Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites


Abstract
:1. Introduction
2. Existing Prediction Models
3. Datasets Collection and Preparation
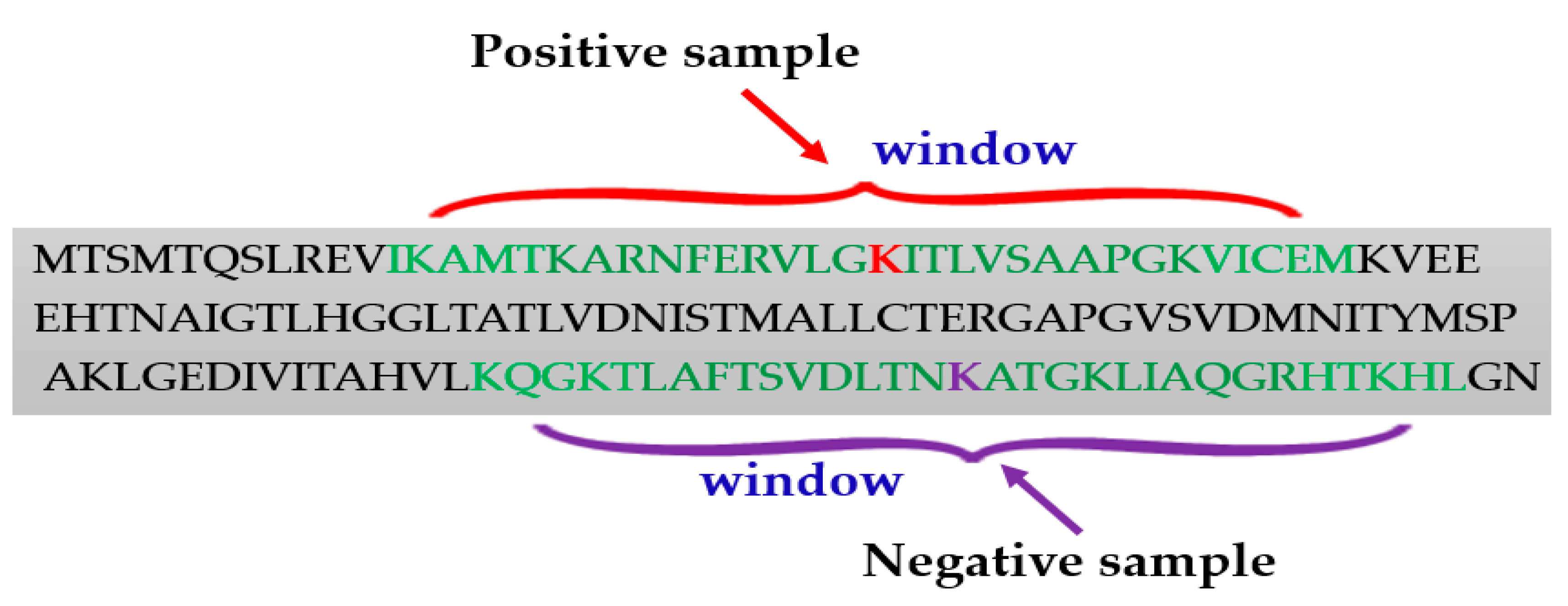
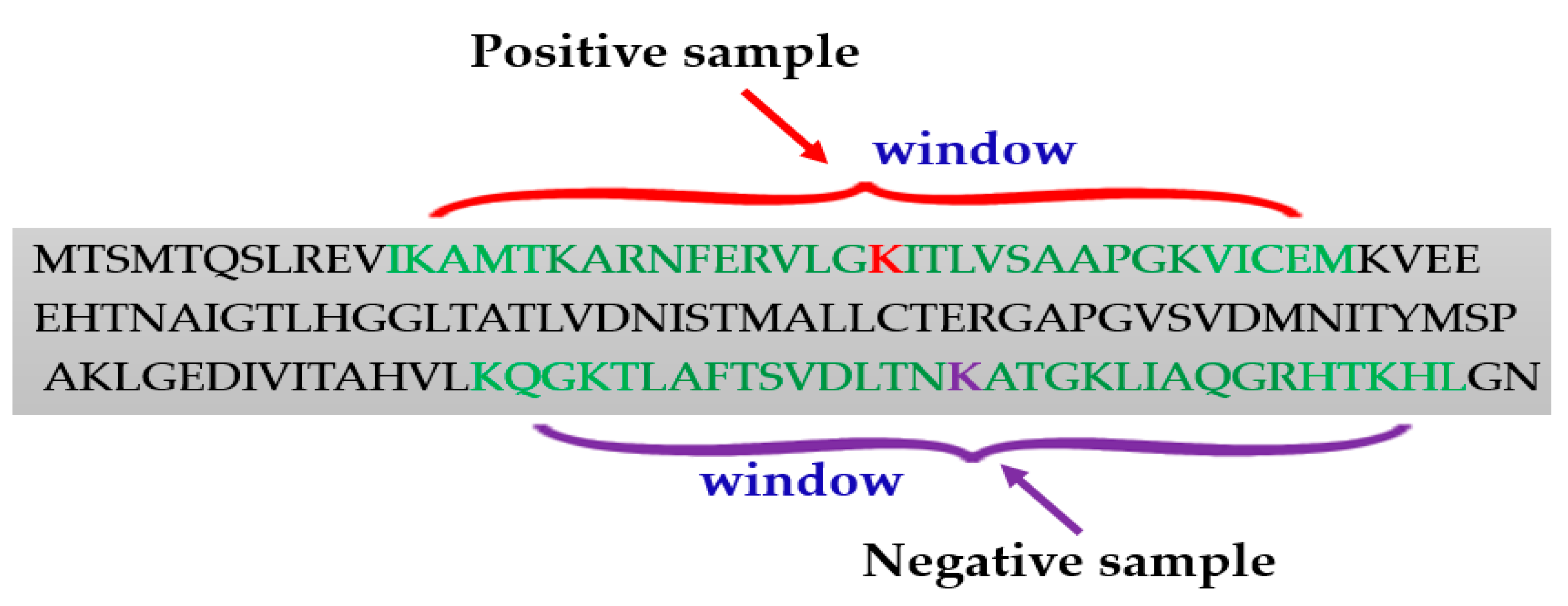
Positive and Negative Samples
4. Algorithms of Predicting Lysine Succinylation Site
4.1. Random Forest
4.2. Support Vector Machine
4.3. Adaptive Boosting
4.4. Decision Trees
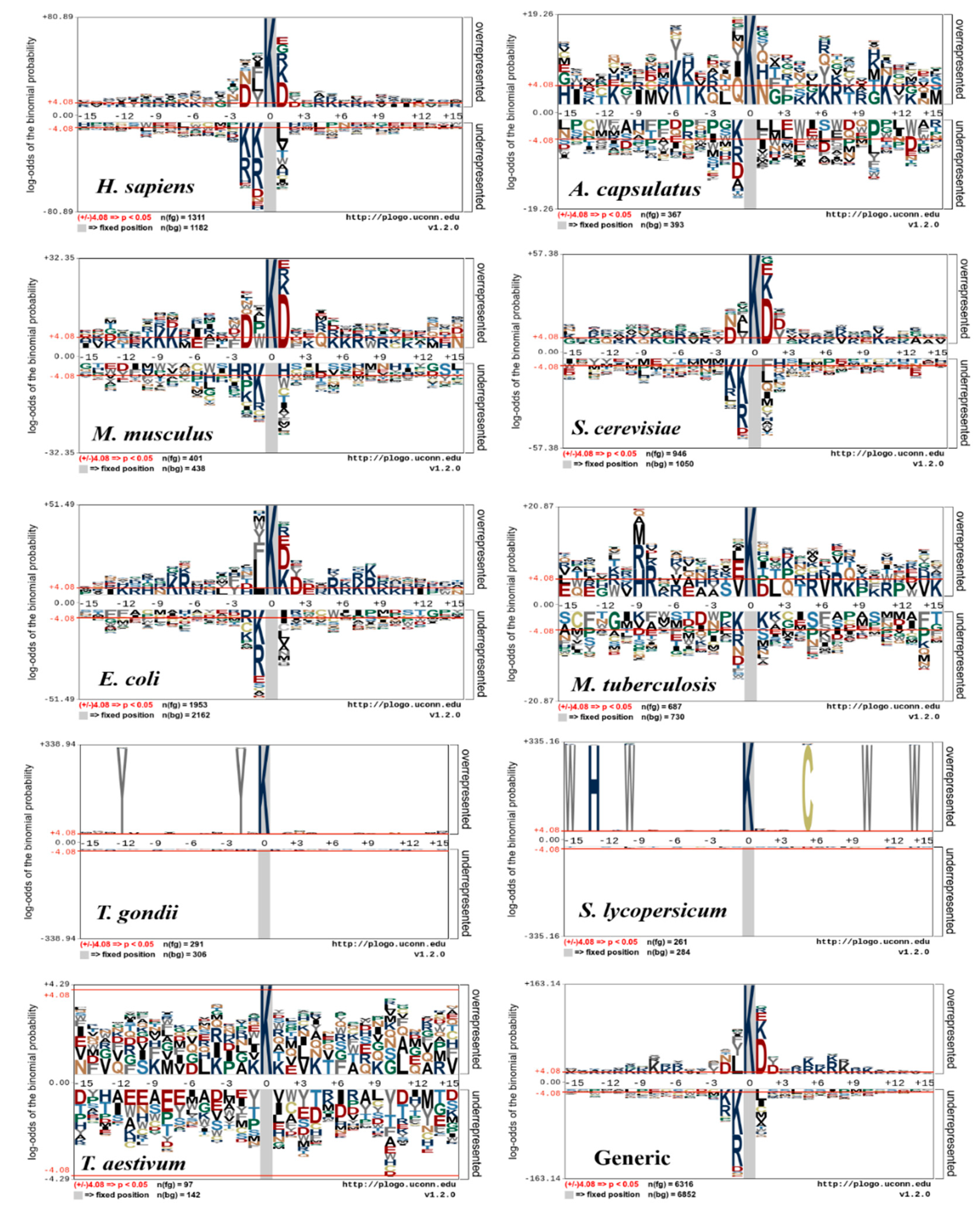
5. Motif Conservation of Species-Specific and Generic Succinylation Sites
6. Important Descriptors for Predicting Succinylation Sites
7. Features Assessment of Species-specific Succinylation Sites
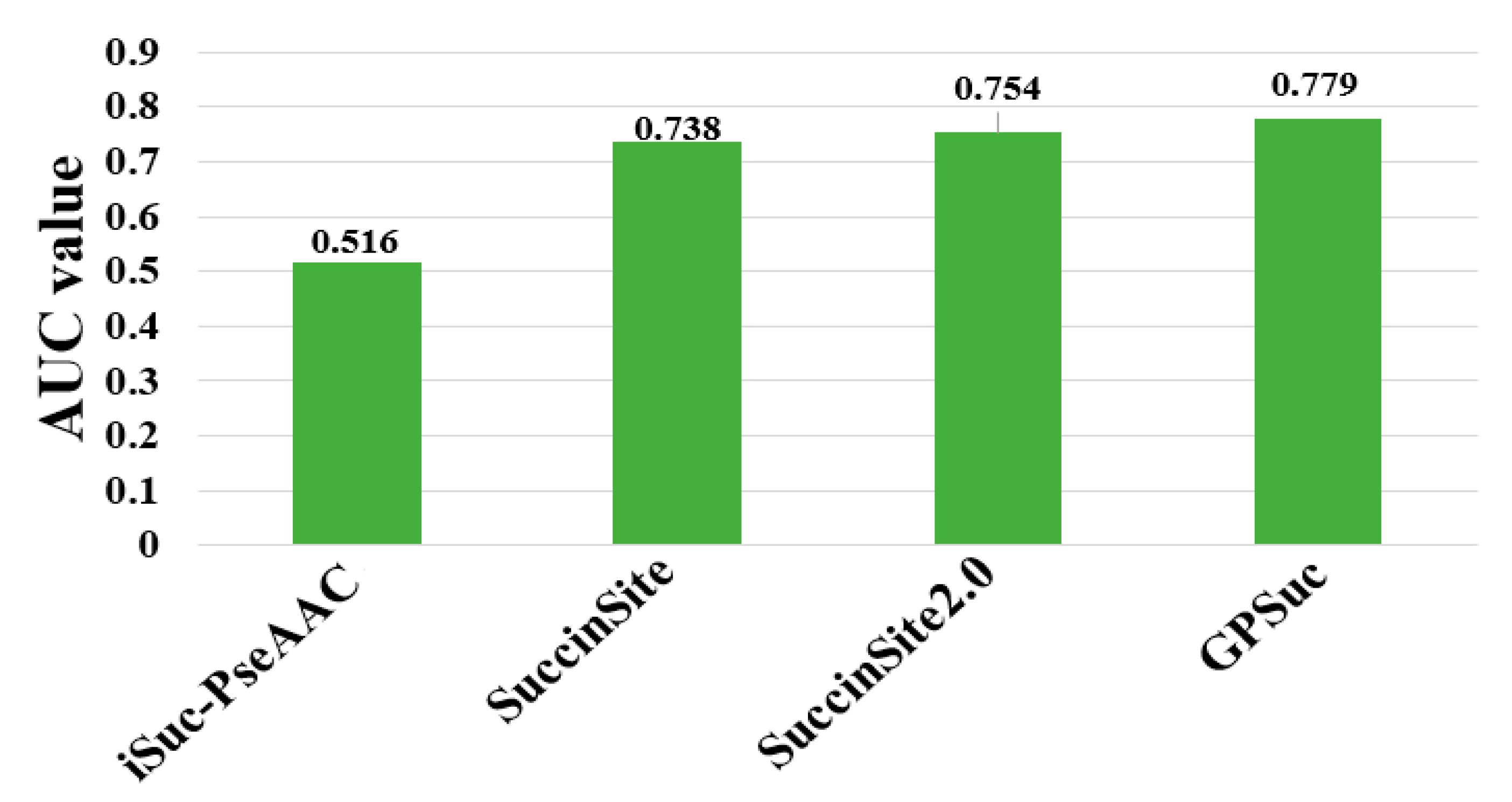
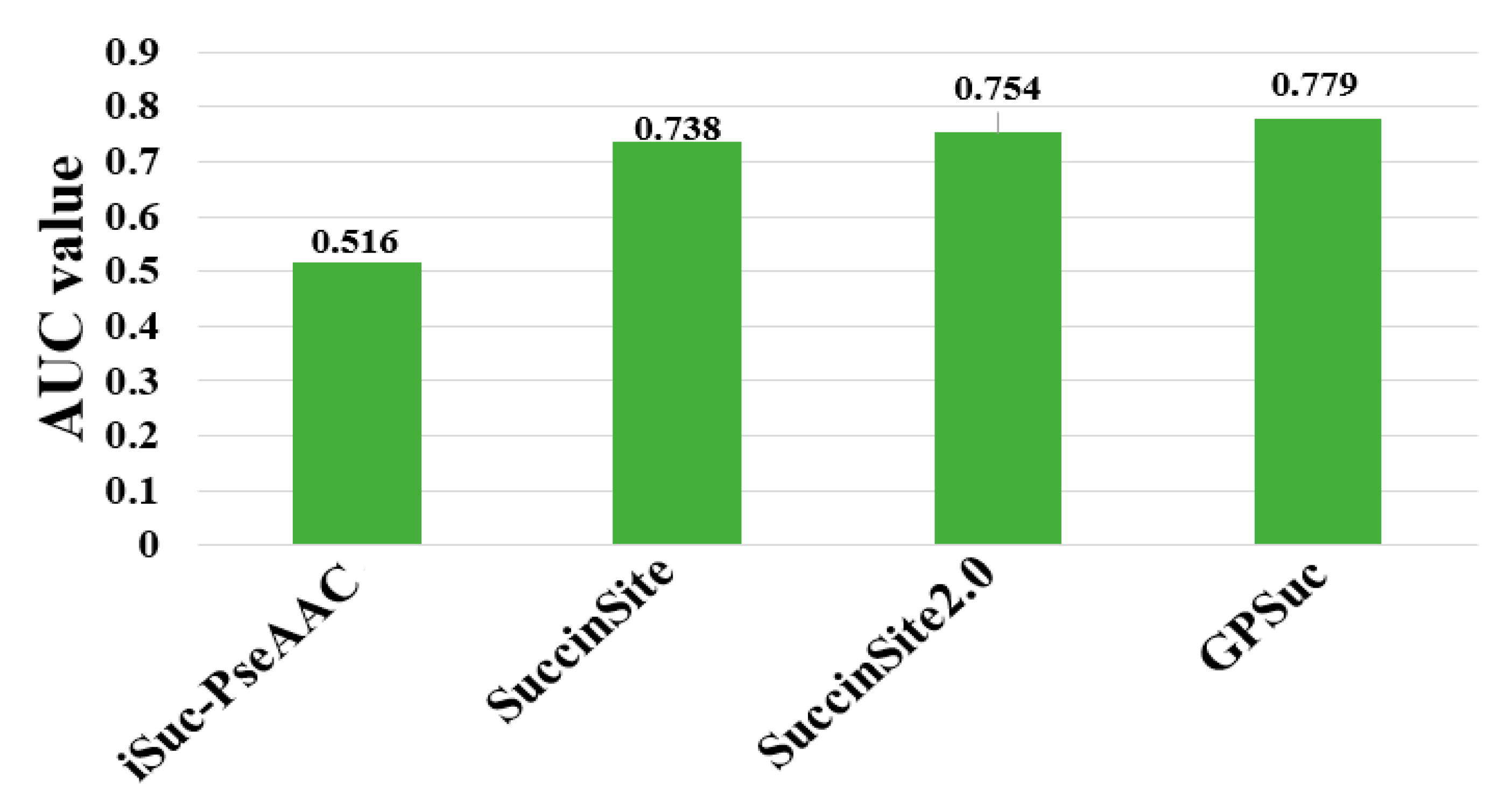
8. Comparative Analysis of Different Predictors
9. The Online Employment Services
10. Perceptions for Prediction Models
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, X.; Hu, X.; Wan, Y.; Xie, G.; Li, X.; Chen, D.; Cheng, Z.; Yi, X.; Liang, S.; Tan, F. Systematic identification of the lysine succinylation in the protozoan parasite Toxoplasma gondii. J. Proteome Res. 2014, 13, 6087–6095. [Google Scholar] [CrossRef] [PubMed]
- Colak, G.; Xie, Z.; Zhu, A.Y.; Dai, L.; Lu, Z.; Zhang, Y.; Wan, X.; Chen, Y.; Cha, Y.H.; Lin, H.; et al. Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli. Mol. Cell Proteomics 2013, 12, 3509–3520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Ariyantoro, A.R.; Katsuno, N.; Nishizu, T. Effects of dual modification with succinylation and annealing on physicochemical, thermal and morphological properties of corn starch. Foods 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Alleyn, M.; Breitzig, M.; Lockey, R.; Kolliputi, N. The dawn of succinylation: a posttranslational modification. Am. J. Physiol., Cell Physiol. 2018, 314, C228–C232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xie, L.; Yang, Z.; Zhou, J.; Xie, J. Lysine succinylation of Mycobacterium tuberculosis isocitrate lyase (ICL) fine-tunes the microbial resistance to antibiotics. J. Biomol. Struct. Dyn. 2017, 35, 1030–1041. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, G.; Song, L.; Mu, P.; Wang, S.; Liang, W.; Lin, Q. Global analysis of protein lysine succinylation profiles in common wheat. BMC Genomics 2017, 18, 309. [Google Scholar] [CrossRef]
- Yokoyama, A.; Katsura, S.; Sugawara, A. Biochemical analysis of histone succinylation. Biochem. Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Xu, X.; Liu, T.; Yang, J.; Chen, L.; Liu, B.; Wei, C.; Wang, L.; Jin, Q. The first succinylome profile of Trichophyton rubrum reveals lysine succinylation on proteins involved in various key cellular processes. BMC Genomics 2017, 18. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.M.; et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 4320–4325. [Google Scholar] [CrossRef]
