Differential Expression of Mitochondrial Biogenesis Markers in Mouse and Human SHH-Subtype Medulloblastoma

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cerebellar Granule Cell Progenitor Culture
2.2. Real-Time RT-qPCR
| Forward | Reverse | |
| GAPDH | ggccttccgtgttcctac | tgtcatcatacttggcaggtt |
| Gli1 | atctctctttcctcctcctcc | cgaggctggcatcagaa |
| Gpr153 | ctcagagcctgccagaactt | aagctcaccaccagcacag |
| Shisa2 | ctcggcagtccccatctac | cgtagacatcggcaacagc |
2.3. Animal Husbandry
2.4. In-Situ Hybridization
2.5. Analysis of Medulloblastoma Gene Expression Datasets
2.6. Gene Set Enrichment Analysis
2.7. Immunohistochemistry
3. Results
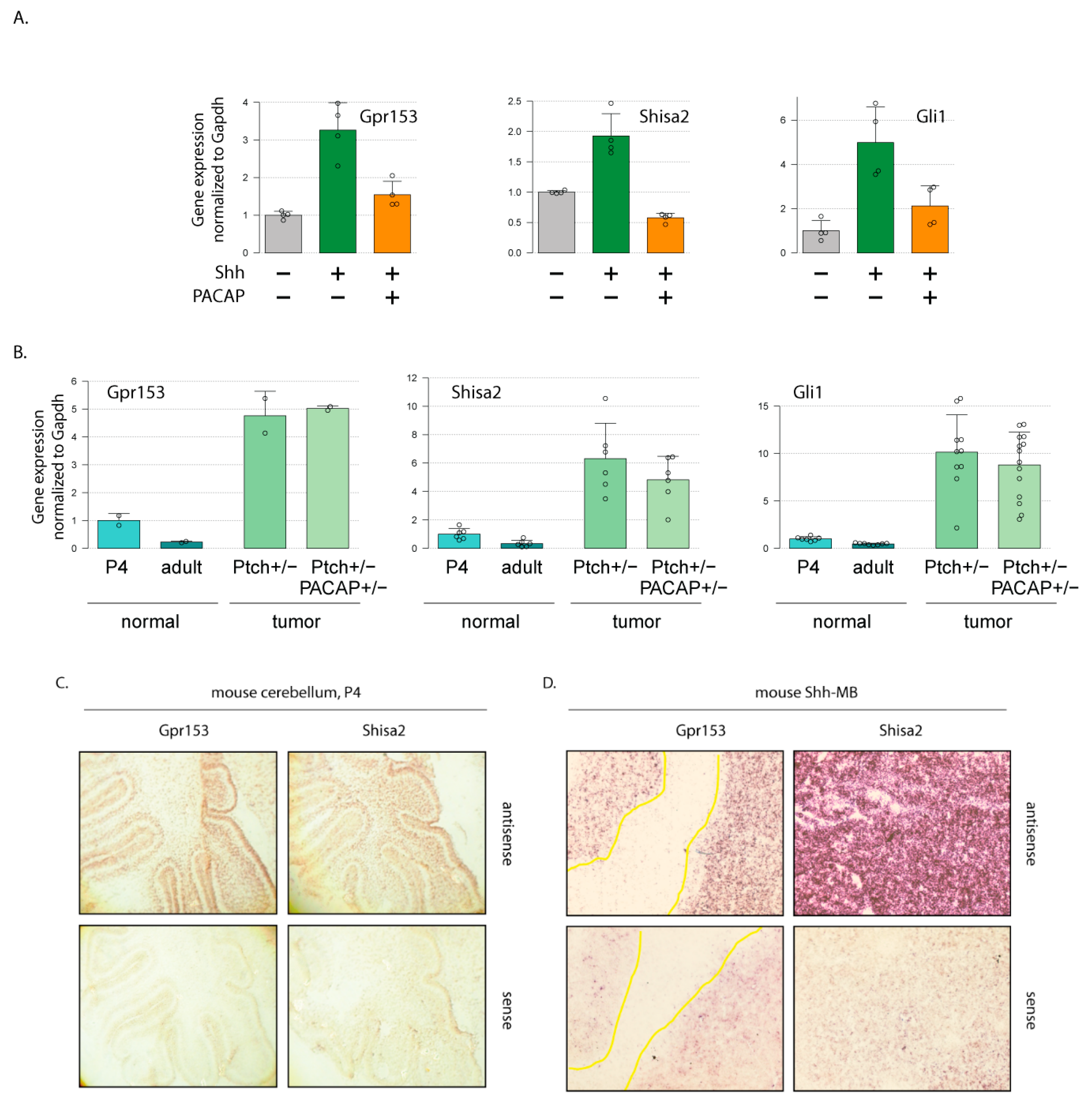
3.1. Gpr153 and Shisa2 are Shh Targets in Murine Cerebellar Granule Cell Progenitors and Mouse Shh-Type Medulloblastoma
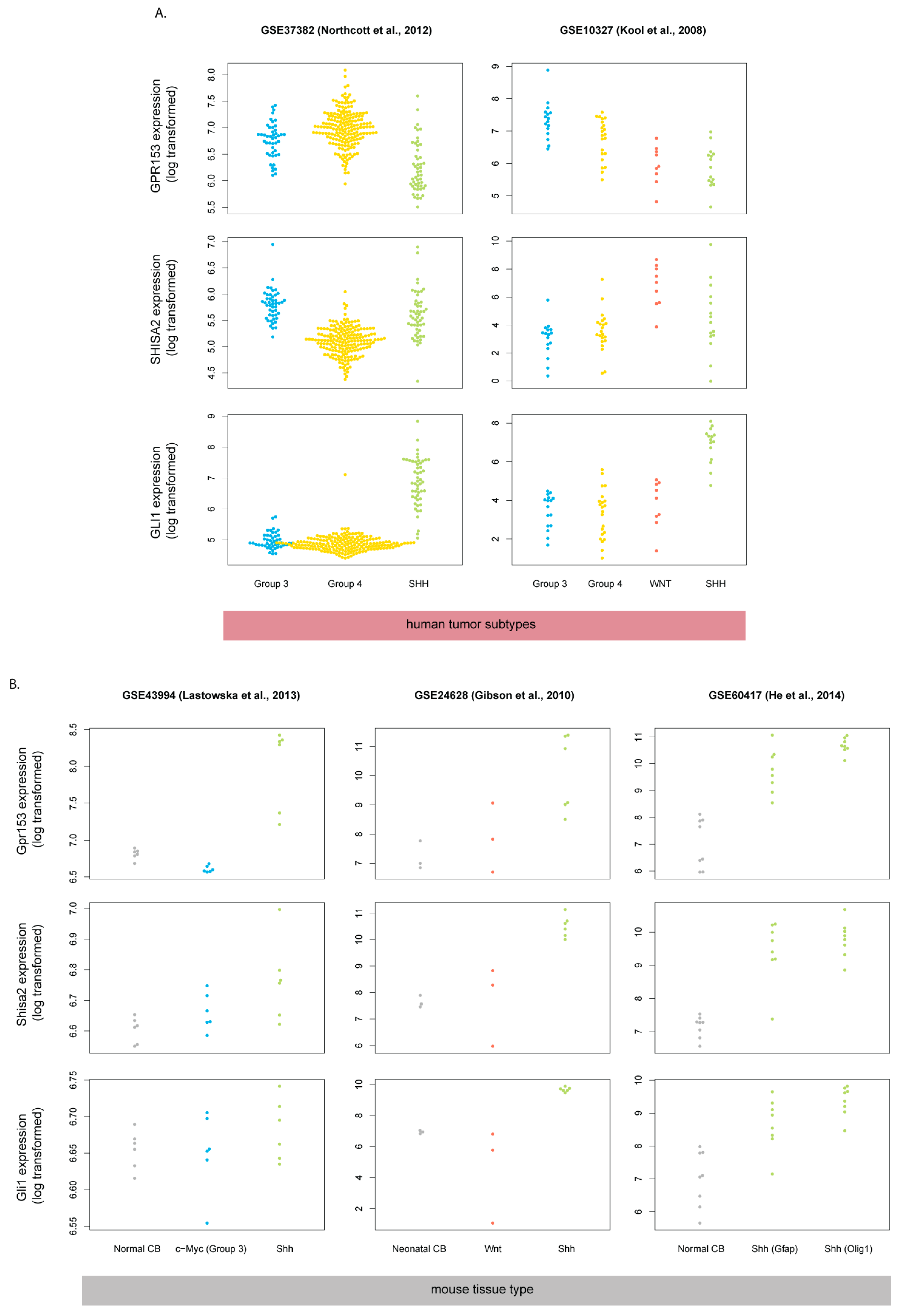
3.2. Gpr153 and Shisa2 Are Not Upregulated in Human SHH-MB
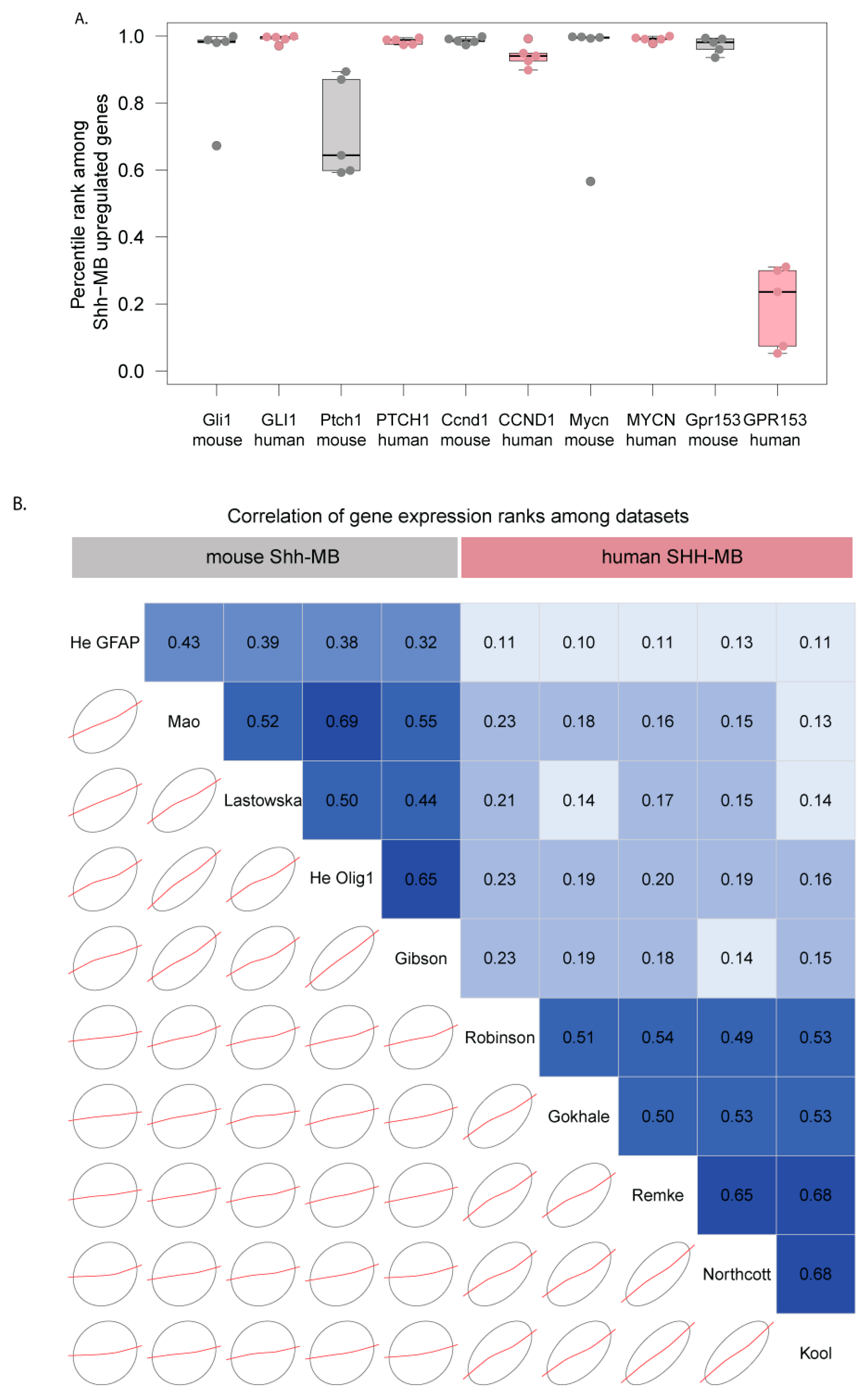
3.3. Mouse Shh-MB Have Different Gene Expression Profiles from Human SHH-MB
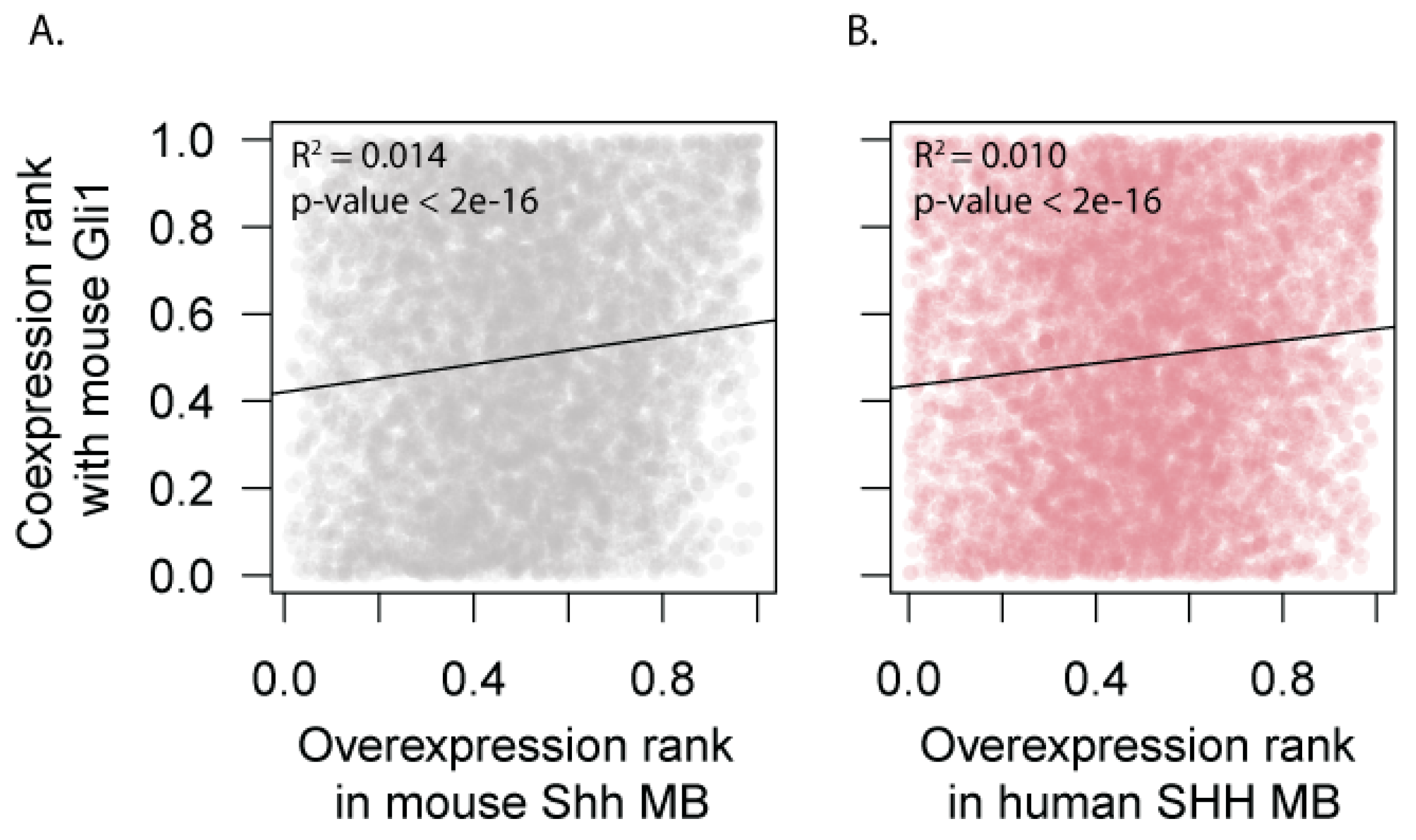
3.4. Expression of HH Target Genes Is Similar Between Mouse and Human SHH-MB
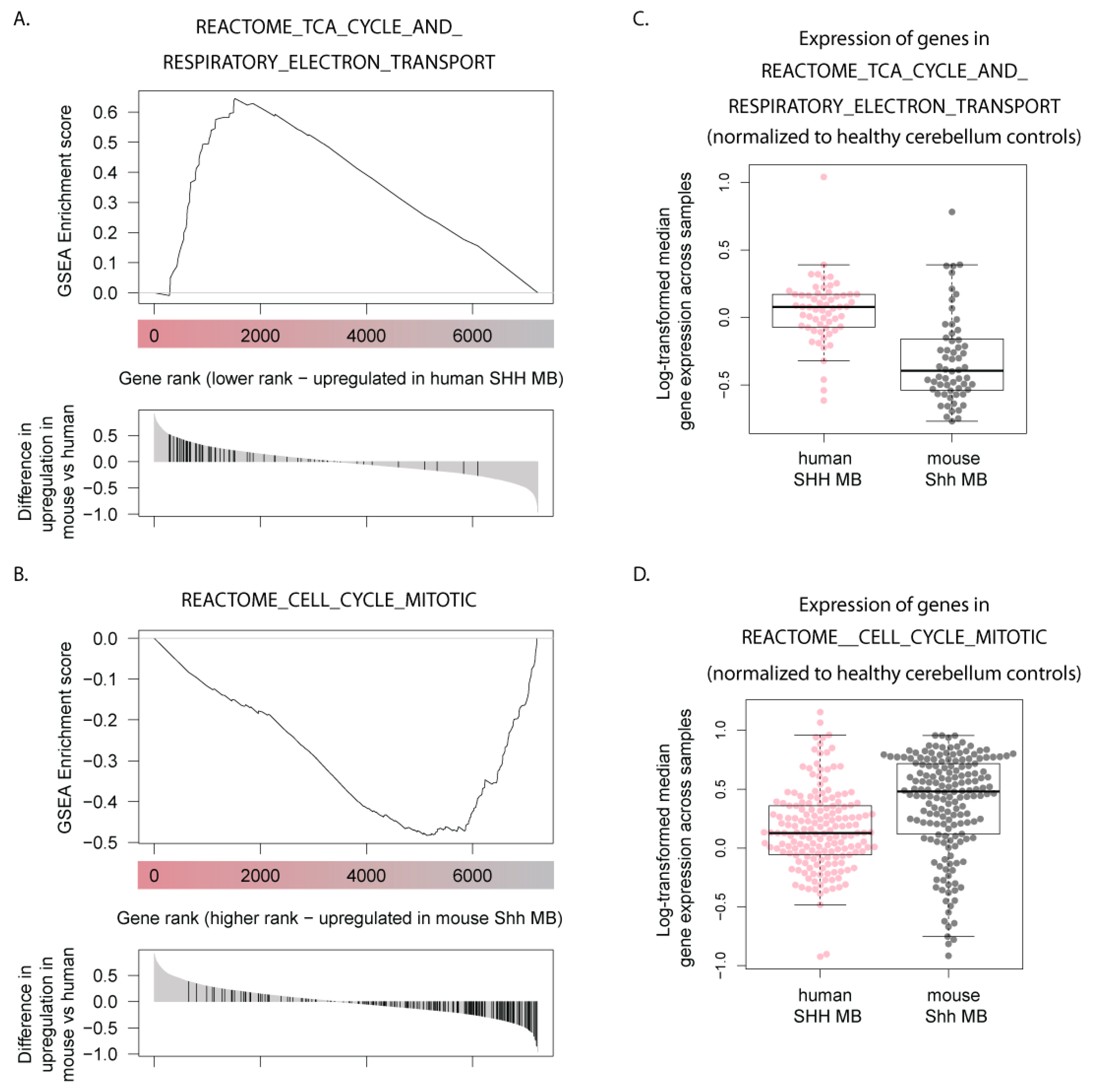
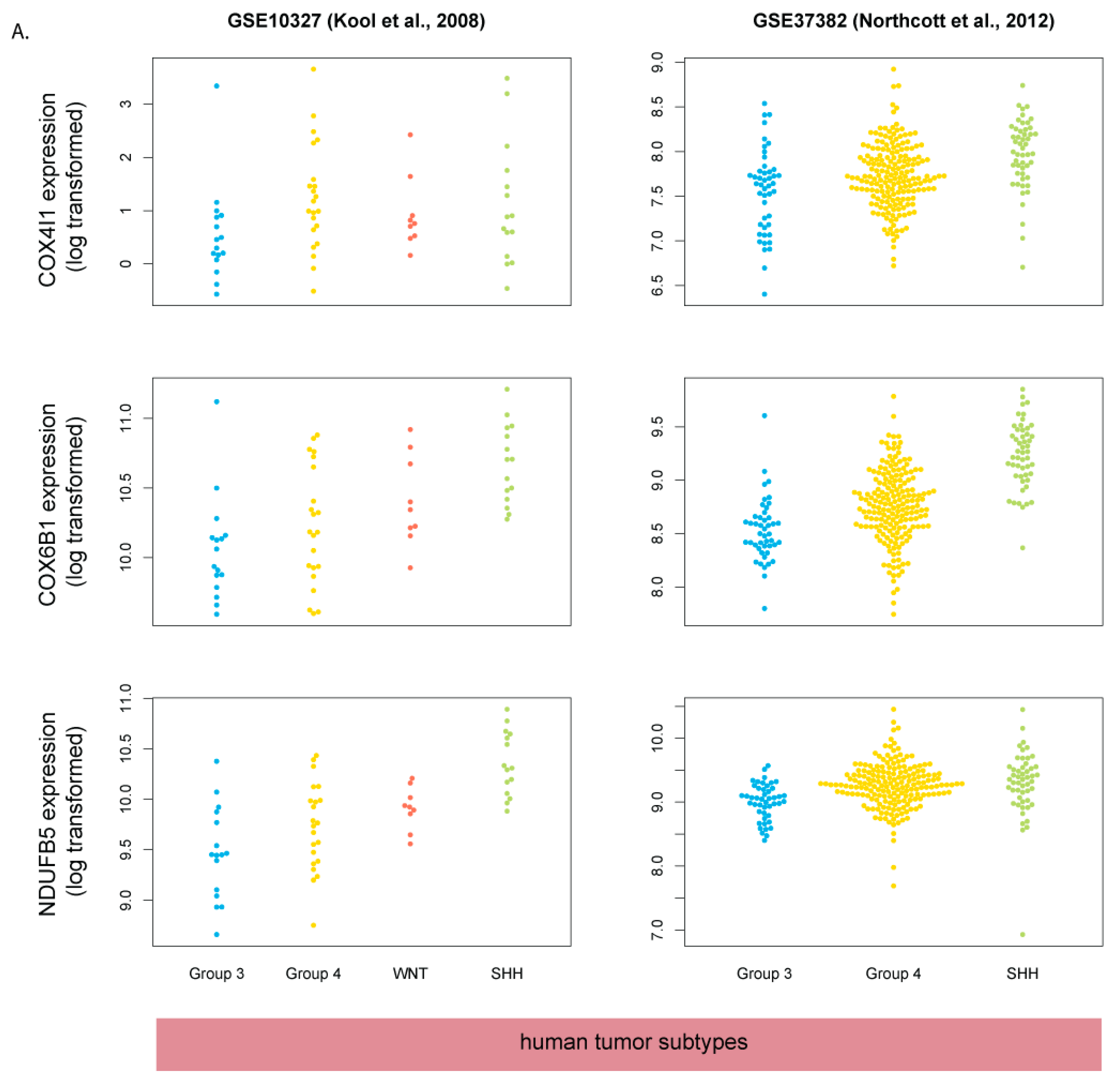
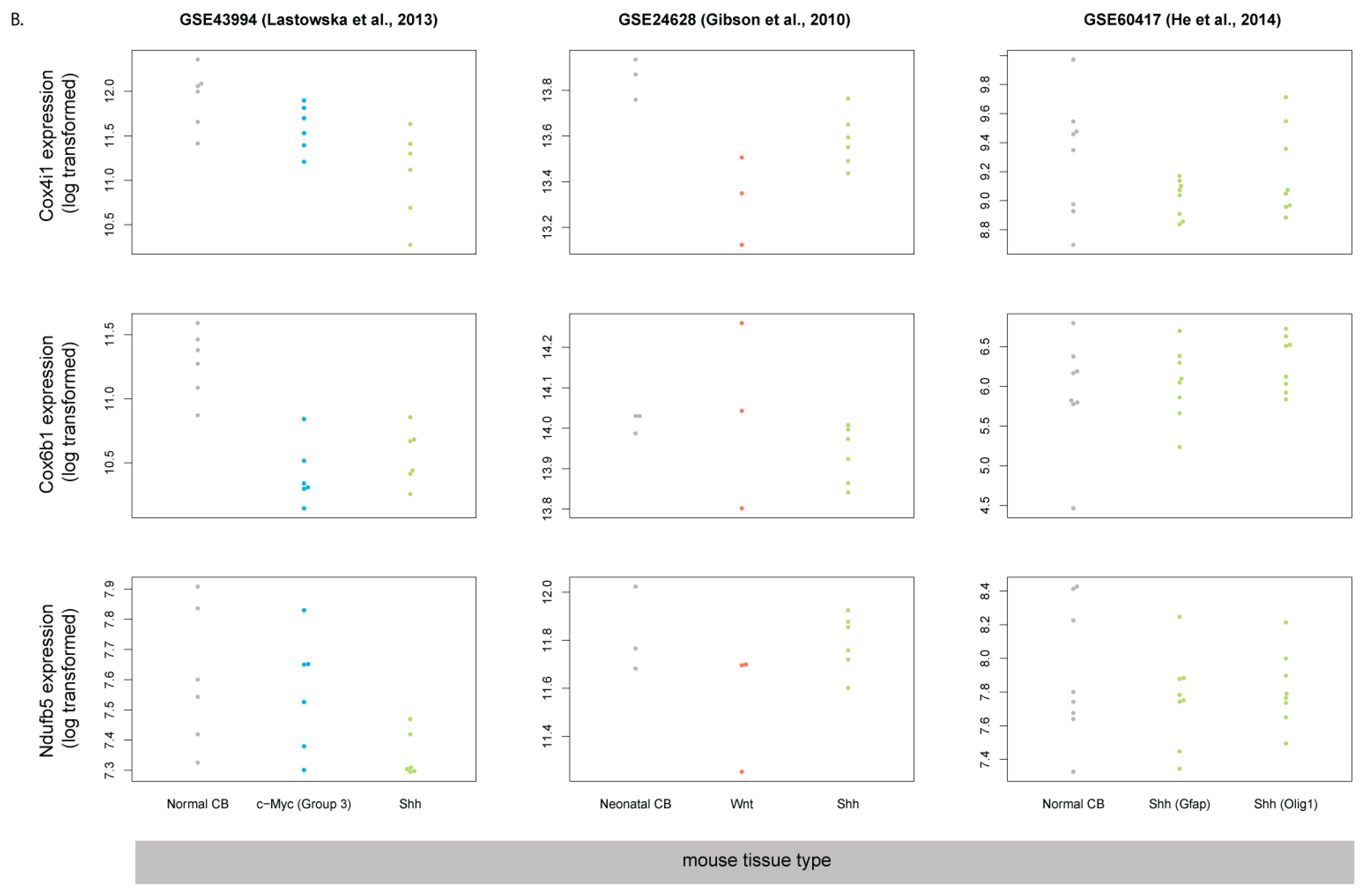
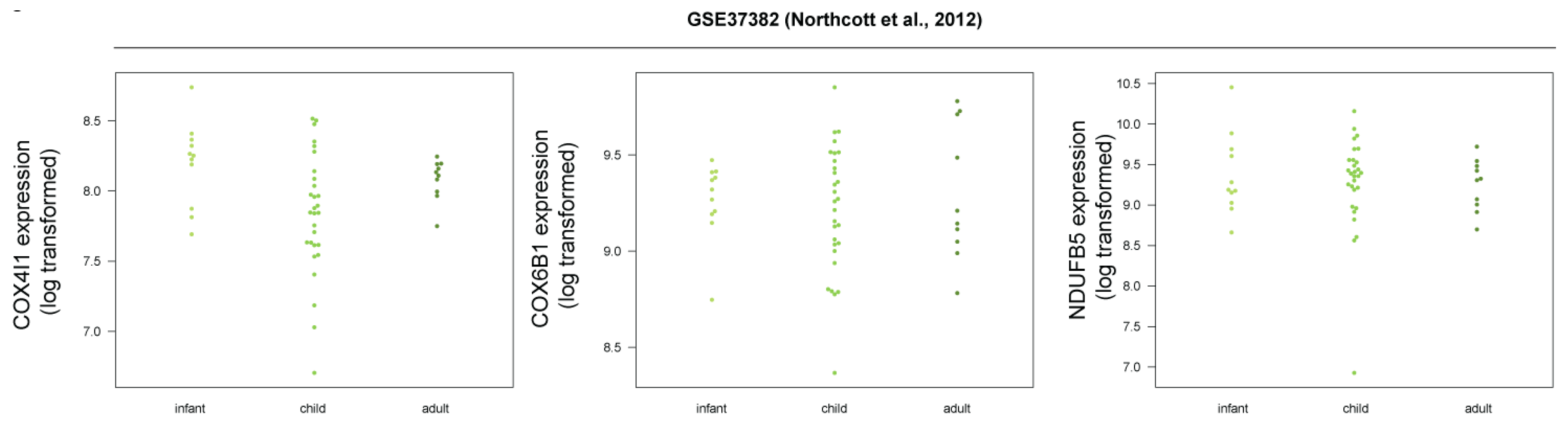
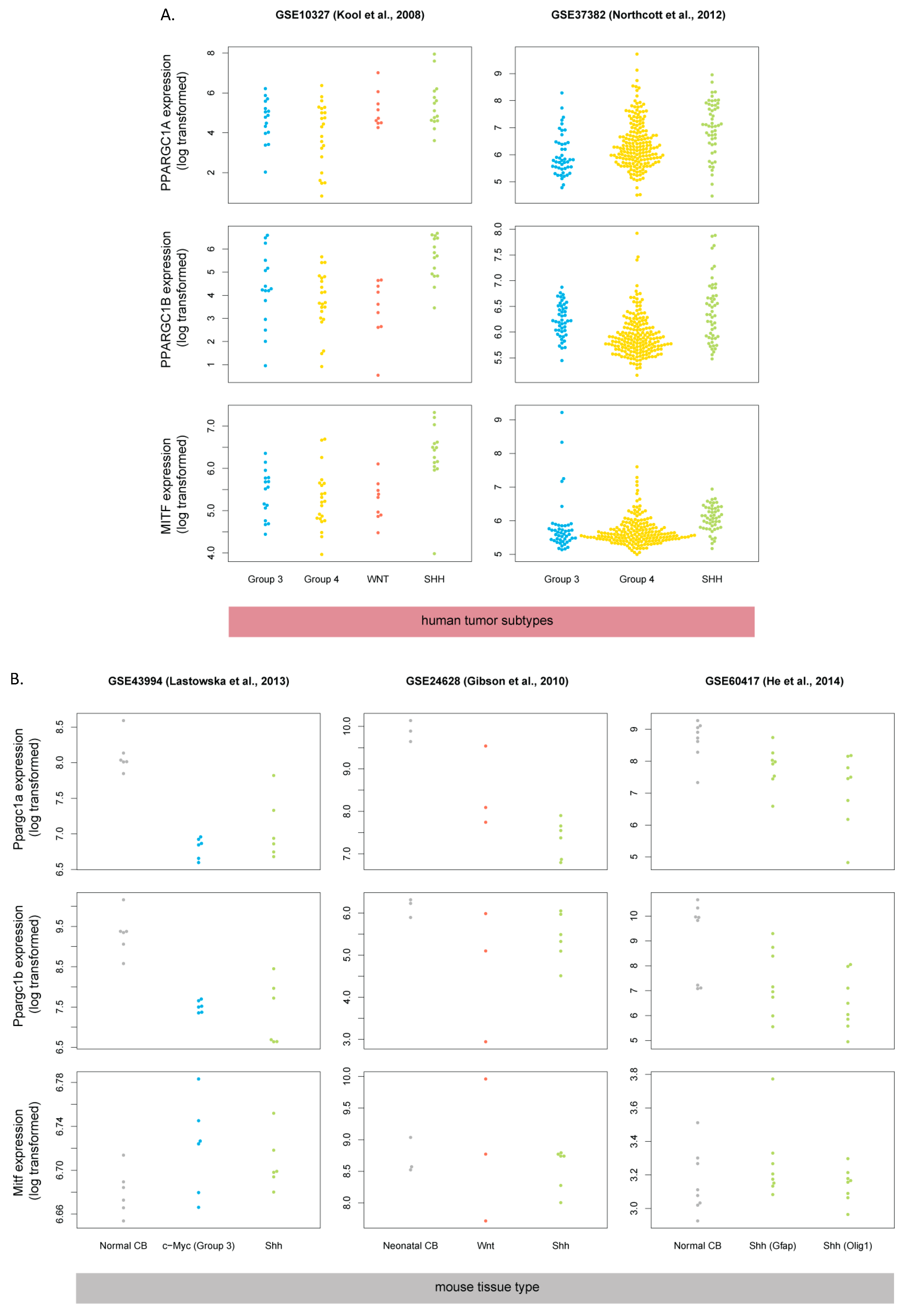
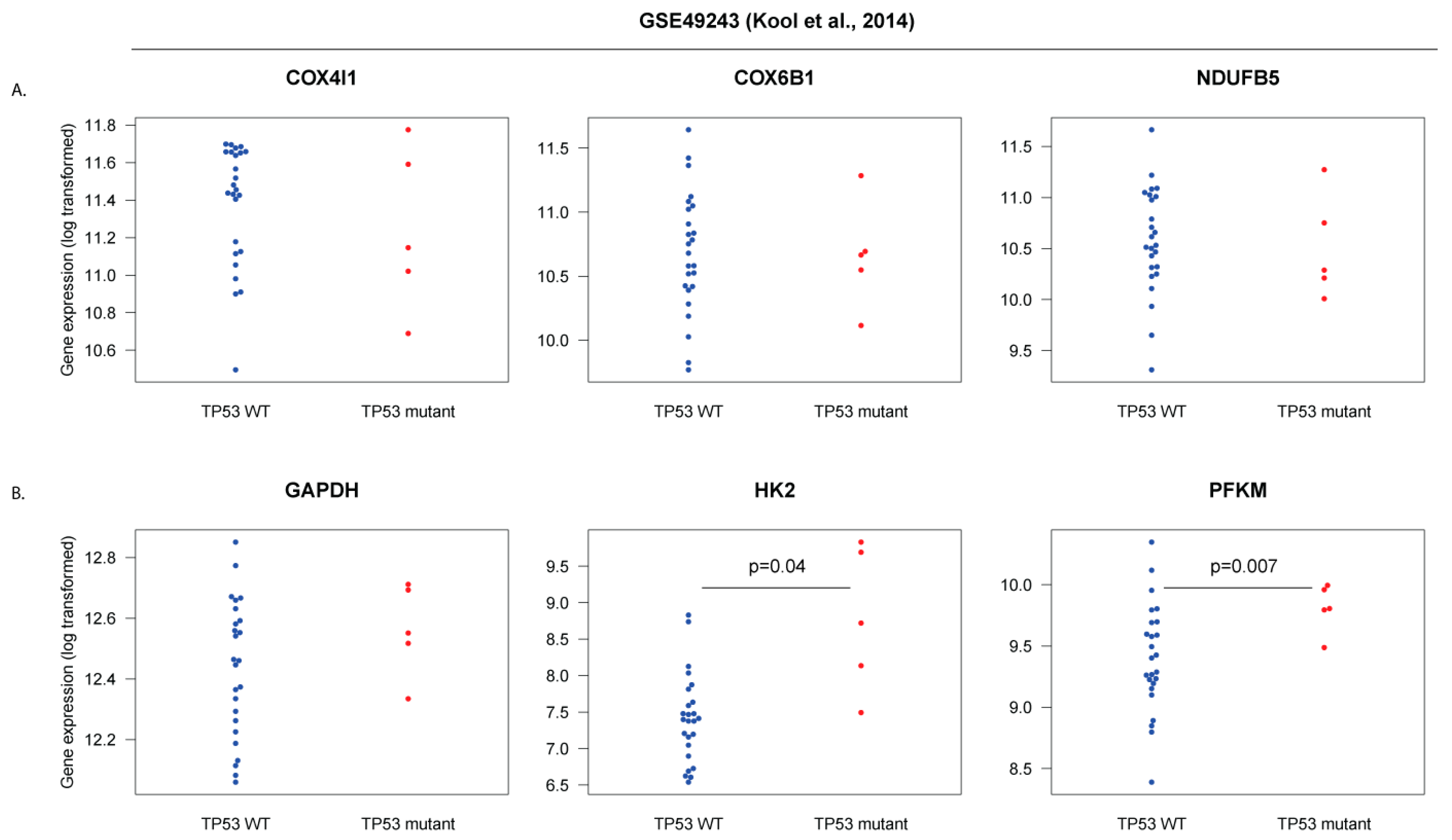
3.5. Human SHH-MB, But Not Mouse Shh-MB, Have High Expression of Genes Associated with Mitochondrial Oxidative Phosphorylation
3.6. Master Regulators of Mitochondrial Biogenesis Are Overexpressed in Human SHH-MB
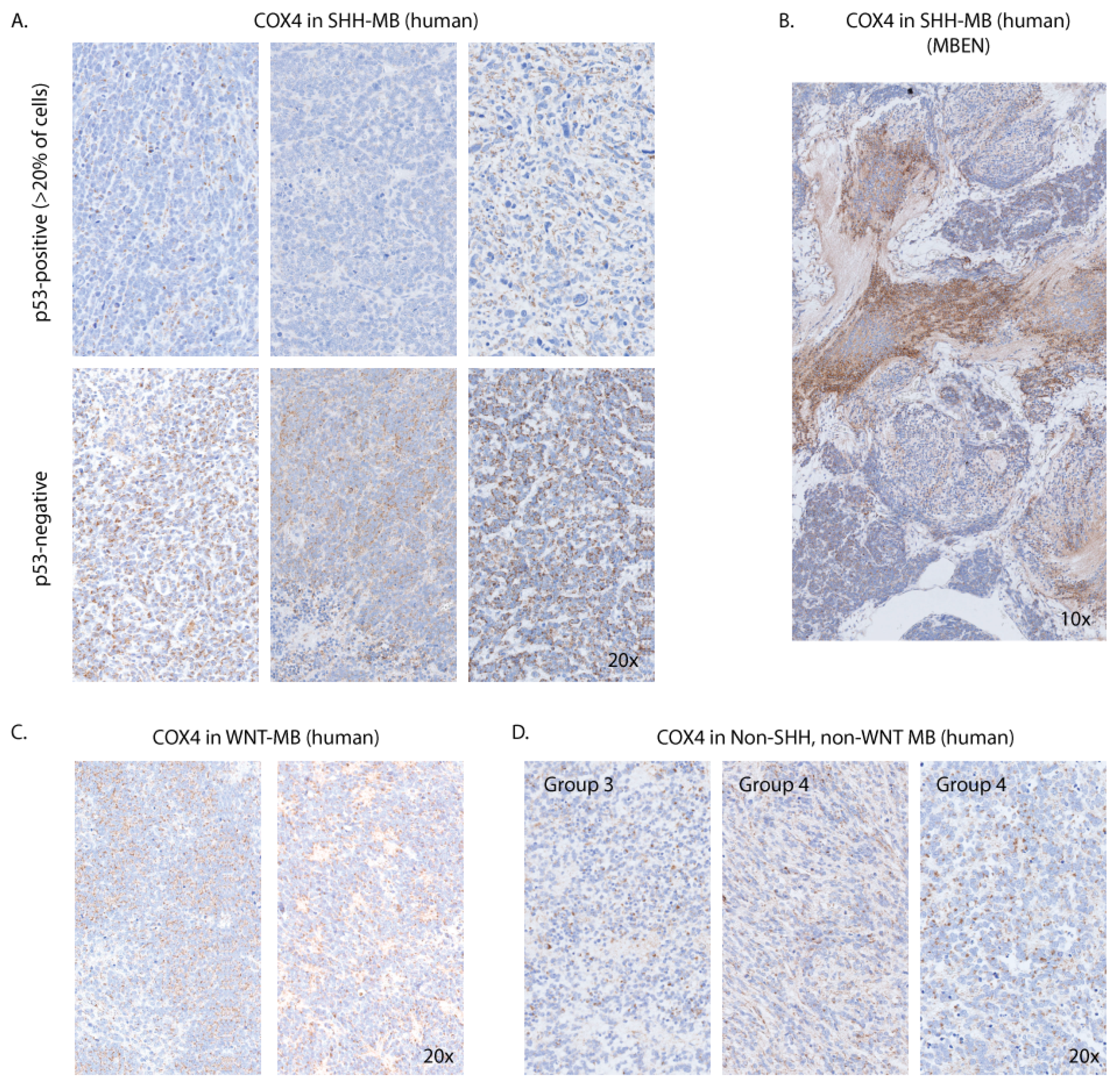
3.7. COX4 Is Highly Expressed in Human p53-Negative SHH-MB
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Northcott, P.A.; Jones, D.T.W.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.