GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression

 ,
,  ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Cultures
2.3. Gene Expression Studies
2.4. Gene silencing Experiments and Plasmids
2.5. CRISPR/Cas9-Mediated FGFR1 Knockout
2.6. Immunofluorescence Microscopy
2.7. Conditioned Medium Derived from CAFs
2.8. Western Blot Analysis
2.9. Enzyme-Linked Immunosorbent Assay
2.10. Chromatin Immunoprecipitation (ChIP) Assay
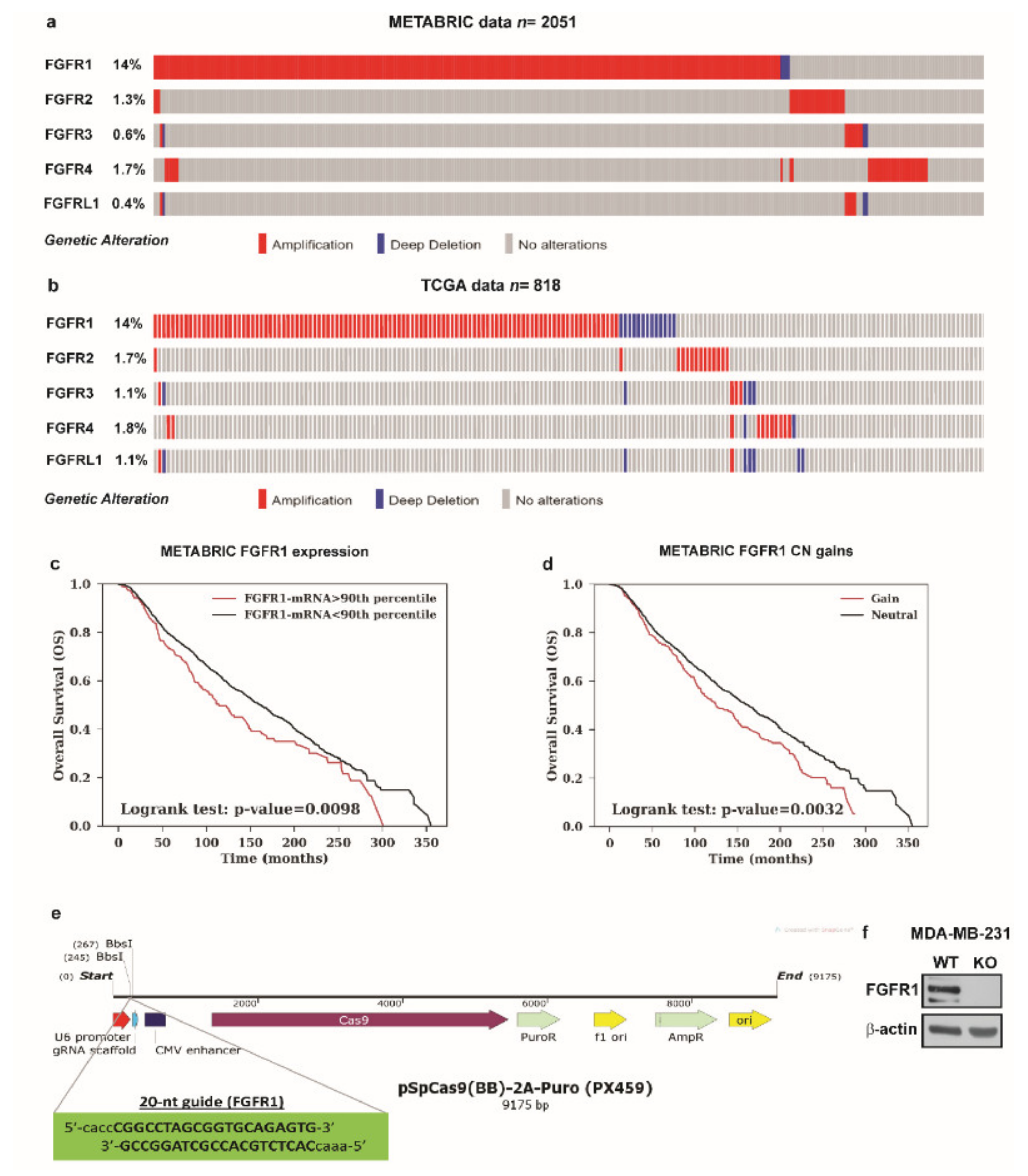
2.11. Analysis of Public Datasets from METABRIC and TCGA and Kaplan-Meier Plotter
2.12. Polarization Assay
2.13. Scratch Assay
2.14. Transwell Migration and Invasion Assays
2.15. Statistical Analysis
2.16. Ethics Approval and Consent to Participate
3. Results
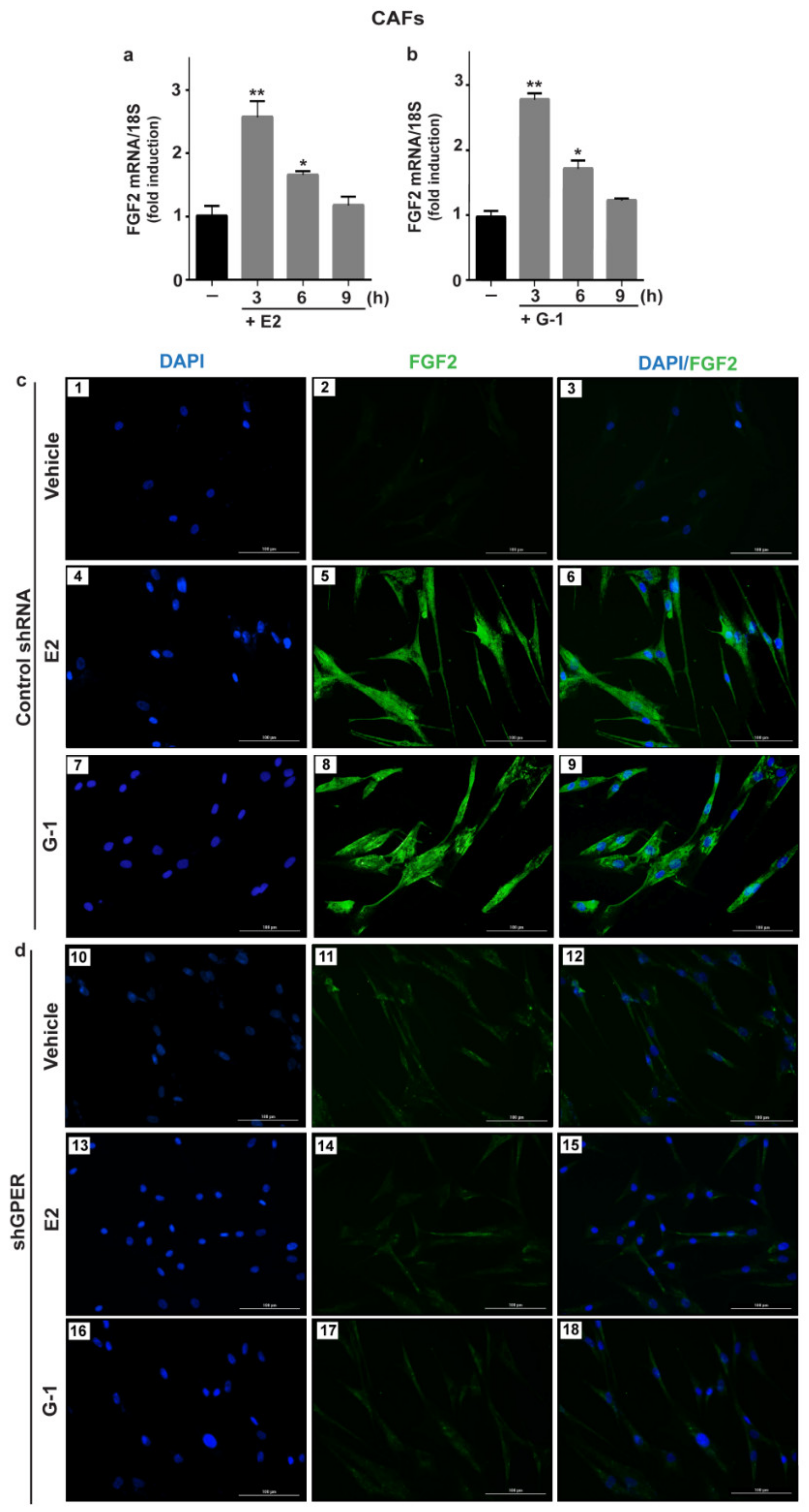
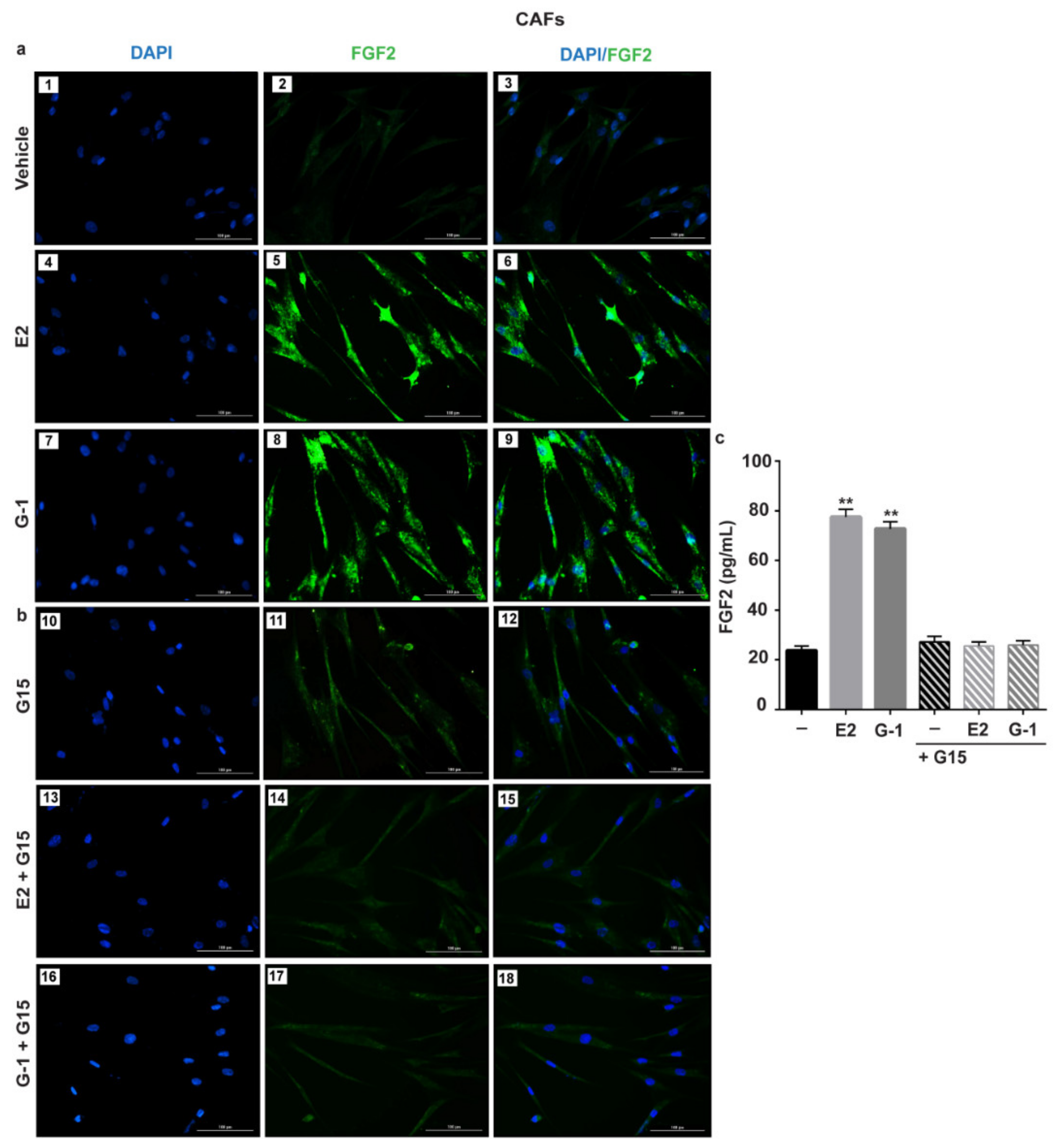
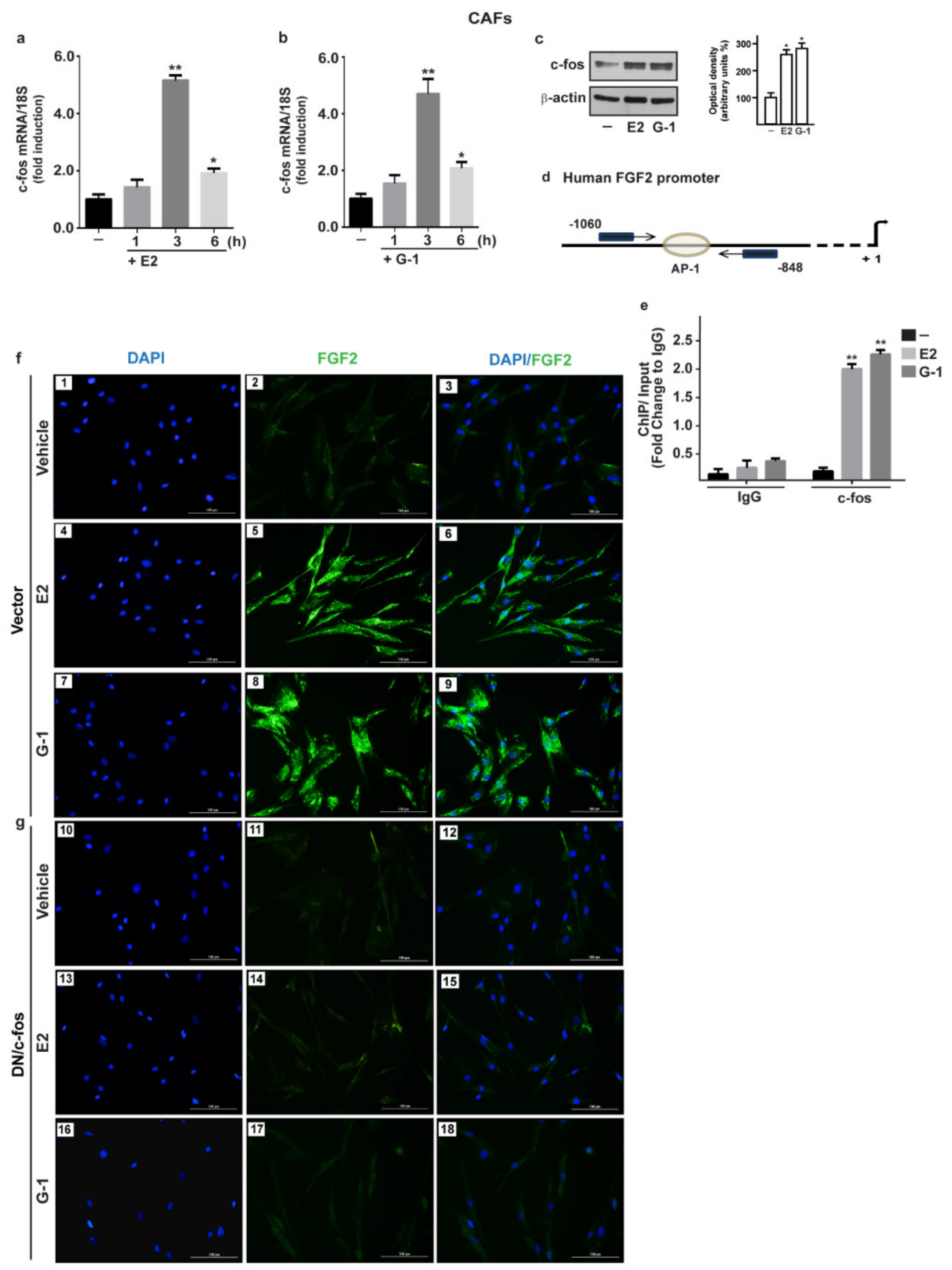
3.1. GPER Mediates the Induction of FGF2 Expression by E2 and G-1 in Breast Cancer-Associated Fibroblasts (CAFs)
3.2. c-fos is Involved in the FGF2 up-Regulation Induced by Estrogenic GPER Signaling in CAFs
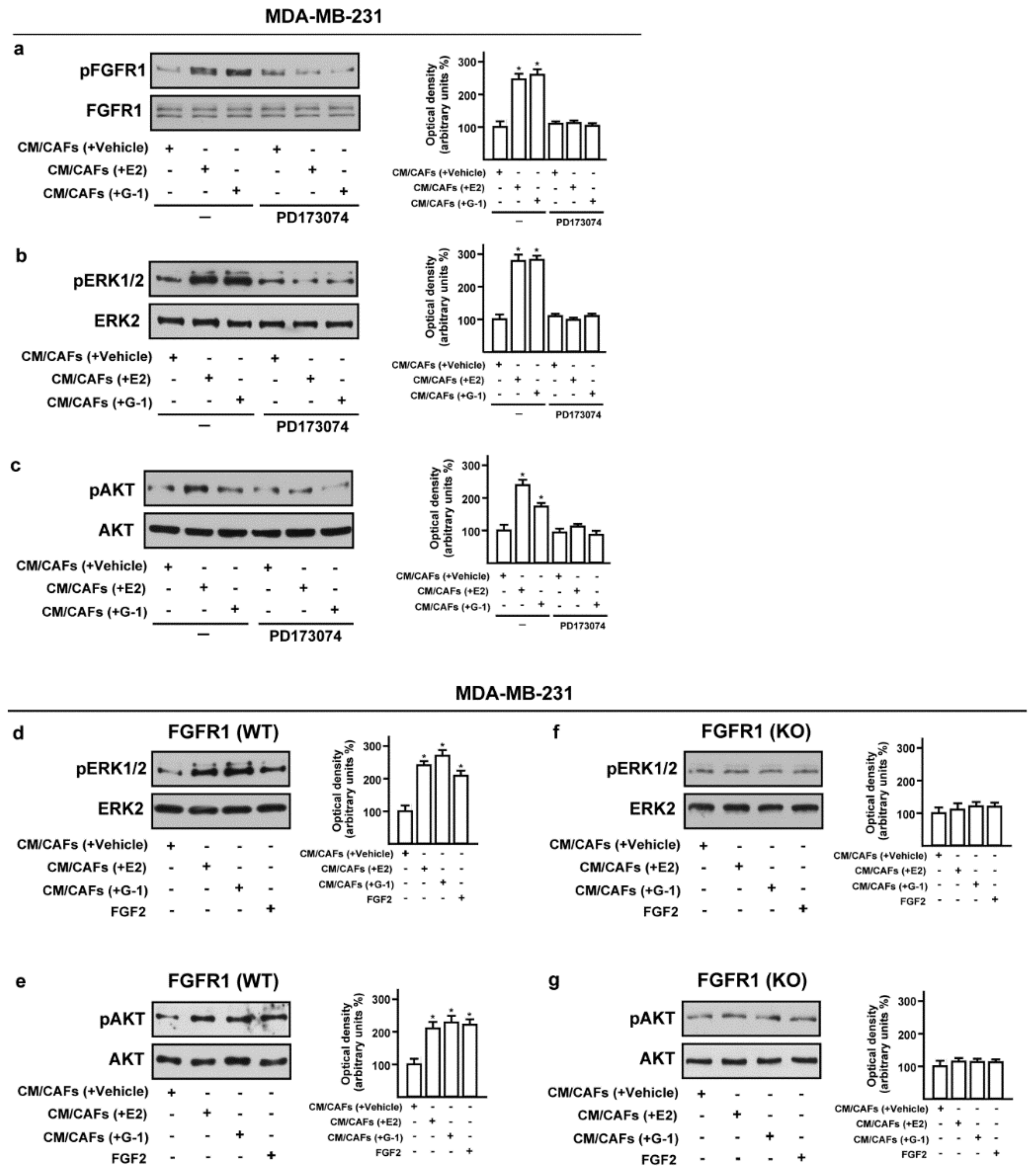
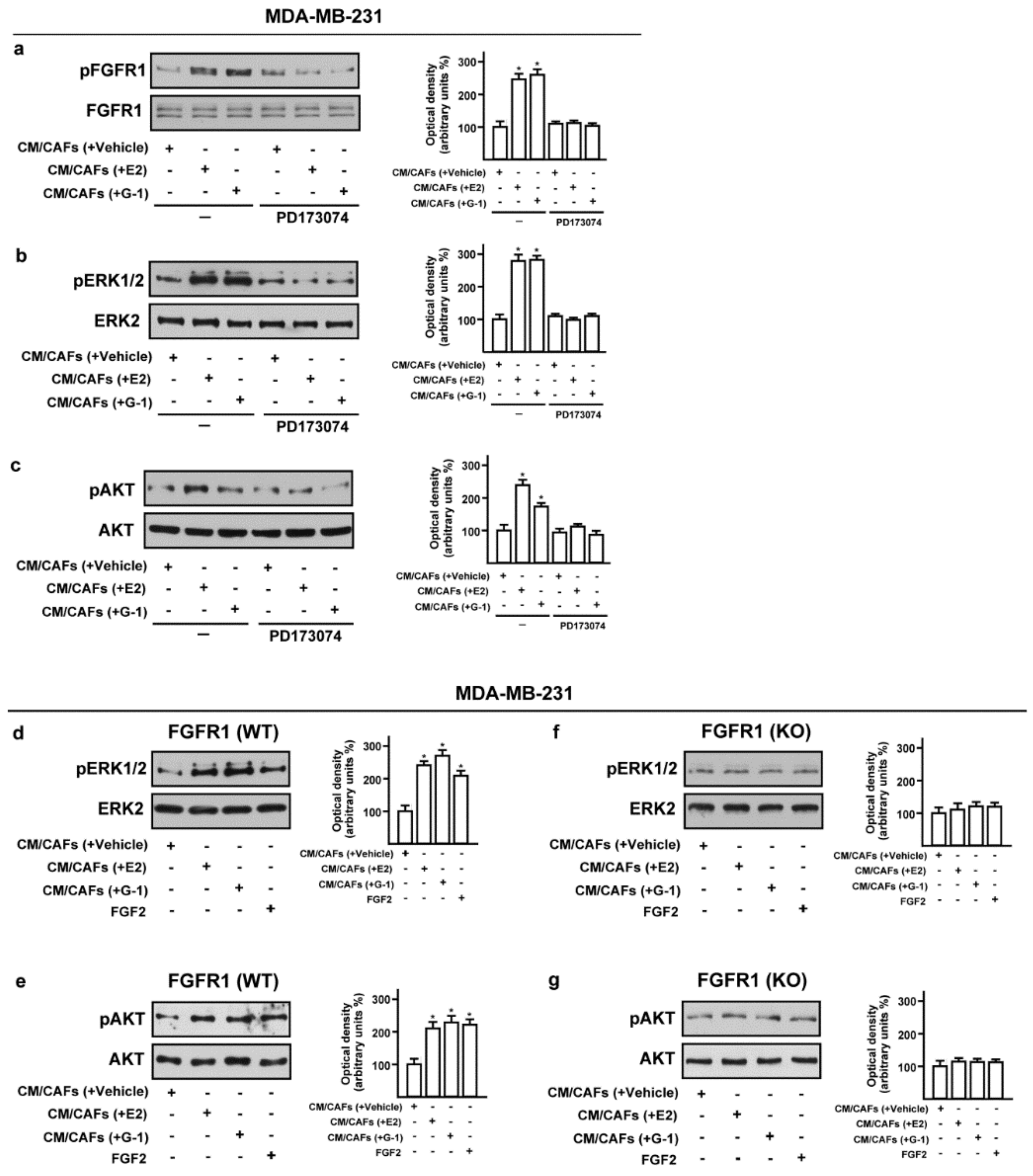
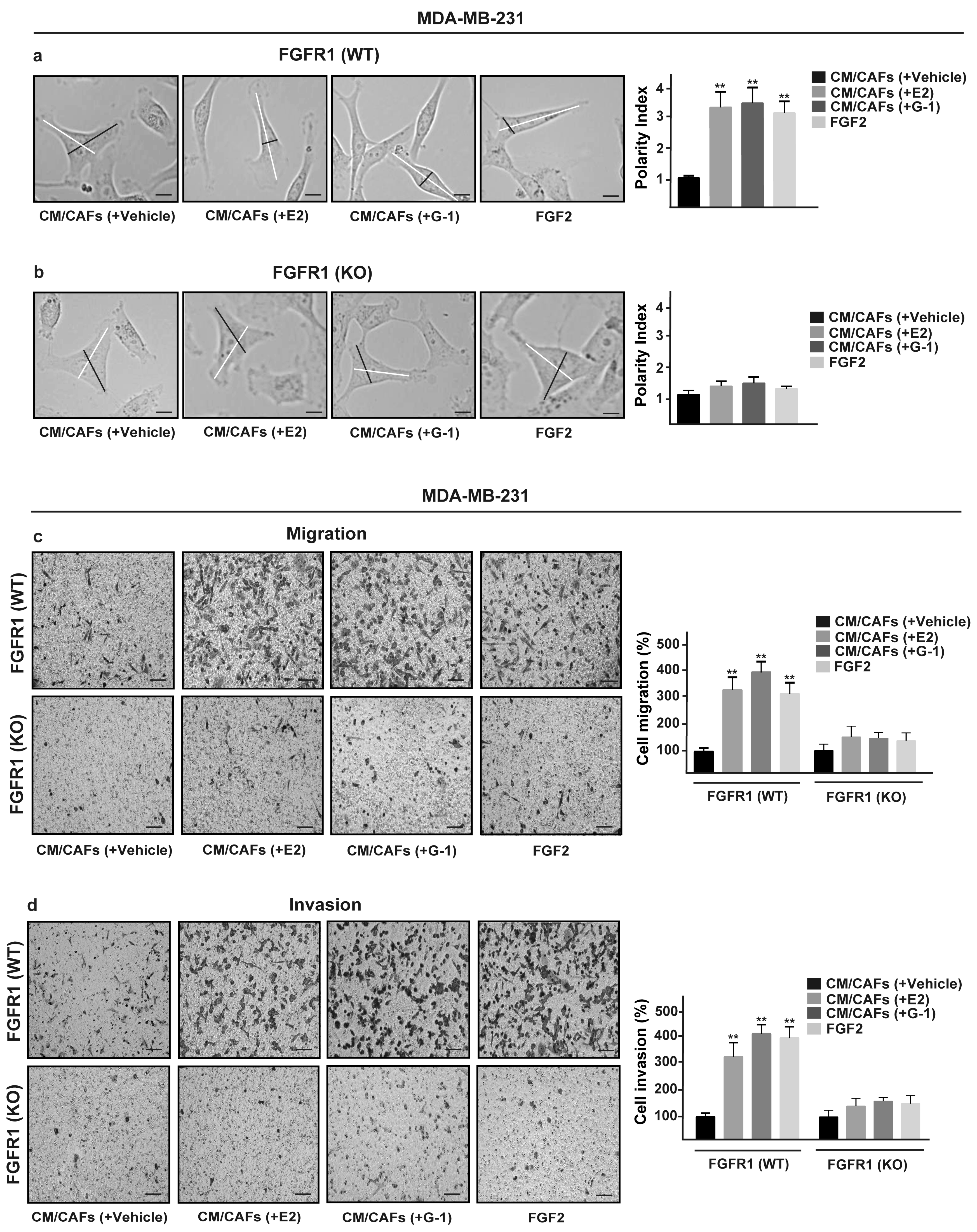
3.3. Conditioned Medium (CM) from Estrogens-Stimulated CAFs Activates the FGFR1-ERK1/2-AKT Transduction Pathway in MDA-MB-231 Cells
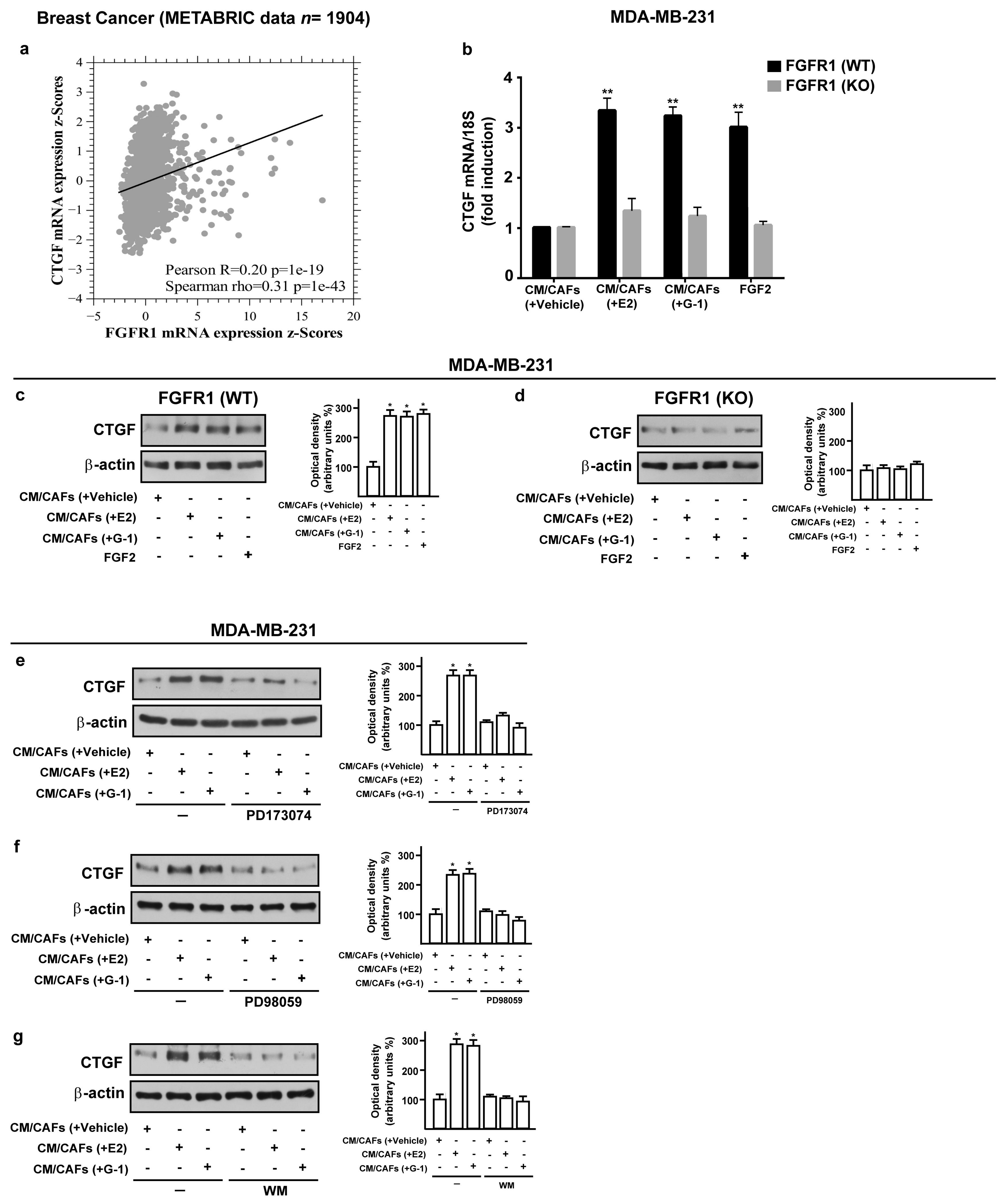
3.4. FGF2/FGFR1 Paracrine Activation Up-Regulates CTGF Expression in MDA-MB-231 Cells
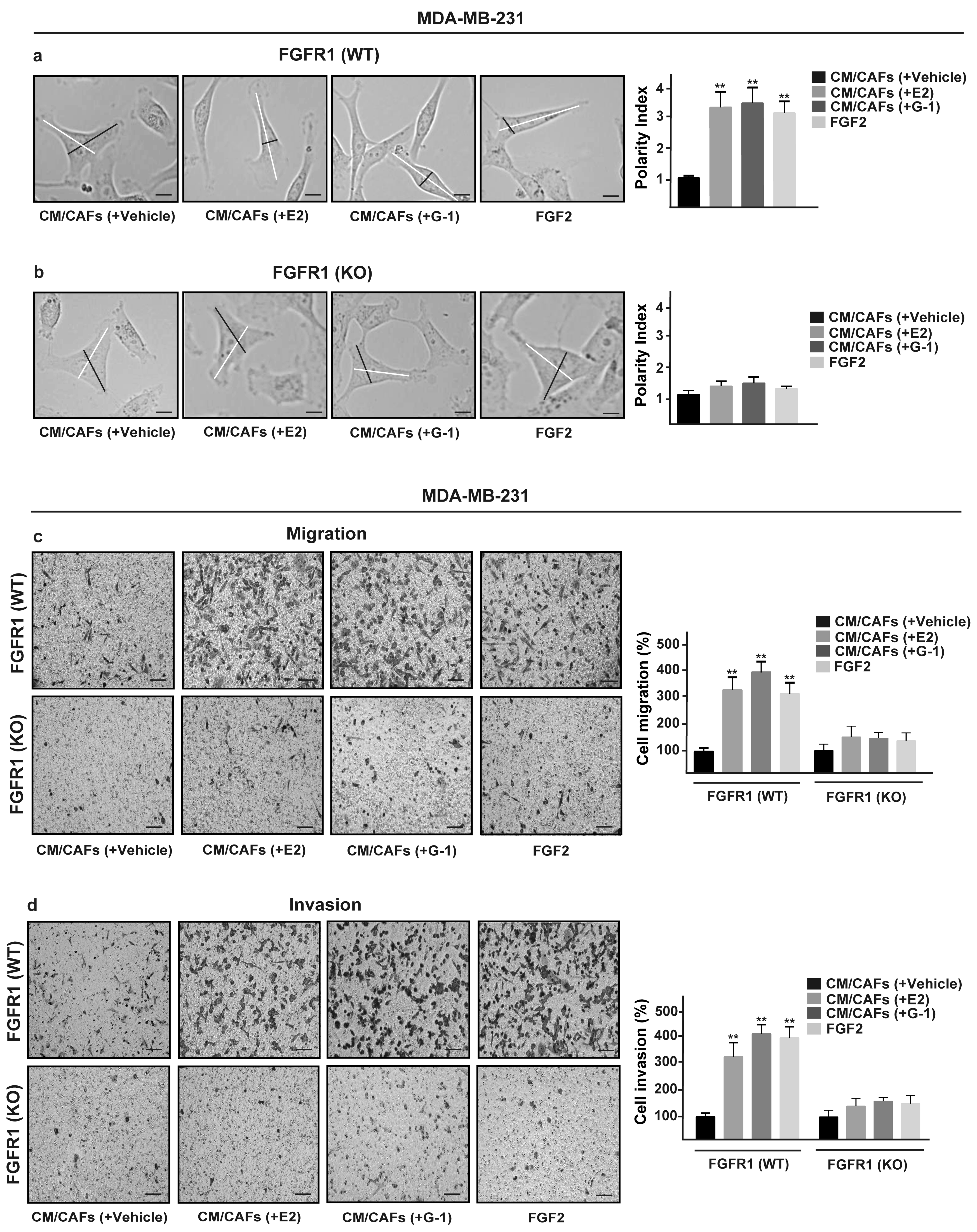
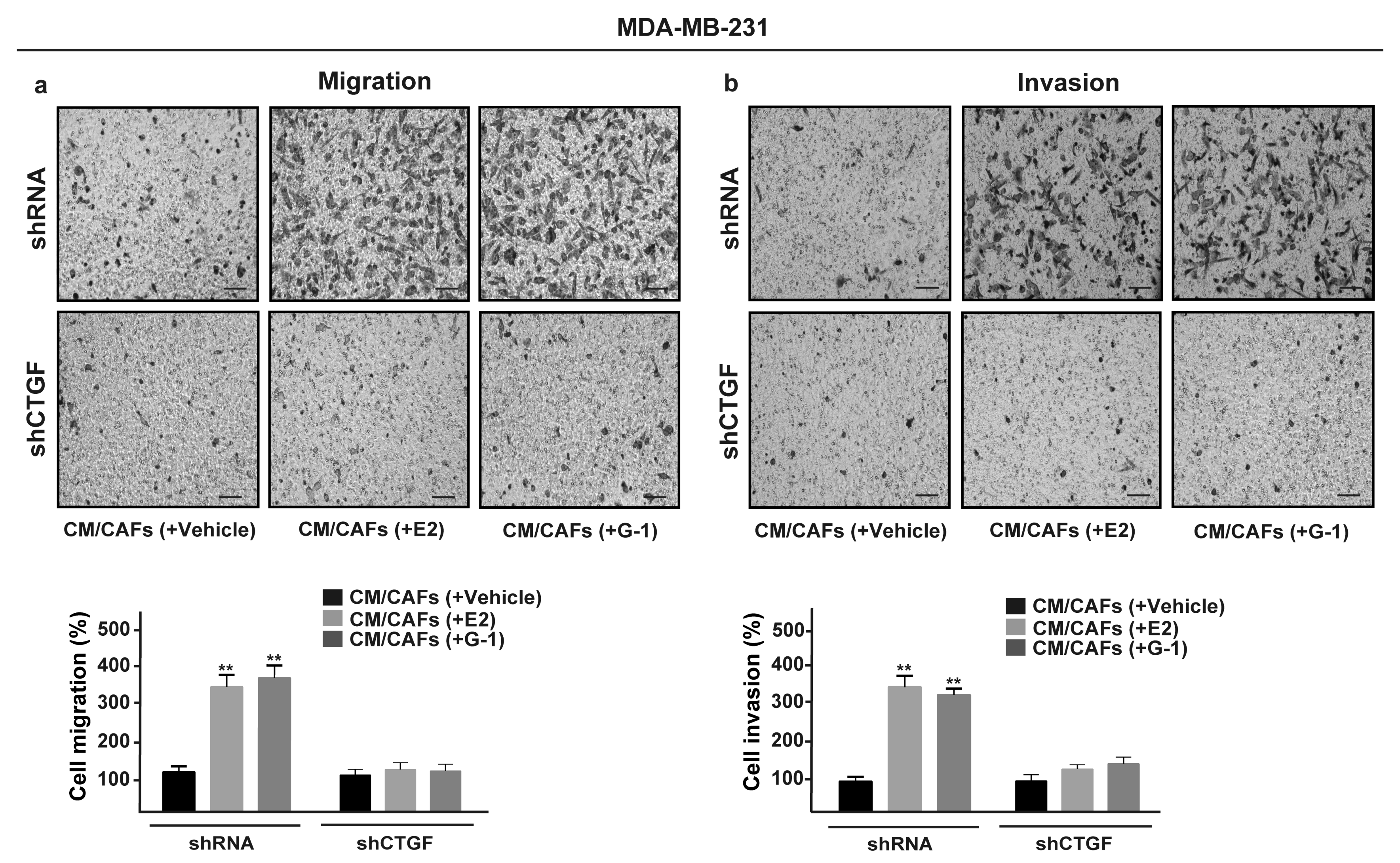
3.5. FGF2/FGFR1 Paracrine Activation Induces Cell Migration and Invasion Through CTGF in MDA-MB-231 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAFs | cancer associated fibroblasts |
| FGFR1 | fibroblast growth factor receptor 1 |
| FGF2 | fibroblast growth factor 2 |
| GPER | G protein estrogen receptor |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| sgRNA | single guide RNA |
| WT | Wild type |
| KO | Knockout ERK, extracellular signal-regulated kinase |
| AP-1 | activator protein 1 |
| PI3K | phosphatidylinositol 3-kinase |