- Moin, A.; Ali, T.M.; Hasnain, A. Effect of succinylation on functional and morphological properties of starches from broken kernels of Pakistani Basmati and Irri rice cultivars. Food Chem. 2016, 191, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Nagano-Shoji, M.; Kubo, S.; Kawamura, Y.; Yoshida, A.; Kawasaki, H.; Nishiyama, M.; Yoshida, M.; Kosono, S. Altered acetylation and succinylation profiles in Corynebacterium glutamicum in response to conditions inducing glutamate overproduction. Microbiol. Open 2016, 5, 152–173. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Dai, J.; Dai, L.; Tan, M.; Cheng, Z.; Wu, Y.; Boeke, J.D.; Zhao, Y. Lysine succinylation and lysine malonylation in histones. Mol. Cell Proteomics 2012, 11, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Payne, R.M. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Lu, Z.; Xie, Z.; Cheng, Z.; Chen, Y.; Tan, M.; Luo, H.; Zhang, Y.; He, W.; Yang, K.; et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Lawal, O.S.; Adebowale, K.O. Effect of acetylation and succinylation on solubility profile, water absorption capacity, oil absorption capacity and emulsifying properties of mucuna bean (Mucuna pruriens) protein concentrate. Nahrung 2004, 48, 129–136. [Google Scholar] [CrossRef]
- Zaghloul, M.; Prakash, V. Effect of succinylation on the functional and physicochemical properties of alpha-globulin, the major protein fraction from Sesamum indicum L. Nahrung 2002, 46, 364–369. [Google Scholar] [CrossRef]
- Xu, C.; Ge, L.; Zhang, Y.; Dehmer, M.; Gutman, I. Prediction of therapeutic peptides by incorporating q-Wiener index into Chou’s general PseAAC. J. Biomed. Inform. 2017. [Google Scholar] [CrossRef]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Li, J.; Deng, W.; Yu, Z.; Fang, W.; Chen, M.; Liao, W.; Xie, J.; Pan, W. Proteomic analysis of lysine succinylation of the human pathogen Histoplasma capsulatum. J. Proteomics 2017, 154, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Sankari, E.S.; Manimegalai, D. Predicting membrane protein types using various decision tree classifiers based on various modes of general PseAAC for imbalanced datasets. J. Theor. Biol. 2017, 435, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Chen, R.; Li, C.; Li, W.; Ye, Z. Global analysis of protein lysine succinylation profiles and their overlap with lysine acetylation in the marine bacterium Vibrio parahemolyticus. J. Proteome Res. 2015, 14, 4309–4318. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Scholz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Dennison, J.B.; Ayres, M.L.; Kaluarachchi, K.; Plunkett, W.; Gandhi, V. Intracellular succinylation of 8-chloroadenosine and its effect on fumarate levels. J. Biol. Chem. 2010, 285, 8022–8030. [Google Scholar] [CrossRef]
- Xu, H.; Chen, X.; Xu, X.; Shi, R.; Suo, S.; Cheng, K.; Zheng, Z.; Wang, M.; Wang, L.; Zhao, Y.; et al. Lysine acetylation and succinylation in hela cells and their essential roles in response to UV-induced stress. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- He, D.; Wang, Q.; Li, M.; Damaris, R.N.; Yi, X.; Cheng, Z.; Yang, P. Global proteome analyses of lysine acetylation and succinylation reveal the widespread involvement of both modification in metabolism in the embryo of germinating rice seed. J. Proteome Res. 2016, 15, 879–890. [Google Scholar] [CrossRef]