-J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.L. Neurodevelopmental impact on children treated for medulloblastoma: A review and proposed conceptual model. Dev. Disabil. Res. Rev. 2008, 14, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.E.; Swartling, F.J.; Schüller, U. Medulloblastoma: Experimental models and reality. Acta Neuropathol. 2017, 134, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.-W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of Medulloblastoma with Hedgehog Pathway Inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.W.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, D.P.; Coyle, B.; Walker, D.A.; Grabowska, A.M. In vitro models of medulloblastoma: Choosing the right tool for the job. J. Biotechnol. 2016, 236, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Milenković, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Hallahan, A.R.; Pritchard, J.I.; Hansen, S.; Benson, M.; Stoeck, J.; Hatton, B.A.; Russell, T.L.; Ellenbogen, R.G.; Bernstein, I.D.; Beachy, P.A.; et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004, 64, 7794–7800. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Villavicencio, E.H.; Tsuchiya, K.D.; Pritchard, J.I.; Ditzler, S.; Pullar, B.; Hansen, S.; Knoblaugh, S.E.; Lee, D.; Eberhart, C.G.; et al. The Smo/Smo model: Hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008, 68, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, L.; Chen, Y.; Remke, M.; Shih, D.; Lu, F.; Wang, H.; Deng, Y.; Yu, Y.; Xia, Y.; et al. The G protein α subunit Gαs is a tumor suppressor in Sonic hedgehog−driven medulloblastoma. Nat. Med. 2014, 20, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Pöschl, J.; Stark, S.; Neumann, P.; Gröbner, S.; Kawauchi, D.; Jones, D.T.W.; Northcott, P.A.; Lichter, P.; Pfister, S.M.; Kool, M.; et al. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol. 2014, 128, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, P.; Zhujiang, A.; Youssef, M.; Waschek, J.A. Interaction of PACAP with Sonic hedgehog reveals complex regulation of the hedgehog pathway by PKA. Cell. Signal. 2013, 25, 2222–2230. [Google Scholar] [CrossRef] [PubMed]