| CTGF/CCN2 | connective tissue growth factor |
References
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.W.; Wang, Y.; Cao, M.; Hom, Y.K.; Zhang, B.; Grossfeld, G.D.; Sudilovsky, D.; Cunha, G.R. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001, 61, 8135–8142. [Google Scholar] [PubMed]
- Procacci, P.; Moscheni, C.; Sartori, P.; Sommariva, M.; Gagliano, N. Tumor–stroma cross-talk in human pancreatic ductal adenocarcinoma: A focus on the effect of the extracellular matrix on tumor cell phenotype and invasive potential. Cells 2018, 7, 158. [Google Scholar] [CrossRef] [PubMed]
- Marsh, T.; Pietras, K.; McAllister, S.S. Fibroblasts as architects of cancer pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, Y.; Jia, T.; Sun, Y. Molecular mechanism underlying the tumor-promoting functions of carcinoma-associated fibroblasts. Tumor Biol. 2015, 36, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.P.; Fearon, A.E.; Grose, R.P. Careless talk costs lives: Fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015, 25, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, M.; Fraser, J.; McDonald, M.; Yuan, S.; White, D.; Grandison, P.; Kumble, K.; Watson, J.D.; Murison, J.G. Identification of a new fibroblast growth factor receptor, FGFR5. Gene 2001, 271, 171–182. [Google Scholar] [CrossRef]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Arnedos, M.; Andre, F.; Soria, J.C. Fibroblast growth factor receptor inhibitors as a cancer treatment: From a biologic rationale to medical perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Englinger, B.; Kallus, S.; Senkiv, J.; Laemmerer, A.; Moser, P.; Gabler, L.; Groza, D.; Kowol, C.; Heffeter, P.; Grusch, M.; et al. Lysosomal sequestration impairs the activity of the preclinical FGFR inhibitor PD173074. Cells 2018, 7, 259. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Young, C.D.; Bhola, N.; Giltnane, J.M.; Estrada, M.V.; Arteaga, C.L. FGFR1 is associated with resistance to interaction with estrogen receptor (ER) α endocrine therapy in ER+/FGFR1-amplified breast cancer. Cancer Res. 2015, 75, 2435. [Google Scholar] [CrossRef]