- Chen, Y. Quantitative analysis of the Sirt5-regulated lysine succinylation proteome in mammalian cells. Methods Mol. Biol. 2016, 1410, 23–37. [Google Scholar] [CrossRef]
- Bontemps-Gallo, S.; Madec, E.; Robbe-Masselot, C.; Souche, E.; Dondeyne, J.; Lacroix, J.M. The opgC gene is required for OPGs succinylation and is osmoregulated through RcsCDB and EnvZ/OmpR in the phytopathogen Dickeya dadantii. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Atila, M.; Katselis, G.; Chumala, P.; Luo, Y. Characterization of N-succinylation of l-lysylphosphatidylglycerol in Bacillus subtilis using tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Tayyab, S.; Qasim, M.A. A correlation between changes in conformation and molecular properties of bovine serum albumin upon succinylation. J. Biochem. 1986, 100, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Jou, Y.H.; Johnson, G.; Pressman, D. Succinylation of hapten-protein conjugates facilitates coupling to erythrocytes by water soluble carbodiimide: Preparation of stable and sensitive target cells for use in hemolytic assays. J. Immunol. Methods 1981, 42, 79–92. [Google Scholar] [CrossRef]
- Thuy, L.P.; Brown, J.E.; Baugh, R.F.; Hougie, C. Effects of succinylation and dodecyl, succinylation on bovine factor VIII/von Willebrand factor complex. Thromb. Res. 1980, 18, 305–313. [Google Scholar] [CrossRef]
- Rosen, R.; Becher, D.; Buttner, K.; Biran, D.; Hecker, M.; Ron, E.Z. Probing the active site of homoserine trans-succinylase. FEBS Lett. 2004, 577, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Bochmann, S.M.; Spiess, T.; Kotter, P.; Entian, K.D. Synthesis and succinylation of subtilin-like lantibiotics are strongly influenced by glucose and transition state regulator AbrB. Appl. Environ. Microbiol. 2015, 81, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, J.; Cheng, Z.; Gao, P.; Sun, J.; Chen, X.; Chen, C.; Wang, Y.; Wang, Z. Quantitative global proteome and lysine succinylome analyses provide insights into metabolic regulation and lymph node metastasis in gastric cancer. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.L.; Xing, J.; Chong, C.K.; Liu, H.; Corke, H. Determination of the degree of succinylation in diverse modified starches by raman spectroscopy. J. Agric. Food Chem. 2000, 48, 5105–5108. [Google Scholar] [CrossRef] [PubMed]
- Alcalde, M.; Plou, F.J.; Teresa Martin, M.; Valdes, I.; Mendez, E.; Ballesteros, A. Succinylation of cyclodextrin glycosyltransferase from Thermoanaerobacter sp. 501 enhances its transferase activity using starch as donor. J. Biotechnol. 2001, 86, 71–80. [Google Scholar] [CrossRef]
- Wan, Y.; Liu, J.; Guo, S. Effects of succinylation on the structure and thermal aggregation of soy protein isolate. Food Chem. 2018, 245, 542–550. [Google Scholar] [CrossRef]