- Lelievre, V.; Seksenyan, A.; Nobuta, H.; Yong, W.H.; Chhith, S.; Niewiadomski, P.; Cohen, J.R.; Dong, H.; Flores, A.; Liau, L.M.; et al. Disruption of the PACAP gene promotes medulloblastoma in ptc1 mutant mice. Dev. Biol. 2008, 313, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Ji, H.; Ouyang, Z.; Zhou, B.; Ma, W.; Vokes, S.A.; McMahon, A.P.; Wong, W.H.; Scott, M.P. Hedgehog pathway-regulated gene networks in cerebellum development and tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 9736–9741. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Collin, F.; Brettschneider, J.; Simpson, K.; Cope, L.; Irizarry, R.A.; Speed, T.P. Quality Assessment of Affymetrix GeneChip Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V.J., Huber, W., Irizarry, R.A., Dudoit, S., Eds.; Statistics for Biology and Health; Springer: New York, NY, USA, 2005; pp. 33–47. ISBN 978-0-387-29362-2. [Google Scholar]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, H.; Johansson, P.; Sundström, A.; Lubovac-Pilav, Z.; Olsson, B.; Nelander, S.; Swartling, F.J. Batch-normalization of cerebellar and medulloblastoma gene expression datasets utilizing empirically defined negative control genes. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Łastowska, M.; Trubicka, J.; Niemira, M.; Paczkowska-Abdulsalam, M.; Karkucińska-Więckowska, A.; Kaleta, M.; Drogosiewicz, M.; Tarasińska, M.; Perek-Polnik, M.; Krętowski, A.; et al. ALK Expression Is a Novel Marker for the WNT-activated Type of Pediatric Medulloblastoma and an Indicator of Good Prognosis for Patients. Am. J. Surg. Pathol. 2017, 41, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-J.; Ellis, T.; Markant, S.L.; Read, T.-A.; Kessler, J.D.; Bourboulas, M.; Schüller, U.; Machold, R.; Fishell, G.; Rowitch, D.H.; et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 2008, 14, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.R.; Resnick, D.Z.; Niewiadomski, P.; Dong, H.; Liau, L.M.; Waschek, J.A. Pituitary adenylyl cyclase activating polypeptide inhibits gli1 gene expression and proliferation in primary medulloblastoma derived tumorsphere cultures. BMC Cancer 2010, 10, 676. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; van Sluis, P.; Troost, D.; Meeteren, N.S.; Caron, H.N.; Cloos, J.; et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Ligon, K.L.; Rakhlin, E.Y.; Thayer, S.P.; Bronson, R.T.; Rowitch, D.; McMahon, A.P. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006, 66, 10171–10178. [Google Scholar] [CrossRef] [PubMed]
- Lastowska, M.; Al-Afghani, H.; Al-Balool, H.H.; Sheth, H.; Mercer, E.; Coxhead, J.M.; Redfern, C.P.F.; Peters, H.; Burt, A.D.; Santibanez-Koref, M.; et al. Identification of a neuronal transcription factor network involved in medulloblastoma development. Acta Neuropathol. Commun. 2013, 1, 35. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, A.; Kunder, R.; Goel, A.; Sarin, R.; Moiyadi, A.; Shenoy, A.; Mamidipally, C.; Noronha, S.; Kannan, S.; Shirsat, N.V. Distinctive microRNA signature of medulloblastomas associated with the WNT signaling pathway. J. Cancer Res. Ther. 2010, 6, 521–529. [Google Scholar] [PubMed]
- Remke, M.; Hielscher, T.; Korshunov, A.; Northcott, P.A.; Bender, S.; Kool, M.; Westermann, F.; Benner, A.; Cin, H.; Ryzhova, M.; et al. FSTL5 is a marker of poor prognosis in non-WNT/non-SHH medulloblastoma. J. Clin. Oncol. 2011, 29, 3852–3861. [Google Scholar] [CrossRef] [PubMed]
- Friendly, M. Corrgrams. Am. Stat. 2002, 56, 316–324. [Google Scholar] [CrossRef]
- Obayashi, T.; Okamura, Y.; Ito, S.; Tadaka, S.; Motoike, I.N.; Kinoshita, K. COXPRESdb: A database of comparative gene coexpression networks of eleven species for mammals. Nucleic Acids Res. 2013, 41, D1014–D1020. [Google Scholar] [CrossRef] [PubMed]
- Okamura, Y.; Aoki, Y.; Obayashi, T.; Tadaka, S.; Ito, S.; Narise, T.; Kinoshita, K. COXPRESdb in 2015: Coexpression database for animal species by DNA-microarray and RNAseq-based expression data with multiple quality assessment systems. Nucleic Acids Res. 2015, 43, D82–D86. [Google Scholar] [CrossRef] [PubMed]
