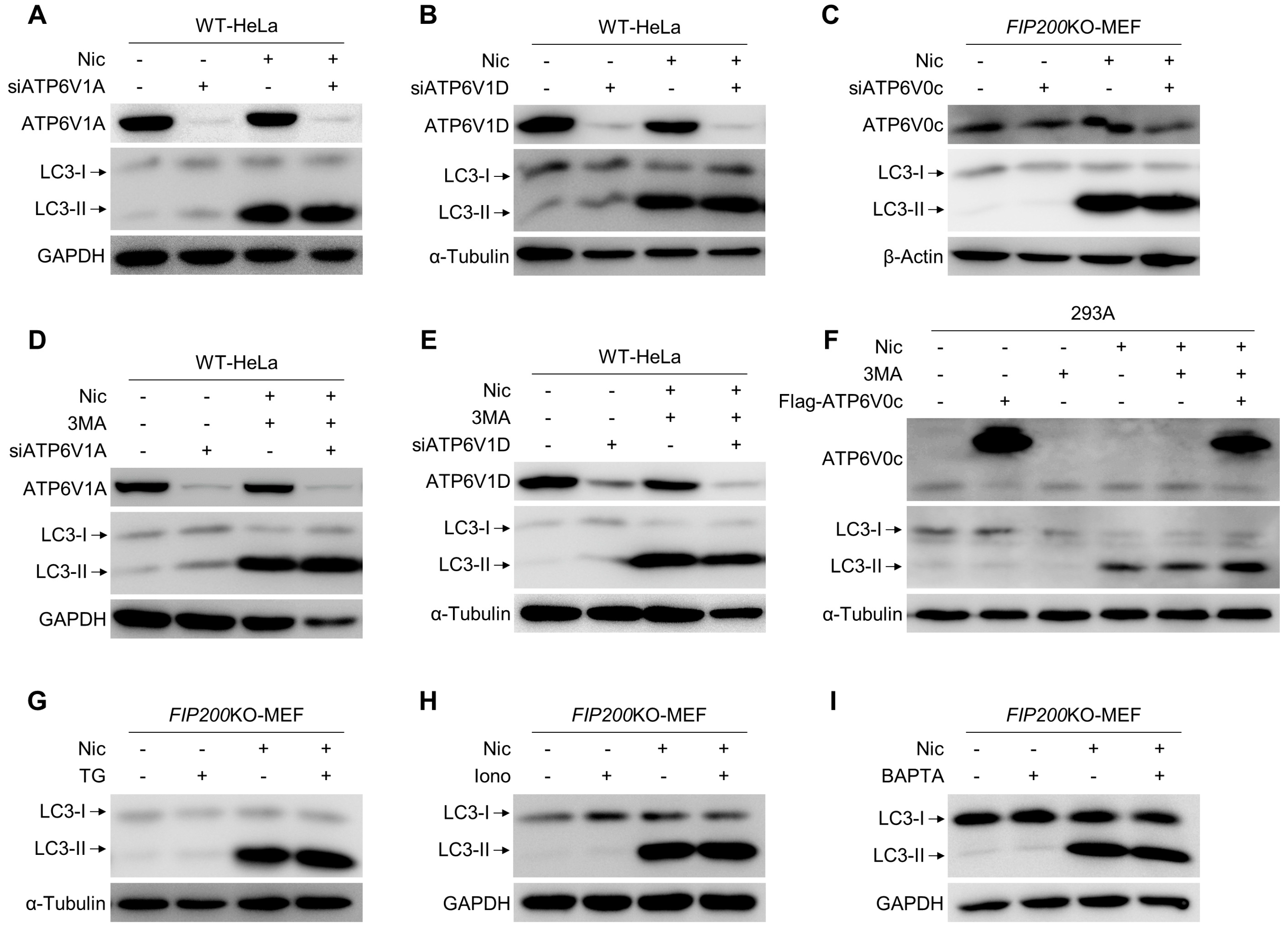
3.4. Nic-Induced NCLL Is Independent of V-ATPase and Ca2+
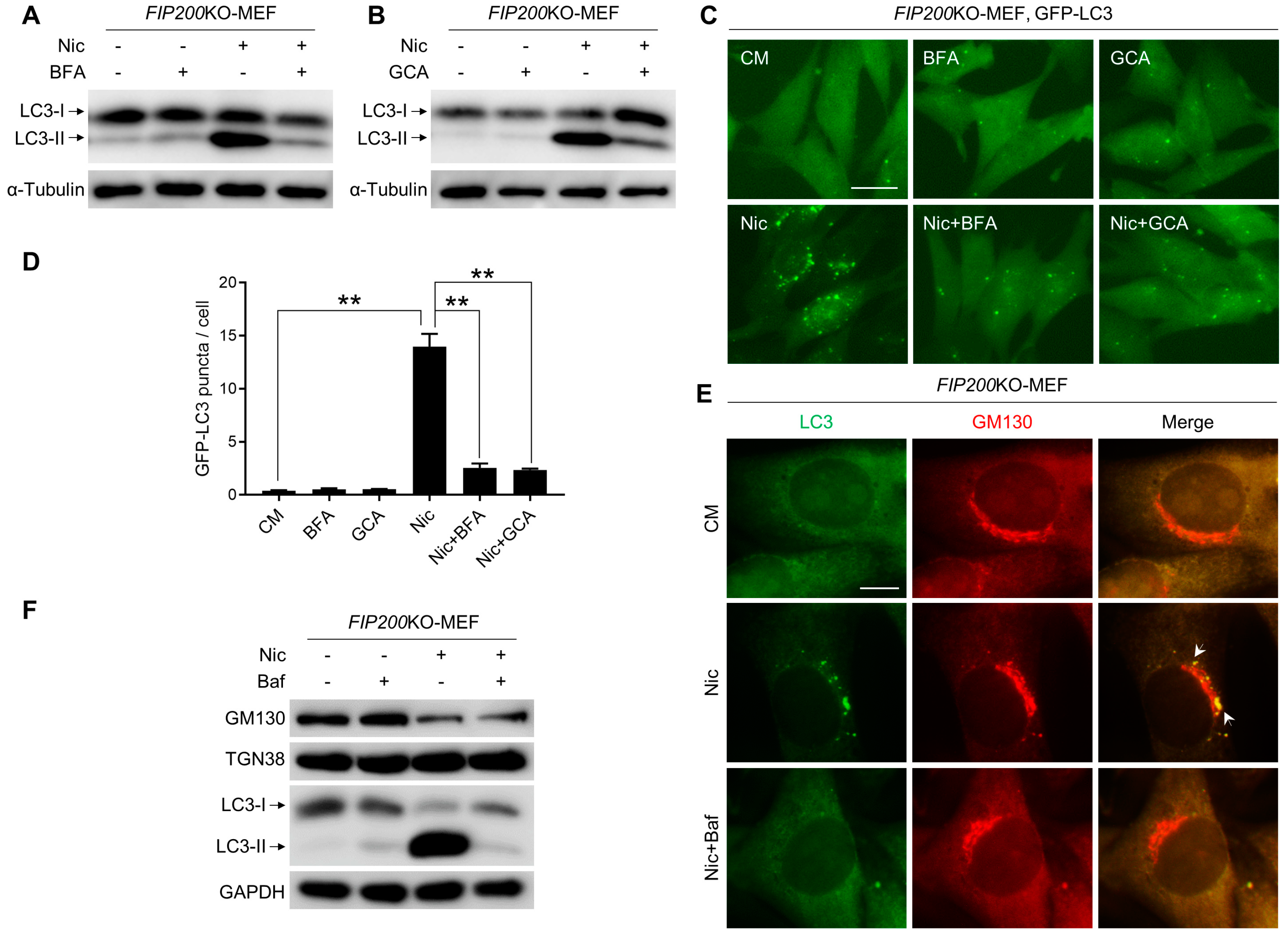
3.5. Nic-Induced NCLL Requires Intact Golgi Complex
3.6. Vimentin Is Involved in Nic-Induced NCLL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug. Discov. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, P.K.; Roberts, M.J.; Arend, R.C.; Samant, R.S.; Buchsbaum, D.J. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014, 349, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Khambu, B.; Zhang, H.; Kang, J.H.; Chen, X.; Chen, D.; Vollmer, L.; Liu, P.Q.; Vogt, A.; Yin, X.M. Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J. Biol. Chem. 2013, 288, 35769–35780. [Google Scholar] [CrossRef] [PubMed]
- Balgi, A.D.; Fonseca, B.D.; Donohue, E.; Tsang, T.C.; Lajoie, P.; Proud, C.G.; Nabi, I.R.; Roberge, M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS ONE 2009, 4, e7124. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ren, X.R.; Piao, H.; Zhao, S.; Osada, T.; Premont, R.T.; Mook, R.A., Jr.; Morse, M.A.; Lyerly, H.K.; Chen, W. Niclosamide-induced wnt signaling inhibition in colorectal cancer is mediated by autophagy. Biochem. J. 2019, 476, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Bio. 2012, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Horbinski, C.; Guo, F.; Watkins, S.; Uchiyama, Y.; Chu, C.T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyI-4-phenylpyridinium-induced cell death. Am. J. Pathol. 2007, 170, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Lin, J.; Zhou, J.; Wang, X.; Li, Y.; Ren, X.; Yu, W.; Zhong, W.; Xiao, J.; Sheng, F.; et al. Beclin 1-independent autophagy induced by a Bcl-XL/Bcl-2 targeting compound, Z18. Autophagy 2010, 6, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Bauvy, C.; Souquere, S.; Tonelli, G.; Liu, L.; Zhu, Y.; Qiao, Z.; Bakula, D.; Proikas-Cezanne, T.; Pierron, G.; et al. The Bcl-2 homology domain 3 mimetic gossypol induces both Beclin 1-dependent and Beclin 1-independent cytoprotective autophagy in cancer cells. J. Biol. Chem. 2010, 285, 25570–25581. [Google Scholar] [CrossRef] [PubMed]