- Smestad, J.; Erber, L.; Chen, Y.; Maher, L.J., III. Chromatin succinylation correlates with active gene expression and is perturbed by defective TCA cycle metabolism. iScience 2018, 2, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Yang, M.; Yue, Y.; Ge, F.; Li, Y.; Guo, X.; Zhang, J.; Zhang, F.; Nie, X.; Wang, S. Lysine succinylation contributes to aflatoxin production and pathogenicity in Aspergillus flavus. Mol. Cell. Proteomics 2018, 17, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Mujahid, H.; Meng, X.; Xing, S.; Peng, X.; Wang, C.; Peng, Z. Malonylome analysis in developing rice (Oryza sativa) seeds suggesting that protein lysine malonylation is well-conserved and overlaps with acetylation and succinylation substantially. J. Proteomics 2018, 170, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.Q.; Li, G.Y.; Xie, Q.T.; Zhang, B.; Li, X.M.; Pan, Y.; Chen, H.Q. Evaluation studies on the combined effect of hydrothermal treatment and octenyl succinylation on the physic-chemical, structural and digestibility characteristics of sweet potato starch. Food Chem. 2018, 256, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, Y.; Chen, Y.; Cheng, Z.; Gu, J.; Deng, J.; Bi, L.; Chen, C.; Mo, R.; Wang, X.; et al. Succinylome analysis reveals the involvement of lysine succinylation in metabolism in pathogenic Mycobacterium tuberculosis. Mol. Cell. Proteomics 2015, 14, 796–811. [Google Scholar] [CrossRef]
- Jin, W.; Wu, F. Proteome-wide identification of lysine succinylation in the proteins of tomato (Solanum lycopersicum). PLoS ONE 2016, 11, e0147586. [Google Scholar] [CrossRef]
- Komine-Abe, A.; Nagano-Shoji, M.; Kubo, S.; Kawasaki, H.; Yoshida, M.; Nishiyama, M.; Kosono, S. Effect of lysine succinylation on the regulation of 2-oxoglutarate dehydrogenase inhibitor, OdhI, involved in glutamate production in Corynebacterium glutamicum. Biosci. Biotechnol. Biochem. 2017, 81, 2130–2138. [Google Scholar] [CrossRef]
- Feng, S.; Jiao, K.; Guo, H.; Jiang, M.; Hao, J.; Wang, H.; Shen, C. Succinyl-proteome profiling of Dendrobium officinale, an important traditional Chinese orchid herb, revealed involvement of succinylation in the glycolysis pathway. BMC Genomics 2017, 18. [Google Scholar] [CrossRef]
- Okanishi, H.; Kim, K.; Fukui, K.; Yano, T.; Kuramitsu, S.; Masui, R. Proteome-wide identification of lysine succinylation in thermophilic and mesophilic bacteria. Biochim. Biophys. Acta 2017, 1865, 232–242. [Google Scholar] [CrossRef]
- Xie, L.; Liu, W.; Li, Q.; Chen, S.; Xu, M.; Huang, Q.; Zeng, J.; Zhou, M.; Xie, J. First succinyl-proteome profiling of extensively drug-resistant Mycobacterium tuberculosis revealed involvement of succinylation in cellular physiology. J. Proteome Res. 2015, 14, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Liu, Z.; Tian, G.; Bao, X.; Ishibashi, T.; Li, X.D. Site-specific installation of succinyl lysine analog into histones reveals the effect of h2bk34 succinylation on nucleosome dynamics. Cell Chem. Biol. 2018, 25, 166–174.e7. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, K.A.; Abraham, D.M.; Liu, J.; Locasale, J.W.; Grimsrud, P.A.; Hirschey, M.D. Ablation of Sirtuin5 in the postnatal mouse heart results in protein succinylation and normal survival in response to chronic pressure overload. J. Biol. Chem. 2018, 293, 10630–10645. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ning, Q.; Chai, H.; Ma, Z. Accurate in silico identification of protein succinylation sites using an iterative semi-supervised learning technique. J. Theor. Biol. 2015, 374, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ding, Y.X.; Ding, J.; Lei, Y.H.; Wu, L.Y.; Deng, N.Y. iSuc-PseAAC: predicting lysine succinylation in proteins by incorporating peptide position-specific propensity. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.D.; Shi, S.P.; Wen, P.P.; Qiu, J.D. SuccFind: A novel succinylation sites online prediction tool via enhanced characteristic strategy. Bioinformatics 2015. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; Lopez, Y.; Lal, S.P.; Taherzadeh, G.; Michaelson, J.; Sattar, A.; Tsunoda, T.; Sharma, A. PSSM-Suc: Accurately predicting succinylation using position specific scoring matrix into bigram for feature extraction. J. Theor. Biol. 2017, 425, 97–102. [Google Scholar] [CrossRef]
- Lopez, Y.; Dehzangi, A.; Lal, S.P.; Taherzadeh, G.; Michaelson, J.; Sattar, A.; Tsunoda, T.; Sharma, A. SucStruct: Prediction of succinylated lysine residues by using structural properties of amino acids. Anal. Biochem. 2017, 527, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Yang, S.; Zhou, Y.; Mollah, M.N. SuccinSite: a computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties. Mol. Biosyst. 2016, 12, 786–795. [Google Scholar] [CrossRef]
- Thanamani, B.; Thanamani Selvados, A. Feature Selection based on Information Gain. Int. J. Innov. Technol. Expl. Eng. 2013, 2, 2278–3075. [Google Scholar]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. pSuc-Lys: Predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J. Theor. Biol. 2016, 394, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Mollah, M.N.; Cao, Y.; Guo, D. A systematic identification of species-specific protein succinylation sites using joint element features information. Int. J. Nanomed. 2017, 12, 6303–6315. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.; Zhao, X.; Bao, L.; Ma, Z.; Zhao, X. Detecting succinylation sites from protein sequences using ensemble support vector machine. BMC Bioinform. 2018, 19, 237. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, M.; Wang, Y.; Chen, Z.; Zhang, J.; Lin, X.; Ge, F.; Zhao, J. Effects of PSII manganese-stabilizing protein succinylation on photosynthesis in the model cyanobacterium Synechococcus sp. PCC 7002. Plant Cell Physiol. 2018, 59, 1466–1482. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Kurata, H. GPSuc: Global prediction of generic and species-specific succinylation sites by aggregating multiple sequence features. PloS One 2018, 13, e0200283. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; Lopez, Y.; Lal, S.P.; Taherzadeh, G.; Sattar, A.; Tsunoda, T.; Sharma, A. Improving succinylation prediction accuracy by incorporating the secondary structure via helix, strand and coil, and evolutionary information from profile bigrams. PLoS ONE 2018, 13, e0191900. [Google Scholar] [CrossRef] [PubMed]