- Wareski, P.; Vaarmann, A.; Choubey, V.; Safiulina, D.; Liiv, J.; Kuum, M.; Kaasik, A. PGC-1α and PGC-1β Regulate Mitochondrial Density in Neurons. J. Biol. Chem. 2009, 284, 21379–21385. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.A.; McCluggage, W.G. Assessing p53 in clinical contexts: Unlearned lessons and new perspectives. J. Pathol. 2006, 208, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Day, C.-P.; Merlino, G.; Van Dyke, T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Yemelyanova, A.; Vang, R.; Kshirsagar, M.; Lu, D.; Marks, M.A.; Shih, I.M.; Kurman, R.J. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: An immunohistochemical and nucleotide sequencing analysis. Mod. Pathol. 2011, 24, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Jiang, A.M.; Qi, C.X.Y. Aberrant Nuclear p53 Expression Predicts Hemizygous 17p (TP53) Deletion in Chronic Lymphocytic Leukemia. Am. J. Clin. Pathol. 2010, 133, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Murnyák, B.; Hortobágyi, T. Immunohistochemical correlates of TP53 somatic mutations in cancer. Oncotarget 2016, 7, 64910–64920. [Google Scholar] [CrossRef] [PubMed]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.H.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugières, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Nör, C.; Taylor, M.D. p53 and Medulloblastoma. Cold Spring Harb. Perspect. Med. 2016, 6, a026278. [Google Scholar] [CrossRef] [PubMed]
- Harami-Papp, H.; Pongor, L.S.; Munkácsy, G.; Horváth, G.; Nagy, Á.M.; Ambrus, A.; Hauser, P.; Szabó, A.; Tretter, L.; Győrffy, B. TP53 mutation hits energy metabolism and increases glycolysis in breast cancer. Oncotarget 2016, 7, 67183–67195. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.-G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Wenger, S.L.; Senft, J.R.; Sargent, L.M.; Bamezai, R.; Bairwa, N.; Grant, S.G. Comparison of established cell lines at different passages by karyotype and comparative genomic hybridization. Biosci. Rep. 2004, 24, 631–639. [Google Scholar] [CrossRef] [PubMed]