- Wynes, M.W.; Hinz, T.K.; Gao, D.; Martini, M.; Marek, L.A.; Ware, K.E.; Edwards, M.G.; Böhm, D.; Perner, S.; Helfrich, B.A.; et al. FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies. Clin. Cancer Res. 2014, 20, 3299–3309. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, T.; Joensuu, H.; Mattson, K.; Salven, P. High pretreatment serum concentration of basic fibroblast growth factor is a predictor of poor prognosis in small cell lung cancer. Cancer Epidemiol. Biomarkers Prev. 2002, 11, 1492–1495. [Google Scholar] [PubMed]
- Brown, W.S.; Tan, L.; Smith, A.; Gray, N.S.; Wendt, M.K.; Wendt, M.; Taylor, M.; Schiemann, B.; Sossey-Alaoui, K.; Schiemann, W.; et al. Covalent targeting of fibroblast growth factor receptor inhibits metastatic breast cancer. Mol. Cancer Ther. 2016, 15, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Jia, H.H.; Xu, Y.Q.; Zhou, X.; Zhao, X.H.; Wang, Y.F.; Song, X.; Zhu, Z.Y.; Sun, T.; Dou, Y.; et al. Paracrine and epigenetic control of CAF-induced metastasis: The role of HOTAIR stimulated by TGF-β1 secretion. Mol. Cancer 2018, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yuan, P.; Wu, J.; Huang, J. Estrogen regulates excitatory amino acid carrier 1 (EAAC1) expression through sphingosine kinase 1 (SphK1) transacting FGFR-mediated ERK signaling in rat C6 astroglial cells. Neuroscience 2016, 319, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J.A. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Maggiolini, M.; Picard, D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J. Endocrinol. 2010, 204, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Lappano, R.; Albanito, L.; Madeo, A.; Maggiolini, M.; Picard, D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009, 28, 523–532. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Santolla, M.F.; Pupo, M.; Sinicropi, M.S.; Caruso, A.; Rosano, C.; Maggiolini, M. MIBE acts as antagonist ligand of both estrogen receptor α and GPER in breast cancer cells. Breast Cancer Res. 2012, 14, R12. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Rosano, C.; Santolla, M.F.; Pupo, M.; de Francesco, E.M.; de Marco, P.; Ponassi, M.; Spallarossa, A.; Ranise, A.; Maggiolini, M. Two novel GPER agonists induce gene expression changes and growth effects in cancer cells. Curr. Cancer Drug Targets 2012, 12, 531–542. [Google Scholar] [CrossRef] [PubMed]