- Lopez, Y.; Sharma, A.; Dehzangi, A.; Lal, S.P.; Taherzadeh, G.; Sattar, A.; Tsunoda, T. Success: evolutionary and structural properties of amino acids prove effective for succinylation site prediction. BMC Genomics 2018, 19. [Google Scholar] [CrossRef]
- Gaviard, C.; Broutin, I.; Cosette, P.; De, E.; Jouenne, T.; Hardouin, J. Lysine succinylation and acetylation in Pseudomonas aeruginosa. J. Proteome Res. 2018, 17, 2449–2459. [Google Scholar] [CrossRef]
- Ai, H.; Wu, R.; Zhang, L.; Wu, X.; Ma, J.; Hu, H.; Huang, L.; Chen, W.; Zhao, J.; Liu, H. pSuc-PseRat: Predicting lysine succinylation in proteins by exploiting the ratios of sequence coupling and properties. J. Comput. Biol. 2017, 24, 1050–1059. [Google Scholar] [CrossRef]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. iSuc-PseOpt: Identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal. Biochem. 2016, 497, 48–56. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Gao, T.; Pan, Z.; Cheng, H.; Yang, Q.; Cheng, Z.; Guo, A.; Ren, J.; Xue, Y. CPLM: a database of protein lysine modifications. Nucleic Acids Res. 2014, 42, D531–D536. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, J.; Lin, S.; Deng, W.; Zhang, Y.; Xue, Y. PLMD: An updated data resource of protein lysine modifications. J. Genet. Genomics 2017, 44, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, X.; Xu, X.; Shi, R.; Suo, S.; Cheng, K.; Zheng, Z.; Wang, M.; Wang, L.; Zhao, Y.; et al. Corrigendum: Lysine acetylation and succinylation in hela cells and their essential roles in response to UV-induced stress. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Yang, Q.; Li, P.; Wen, Y.; Li, S.; Chen, J.; Liu, X.; Wang, L.; Li, X. Cadmium inhibits lysine acetylation and succinylation inducing testicular injury of mouse during development. Toxicol. Lett. 2018, 291, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Meyer, J.G.; Schilling, B. quantification of site-specific protein lysine acetylation and succinylation stoichiometry using data-independent acquisition mass spectrometry. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Deng, X.; Wang, J.; Zhu, G.; Cao, H.; Yuan, L.; Yan, Y. First comprehensive proteome analyses of lysine acetylation and succinylation in seedling leaves of Brachypodium distachyon L. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Mollah, M.N.H.; Yong, C.; Guo, D. NTyroSite: Computational identification of protein nitrotyrosine sites using sequence evolutionary features. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Kurata, H. Computational modeling of lysine post-translational modification: An overview. Curr. Synthetic Sys. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Hasan, M.M; Khatun, M.S. Prediction of protein post-translational modification sites: An overview. Ann. Proteom. Bioinform. 2018, 2, 49–57. [Google Scholar] [CrossRef]