- De Witt Hamer, P.C.; Van Tilborg, A.A.G.; Eijk, P.P.; Sminia, P.; Troost, D.; Van Noorden, C.J.F.; Ylstra, B.; Leenstra, S. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene 2008, 27, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, Z.; Yu, L.; Zhang, Y.; Baxter, P.; Voicu, H.; Gurusiddappa, S.; Luan, J.; Su, J.M.; Leung, H.E.; et al. Global gene expression profiling confirms the molecular fidelity of primary tumor-based orthotopic xenograft mouse models of medulloblastoma. Neuro-Oncol. 2012, 14, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Gururangan, S.; Hwang, E.; Herndon, J.E.; Fuchs, H.; George, T.; Coleman, R.E. [18F]fluorodeoxyglucose-positron emission tomography in patients with medulloblastoma. Neurosurgery 2004, 55, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Zukotynski, K.; Fahey, F.; Kocak, M.; Kun, L.; Boyett, J.; Fouladi, M.; Vajapeyam, S.; Treves, T.; Poussaint, T.Y. 18F-FDG PET and MR imaging associations across a spectrum of pediatric brain tumors: A report from the pediatric brain tumor consortium. J. Nucl. Med. 2014, 55, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Gershon, T.R.; Crowther, A.J.; Tikunov, A.; Garcia, I.; Annis, R.; Yuan, H.; Miller, C.R.; Macdonald, J.; Olson, J.; Deshmukh, M. Hexokinase-2-mediated aerobic glycolysis is integral to cerebellar neurogenesis and pathogenesis of medulloblastoma. Cancer Metab. 2013, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Magno, L.D.; Manzi, D.; D’Amico, D.; Coni, S.; Macone, A.; Infante, P.; Marcotullio, L.D.; Smaele, E.D.; Ferretti, E.; Screpanti, I.; et al. Druggable glycolytic requirement for Hedgehog-dependent neuronal and medulloblastoma growth. Cell Cycle 2014, 13, 3404–3413. [Google Scholar] [CrossRef] [PubMed]