- De Marco, P.; Lappano, R.; De Francesco, E.M.; Cirillo, F.; Pupo, M.; Avino, S.; Vivacqua, A.; Abonante, S.; Picard, D.; Maggiolini, M. GPER signalling in both cancer-associated fibroblasts and breast cancer cells mediates a feedforward IL1β/IL1R1 response. Sci. Rep. 2016, 6, 24354. [Google Scholar] [CrossRef] [PubMed]
- Santolla, M.F.; Avino, S.; Pellegrino, M.; De Francesco, E.M.; De Marco, P.; Lappano, R.; Vivacqua, A.; Cirillo, F.; Rigiracciolo, D.C.; Scarpelli, A.; et al. SIRT1 is involved in oncogenic signaling mediated by GPER in breast cancer. Cell Death Dis. 2015, 6, e1834. [Google Scholar] [CrossRef] [PubMed]
- De Marco, P.; Bartella, V.; Vivacqua, A.; Lappano, R.; Santolla, M.F.; Morcavallo, A.; Pezzi, V.; Belfiore, A.; Maggiolini, M. Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene 2012, 32, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, A.; Sebastiani, A.; Miglietta, A.; Rigiracciolo, D.; Cirillo, F.; Galli, G.; Talia, M.; Santolla, M.; Lappano, R.; Giordano, F.; et al. miR-338-3p is regulated by estrogens through GPER in breast cancer cells and cancer-associated fibroblasts (CAFs). Cells 2018, 7, 203. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.M.; Gupta, P.B.; Rudnick, J.A.; Caballero, S.; Keller, P.J.; Lander, E.S.; Kuperwasser, C. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21737–21742. [Google Scholar] [CrossRef] [PubMed]
- Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Miller, T.W.; Kuba, M.G.; Meszoely, I.M.; Wagle, N.; Garraway, L.A.; Arteaga, C.L. Discordant cellular response to presurgical letrozole in bilateral synchronous ER+ breast cancers with a KRAS mutation or FGFR1 gene amplification. Mol. Cancer Ther. 2012, 11, 2301–2305. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1alpha/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 2013, 15, R64. [Google Scholar] [CrossRef] [PubMed]