- O’Shea, J.P.; Chou, M.F.; Quader, S.A.; Ryan, J.K.; Church, G.M.; Schwartz, D. pLogo: A probabilistic approach to visualizing sequence motifs. Nat. Methods 2013, 10, 1211–1212. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qiu, J.D.; Shi, S.P.; Suo, S.B.; Huang, S.Y.; Liang, R.P. Incorporating key position and amino acid residue features to identify general and species-specific ubiquitin conjugation sites. Bioinformatics 2013, 29, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Wuyun, Q.; Zheng, W.; Zhang, Y.; Ruan, J.; Hu, G. Improved species-specific lysine acetylation site prediction based on a large variety of features set. PLoS ONE 2016, 11, e0155370. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Wang, H.; Tan, H.; Zhang, Z.; Webb, G.I.; Song, J. Accurate in silico identification of species-specific acetylation sites by integrating protein sequence-derived and functional features. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.P.; Shi, S.P.; Xu, H.D.; Wang, L.N.; Qiu, J.D. Accurate in silico prediction of species-specific methylation sites based on information gain feature optimization. Bioinformatics 2016, 32, 3107–3115. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.D.; Lee, T.Y.; Tzeng, S.W.; Horng, J.T. KinasePhos: A web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 2005, 33, W226–W229. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wang, H.; Wang, J.; Leier, A.; Marquez-Lago, T.; Yang, B.; Zhang, Z.; Akutsu, T.; Webb, G.I.; Daly, R.J. PhosphoPredict: A bioinformatics tool for prediction of human kinase-specific phosphorylation substrates and sites by integrating heterogeneous feature selection. Sci. Rep. 2017, 7, 6862. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.N.; Shi, S.P.; Xu, H.D.; Wen, P.P.; Qiu, J.D. Computational prediction of species-specific malonylation sites via enhanced characteristic strategy. Bioinformatics 2017, 33, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.P.; Xu, H.D.; Wen, P.P.; Qiu, J.D. Progress and challenges in predicting protein methylation sites. Mol. Biosyst. 2015, 11, 2610–2619. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Li, F.; Li, C.; Marquez-Lago, T.; Leier, A.; Akutsu, T.; Webb, G.I.; Xu, D.; Smith, A.I.; et al. Large-scale comparative assessment of computational predictors for lysine post-translational modification sites. Brief. Bioinform. 2018. [Google Scholar] [CrossRef]
- Khatun, M.S.; Hasan, M.M.; Mollah, M.N.H.; Kurata, H. SIPMA: A systematic identification of protein-protein interactions in Zea mays using autocorrelation features in a machine-learning framework. In Proceedings of the IEEE 18th International Conference on Bioinformatics and Bioengineering, Taichung, Taiwan, 29–31 October 2018; IEEE: Piscataway, NJ, USA, 2018; pp. 122–125. [Google Scholar]
- Hasan, M.M.; Kurata, H. iLMS, Computational Identification of lysine-malonylation sites by combining multiple sequence features. In Proceedings of the IEEE 18th International Conference on Bioinformatics and Bioengineering, Taichung, Taiwan, 29–31 October 2018; IEEE: Piscataway, NJ, USA, 2018; pp. 356–359. [Google Scholar]