- Tech, K.; Gershon, T.R. Energy metabolism in neurodevelopment and medulloblastoma. Transl. Pediatr. 2015, 4, 12–19. [Google Scholar] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession Number | Species | Microarray Platform | Ref. | Dataset Description |
|---|---|---|---|---|
| GSE60417 | mouse | Affymetrix Mouse Gene 1.1 ST Array | [12] | Shh-MB model: Gαs cKO in Olig1- or GFAP-expressing cells |
| GSE9299 | mouse | Affymetrix Mouse Expression 430A/B Array Set | [29] | Shh-MB model: postnatal SmoM2 overexpression |
| GSE43994 | mouse | Illumina MouseRef-8 v2.0 expression beadchip | [30] | Shh-MB model: Ptch1+/− |
| GSE24628 | mouse | Affymetrix Mouse Genome 430 2.0 Array | [28] | Shh-MB and Wnt-MB model: Ptch1+/−; Tp53−/− and Blbp-Cre+/−; Ctnnb1+/lox(ex3); Tp53flx/flx |
| GSE37418 | human | Affymetrix Human Genome U133 Plus 2.0 Array | [31] | human tumors of different subtypes (Wnt, Shh, Group 3, Group 4) |
| GSE41842 | human | Affymetrix Human Gene 1.0 ST Array | [32] | human tumors of different subtypes (Wnt, Shh, Group 3, Group 4) |
| GSE10327 | human | Affymetrix Human Genome U133 Plus 2.0 Array | [27] | human tumors of different subtypes (Wnt, Shh, Group 3(C), Group 4(D), Group E) |
| GSE37382 | human | Affymetrix Human Gene 1.1 ST Array | [26] | human tumors of different subtypes (Wnt, Shh, Group 3) |
| GSE28245 | human | Agilent-014850 Whole Human Genome Microarray 4x44K G4112F | [33] | human tumors of different subtypes (Wnt, Shh, Group 3, Group 4) |
| GSE49243 | human | Affymetrix Human Genome U133 Plus 2.0 Array | [7] | human SHH-MB tumors with matched mutation data |
| Gene Set Name | FWER p-val |
|---|---|
| REACTOME_RESPIRATORY_ELECTRON_TRANSPORT_ATP_SYNTHESIS_BY_CHEMIOSMOTIC_COUPLING_AND_HEAT_PRODUCTION_BY_UNCOUPLING_PROTEINS_ | 0 |
| REACTOME_TCA_CYCLE_AND_RESPIRATORY_ELECTRON_TRANSPORT | 0 |
| REACTOME_RESPIRATORY_ELECTRON_TRANSPORT | 0 |
| KEGG_OXIDATIVE_PHOSPHORYLATION | 0 |
| POMEROY_MEDULLOBLASTOMA_DESMOPLASIC_VS_CLASSIC_DN | 0 |
| MOOTHA_VOXPHOS | 0 |
| LEE_TARGETS_OF_PTCH1_AND_SUFU_DN | 0 |
| ANASTASSIOU_MULTICANCER_INVASIVENESS_SIGNATURE | 0 |
| GO_RESPIRATORY_CHAIN | 0 |