- Santolla, M.F.; De Francesco, E.M.; Lappano, R.; Rosano, C.; Abonante, S.; Maggiolini, M. Niacin activates the G protein estrogen receptor (GPER)-mediated signalling. Cell. Signal. 2014, 26, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Pisano, A.; Santolla, M.F.; De Francesco, E.M.; De Marco, P.; Rigiracciolo, D.C.; Perri, M.G.; Vivacqua, A.; Abonante, S.; Cappello, A.R.; Dolce, V.; et al. GPER, IGF-IR, and EGFR transduction signaling are involved in stimulatory effects of zinc in breast cancer cells and cancer-associated fibroblasts. Mol. Carcinog. 2017, 56, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.P.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.S.; Aksoy, B.A.B.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci.Signal. 2013, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Koach, M.A.; Whitmore, L.; Lamers, M.L.; Horwitz, A.F. Segregation and activation of myosin IIB creates a rear in migrating cells. J. Cell Biol. 2008, 183, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, J.M.; Farooqui, M.; Rothenberger, N.J.; Dacic, S.; Stabile, L.P. Interaction between the estrogen receptor and fibroblast growth factor receptor pathways in non-small cell lung cancer. Oncotarget 2017, 8, 24063–24076. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.J.; Chioni, A.-M.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Tanner, Y.; Grose, R.P. Dysregulated FGF signalling in neoplastic disorders. Semin. Cell Dev. Biol. 2016, 53, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ji, W.; Yu, Y.; Li, Z.; Niu, X.; Xia, W.; Lu, S. FGFR1-ERK1/2-SOX2 axis promotes cell proliferation, epithelial–mesenchymal transition, and metastasis in FGFR1-amplified lung cancer. Oncogene 2018, 37, 5340–5354. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, R.; Pearson, A.; Herrera-Abreu, M.T.; Johnson, D.; Mackay, A.; Welti, J.C.; Natrajan, R.; Reynolds, A.R.; Reis-Filho, J.S.; Ashworth, A.; et al. FGFR signaling promotes the growth of triple-negative and basal-like breast cancer cell lines both in vitro and in vivo. Clin. Cancer Res. 2011, 17, 5275–5286. [Google Scholar] [CrossRef] [PubMed]