- Grishchuk, Y.; Ginet, V.; Truttmann, A.C.; Clarke, P.G.; Puyal, J. Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy 2011, 7, 1115–1131. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Lindsten, T.; Wu, J.; Lu, C.; Thompson, C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA 2011, 108, 11121–11126. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J. LAP it up, fuzz ball: A short history of LC3-associated phagocytosis. Curr. Opin. Immunol. 2018, 55, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2018, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, Y.; Hong, L.; Yang, Z.; Cai, X.; Chen, X.; Fu, Y.; Lin, Y.; Wen, W.; Li, S.; et al. Golgi-associated LC3 lipidation requires V-ATPase in noncanonical autophagy. Cell Death Dis. 2016, 7, e2330. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, B.; Yang, M.; Zhu, J.; Li, H. Systematic Profiling of Histone Readers in Arabidopsis thaliana. Cell Rep. 2018, 22, 1090–1102. [Google Scholar] [CrossRef]
- Dall’Armi, C.; Devereaux, K.A.; Di Paolo, G. The role of lipids in the control of autophagy. Curr. Biol. 2013, 23, R33–R45. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Drose, S.; Altendorf, K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J. Exp. Biol. 1997, 200, 1–8. [Google Scholar] [PubMed]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef] [PubMed]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar] [PubMed]
- Ganley, I.G.; Wong, P.M.; Gammoh, N.; Jiang, X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol. Cell 2011, 42, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Niso-Santano, M.; Malik, S.A.; Pietrocola, F.; Bravo-San Pedro, J.M.; Marino, G.; Cianfanelli, V.; Ben-Younes, A.; Troncoso, R.; Markaki, M.; Sica, V.; et al. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015, 34, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Florey, O.; Gammoh, N.; Kim, S.E.; Jiang, X.; Overholtzer, M. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 2015, 11, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Chen, X.; Li, M.; Zhang, H.; Ding, W.X.; Yin, X.M. CCCP-Induced LC3 lipidation depends on Atg9 whereas FIP200/Atg13 and Beclin 1/Atg14 are dispensable. Biochem. Biophys. Res. Commun. 2013, 432, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, L.; Hasegawa, H.; Amin, P.; Han, B.X.; Kaneko, S.; He, Y.; Wang, F. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 9424–9429. [Google Scholar] [CrossRef] [PubMed]