| GO_REGULATION_OF_POSTSYNAPTIC_MEMBRANE_POTENTIAL | 0 |
| LEIN_NEURON_MARKERS | 0.001 |
| VECCHI_GASTRIC_CANCER_EARLY_DN | 0.001 |
| DELYS_THYROID_CANCER_DN | 0.005 |
| GO_CELLULAR_RESPIRATION | 0.007 |
| GO_MITOCHONDRIAL_PROTEIN_COMPLEX | 0.008 |
| Gene Set Name | FWER p-val |
|---|---|
| HALLMARK_E2F_TARGETS | 0 |
| REACTOME_CELL_CYCLE | 0 |
| REACTOME_CELL_CYCLE_MITOTIC | 0 |
| MARSON_BOUND_BY_E2F4_UNSTIMULATED | 0 |
| REACTOME_DNA_REPLICATION | 0 |
| POMEROY_MEDULLOBLASTOMA_DESMOPLASIC_VS_CLASSIC_UP | 0 |
| SOTIRIOU_BREAST_CANCER_GRADE_1_VS_3_UP | 0 |
| ZHANG_TLX_TARGETS_60HR_DN | 0 |
| CHANG_CYCLING_GENES | 0 |
| REACTOME_S_PHASE | 0 |
| REACTOME_SYNTHESIS_OF_DNA | 0 |
| NIKOLSKY_BREAST_CANCER_17Q21_Q25_AMPLICON | 0 |
| GO_POSTREPLICATION_REPAIR | 0 |
| KONG_E2F3_TARGETS | 0.005 |
| PUJANA_XPRSS_INT_NETWORK | 0.005 |
| FISCHER_DREAM_TARGETS | 0.005 |
| ROSTY_CERVICAL_CANCER_PROLIFERATION_CLUSTER | 0.008 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łastowska, M.; Karkucińska-Więckowska, A.; Waschek, J.A.; Niewiadomski, P. Differential Expression of Mitochondrial Biogenesis Markers in Mouse and Human SHH-Subtype Medulloblastoma. Cells 2019, 8, 216. https://doi.org/10.3390/cells8030216
Łastowska M, Karkucińska-Więckowska A, Waschek JA, Niewiadomski P. Differential Expression of Mitochondrial Biogenesis Markers in Mouse and Human SHH-Subtype Medulloblastoma. Cells. 2019; 8(3):216. https://doi.org/10.3390/cells8030216
Chicago/Turabian StyleŁastowska, Maria, Agnieszka Karkucińska-Więckowska, James A. Waschek, and Paweł Niewiadomski. 2019. "Differential Expression of Mitochondrial Biogenesis Markers in Mouse and Human SHH-Subtype Medulloblastoma" Cells 8, no. 3: 216. https://doi.org/10.3390/cells8030216
APA StyleŁastowska, M., Karkucińska-Więckowska, A., Waschek, J. A., & Niewiadomski, P. (2019). Differential Expression of Mitochondrial Biogenesis Markers in Mouse and Human SHH-Subtype Medulloblastoma. Cells, 8(3), 216. https://doi.org/10.3390/cells8030216