- Lundin, L.; Rönnstrand, L.; Cross, M.; Hellberg, C.; Lindahl, U.; Claesson-Welsh, L. Differential tyrosine phosphorylation of fibroblast growth factor (FGF) receptor-1 and receptor proximal signal transduction in response to FGF-2 and heparin. Exp. Cell Res. 2003, 287, 190–198. [Google Scholar] [CrossRef]
- Xu, R.; Zhao, H.; Muhammad, H.; Dong, M.; Besenbacher, F.; Chen, M. Dual-delivery of FGF-2/CTGF from silk fibroin/PLCL-PEO coaxial fibers enhances MSC proliferation and fibrogenesis. Sci. Rep. 2017, 7, 8509. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, E.; Kubota, S.; Takigawa, M. CCN2/CTGF binds to fibroblast growth factor receptor 2 and modulates its signaling. FEBS Lett. 2012, 586, 4270–4275. [Google Scholar] [CrossRef] [PubMed]
- Chujo, S.; Shirasaki, F.; Kondo-Miyazaki, M.; Ikawa, Y.; Takehara, K. Role of connective tissue growth factor and its interaction with basic fibroblast growth factor and macrophage chemoattractant protein-1 in skin fibrosis. J. Cell. Physiol. 2009, 220, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yu, X.; Li, X.; Luo, B.; Yang, W.; Lin, Y.; Li, D.; Gan, Z. TNFAIP3 is required for FGFR1 activation-promoted proliferation and tumorigenesis of premalignant DCIS. COM human mammary epithelial cells. Breast Cancer Res. 2018, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Compagni, A.; Wilgenbus, P.; Impagnatiello, M.A.; Cotten, M.; Christofori, G. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res. 2000, 60, 7163–7169. [Google Scholar] [PubMed]
- Ronca, R.; Giacomini, A.; Rusnati, M.; Presta, M. The potential of fibroblast growth factor/fibroblast growth factor receptor signaling as a therapeutic target in tumor angiogenesis. Expert Opin. Ther. Targets 2015, 19, 1361–1377. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour-host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Mareel, M. Role of tissue stroma in cancer cell invasion. J. Pathol. 2003, 200, 429–447. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, G.; Bos, L.; van der Pluijm, G. Epithelial plasticity, cancer stem cells, and the tumor-supportive stroma in bladder carcinoma. Mol. Cancer Res. 2012, 10, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Hayashi, S. Estrogen-related cancer microenvironment of breast carcinoma. Endocr. J. 2009, 56, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; Ianza, A.; D’Angelo, A.; Roviello, G.; Giudici, F.; Bortul, M.; Zanconati, F.; Bottin, C.; Generali, D. Current status of fibroblast growth factor receptor-targeted therapies in breast cancer. Cells 2018, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.; Ryan, C.J.; Brough, R.; Bajrami, I.; Pemberton, H.N.; Chong, I.Y.; Costa-Cabral, S.; Frankum, J.; Gulati, A.; Holme, H.; et al. Large-scale profiling of kinase dependencies in cancer cell lines. Cell Rep. 2016, 14, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.C. Targeting FGFR signaling in cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Froum, S.; Hamby, J.M.; Schroeder, M.C.; Panek, R.L.; Lu, G.H.; Eliseenkova, V.A.; Green, D.; Schlessinger, J.; Hubbard, S.R. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 1998, 17, 5896–5904. [Google Scholar] [CrossRef] [PubMed]
- Dutt, A.; Salvesen, H.B.; Chen, T.-H.; Ramos, A.H.; Onofrio, R.C.; Hatton, C.; Nicoletti, R.; Winckler, W.; Grewal, R.; Hanna, M.; et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 8713–8717. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, L.J.; Weigelt, B. Road map to metastasis. Nat. Med. 2003, 9, 999–1000. [Google Scholar] [CrossRef] [PubMed]
- Kraljevic, S.P.; Sedic, M.; Bosnjak, H.; Spaventi, S.; Pavelic, K. Metastasis: New perspectives on an old problem. Mol. Cancer 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, V.D.; Gangula, R.D.; Freeman, K.W.; Li, R.; Zhang, Y.; Wang, F.; Ayala, G.E.; Peterson, L.E.; Ittmann, M.; Spencer, D.M. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell 2007, 12, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Abramson, V.G.; Lehmann, B.D.; Ballinger, T.J.; Pietenpol, J.A. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer 2015, 121, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-S.; Wang, M.-Y.; Wu, S.-N.; Su, J.-L.; Hong, C.-C.; Chuang, S.-E.; Chen, M.-W.; Hua, K.-T.; Wu, Y.-L.; Cha, S.-T.; et al. CTGF enhances the motility of breast cancer cells via an integrin-αvβ3-ERK1/2-dependent S100A4-upregulated pathway. J. Cell Sci. 2007, 120, 2053–2065. [Google Scholar] [CrossRef] [PubMed]
- Goltsov, A.; Deeni, Y.; Khalil, H.; Soininen, T.; Kyriakidis, S.; Hu, H.; Langdon, S.; Harrison, D.; Bown, J. Systems analysis of drug-induced receptor tyrosine kinase reprogramming following targeted mono- and combination anti-cancer therapy. Cells 2014, 3, 563–591. [Google Scholar] [CrossRef] [PubMed]
- Cicha, I.; Goppelt-Struebe, M. Connective tissue growth factor: Context-dependent functions and mechanisms of regulation. BioFactors 2009, 35, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santolla, M.F.; Vivacqua, A.; Lappano, R.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Brunetti, G.; Miglietta, A.M.; Belfiore, A.; et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression. Cells 2019, 8, 223. https://doi.org/10.3390/cells8030223
Santolla MF, Vivacqua A, Lappano R, Rigiracciolo DC, Cirillo F, Galli GR, Talia M, Brunetti G, Miglietta AM, Belfiore A, et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression. Cells. 2019; 8(3):223. https://doi.org/10.3390/cells8030223
Chicago/Turabian StyleSantolla, Maria Francesca, Adele Vivacqua, Rosamaria Lappano, Damiano Cosimo Rigiracciolo, Francesca Cirillo, Giulia Raffaella Galli, Marianna Talia, Giuseppe Brunetti, Anna Maria Miglietta, Antonino Belfiore, and et al. 2019. "GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression" Cells 8, no. 3: 223. https://doi.org/10.3390/cells8030223
APA StyleSantolla, M. F., Vivacqua, A., Lappano, R., Rigiracciolo, D. C., Cirillo, F., Galli, G. R., Talia, M., Brunetti, G., Miglietta, A. M., Belfiore, A., & Maggiolini, M. (2019). GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression. Cells, 8(3), 223. https://doi.org/10.3390/cells8030223