- McLeod, I.X.; Zhou, X.; Li, Q.J.; Wang, F.; He, Y.W. The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J. Immunol. 2011, 187, 5051–5061. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Saitoh, T.; Kageyama, S.; Akira, S.; Noda, T.; Yoshimori, T. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: Analysis of the organization of the Atg16L1 complex in fibroblasts. J. Biol. Chem. 2009, 284, 32602–32609. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Florey, O.; Overholtzer, M.; Jiang, X. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat. Struct. Mol. Biol. 2013, 20, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, K.; Ulferts, R.; Jacquin, E.; Veith, T.; Gammoh, N.; Arasteh, J.M.; Mayer, U.; Carding, S.R.; Wileman, T.; Beale, R.; et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.M. The Golgi complex: A common platform for canonical and non-canonical autophagy? Cell Cycle 2013, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Naydenov, N.G.; Harris, G.; Morales, V.; Ivanov, A.I. Loss of a membrane trafficking protein alphaSNAP induces non-canonical autophagy in human epithelia. Cell Cycle 2012, 11, 4613–4625. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mei, M.; Li, Q.; Roboti, P.; Pang, Q.; Ying, Z.; Gao, F.; Lowe, M.; Bao, S. Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 346–351. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Luo, X.; Shan, H.; Fu, Y.; Gu, Q.; Zheng, X.; Dai, Q.; Xia, F.; Zheng, Z.; Liu, P.; et al. Niclosamide Triggers Non-Canonical LC3 Lipidation. Cells 2019, 8, 248. https://doi.org/10.3390/cells8030248
Liu Y, Luo X, Shan H, Fu Y, Gu Q, Zheng X, Dai Q, Xia F, Zheng Z, Liu P, et al. Niclosamide Triggers Non-Canonical LC3 Lipidation. Cells. 2019; 8(3):248. https://doi.org/10.3390/cells8030248
Chicago/Turabian StyleLiu, Yajun, Xia Luo, Hao Shan, Yuanyuan Fu, Qianqian Gu, Xueping Zheng, Qi Dai, Fan Xia, Zhihua Zheng, Peiqing Liu, and et al. 2019. "Niclosamide Triggers Non-Canonical LC3 Lipidation" Cells 8, no. 3: 248. https://doi.org/10.3390/cells8030248
APA StyleLiu, Y., Luo, X., Shan, H., Fu, Y., Gu, Q., Zheng, X., Dai, Q., Xia, F., Zheng, Z., Liu, P., Yin, X.-M., Hong, L., & Li, M. (2019). Niclosamide Triggers Non-Canonical LC3 Lipidation. Cells, 8(3), 248. https://doi.org/10.3390/cells8030248