- Siu, M.; Thompson, L.U. Effect of succinylation on the protein quality and urinary excretion of bound and free amino acids. J. Agric. Food Chem. 1982, 30, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhou, Y.; Zhang, Z.; Song, J. Towards more accurate prediction of ubiquitination sites: A comprehensive review of current methods, tools and features. Brief. Bioinform. 2015, 16, 640–657. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M; Khatun, M.S.; Kurata, H. A comprehensive review of in silico analysis for protein S-sulfenylation sites. Protein Pept. Lett. 2018, 25, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Guo, D.; Kurata, H. Computational identification of protein S-sulfenylation sites by incorporating the multiple sequence features information. Mol. Biosyst. 2017, 13, 2545–2550. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Zhou, Y.; Lu, X.; Li, J.; Song, J.; Zhang, Z. Computational Identification of protein pupylation sites by using profile-based composition of k-spaced amino acid pairs. PLoS ONE 2015, 10, e0129635. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.T. Recent progress and challenges for protein pupylation sites prediction. EC Proteomics Bioinform. 2017, 2, 36–45. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Li, W.; Fu, L.; Niu, B.; Wu, S.; Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinform. 2012, 13, 656–668. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Radi, R. Protein tyrosine nitration: Biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tools | SucPred | iSuc-PseAAC | SuccFind | iSuc-PseOpt | pSuc-Lys | SucStruct | PSSM-Suc | SuccinSite | SuccinSite2.0 | SSEvol-Suc | Success | GPSuc |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | Generic | Generic | Generic | Generic | Generic | Generic | Generic | Generic | Generic and Species-specific | Generic | Generic | Generic and Species-specific |
| Web-server link | http://59.73.198.144:8088/SucPred/ | http://app.aporc.org/iSuc-PseAAC/ | http://bioinfo.ncu.edu.cn/SuccFind.aspx | http://www.jci-bioinfo.cn/iSuc-PseOpt | http://www.jci-bioinfo.cn/pSuc-Lys | https://github.com/YosvanyLopez/ | https://github.com/YosvanyLopez/PSSM-Suc | http://systbio.cau.edu.cn/SuccinSite/ | https://biocomputer.bio.cuhk.edu.hk/SuccinSite2.0/ | https://github.com/YosvanyLopez/SSEvol-Suc | https://github.com/YosvanyLopez/Success | http://kurata14.bio.kyutech.ac.jp/GPSuc/ |
| Working server | No | Yes | No | No | No | No | No | Yes | Yes | No | No | Yes |
| Machine learning | SVM | SVM | SVM | RF | RF | DT | DT | RF | RF | AdaBoost | SVM | RF and LR |
| Dataset size (Protein/succinylated) | 897/2511 | 896/2521 | 1044/2938 | 896/2521 | 896/2521 | 670/1782 | 670 / 1782 | 2322/5004 | 2322/5004 | 670/1782 | 670/1782 | 2322/5004 |
| Training (Pos/Neg) | 1436/18,958 | 1167/3553 | 2713/23598 | 1167/3553 | 1167/3553 | 1782/1872 | 1782/1643 | 4750/9500 | 4750/9500 | 1782/1872 | 1782/1872 | 4750/9500 |
| Independent (Pos/Neg) | 250/- | - | - | - | - | - | - | 254/2977 | 254/2977 | - | - | 254/2977 |
| Homolog redundancy | 35% | 40% | 30% | 40% | 40% | 40% | 40% | 30% | 30% | 40% | 40% | 30% |
| Window size | from −9 to +9 | from −7 to +7 | from −10 to +10 | from −15 to +15 | from −15 to +15 | from −15 to +15 | from −15 to +15 | from −13 to +13 | from −20 to +20 | from −15 to +15 | from −15 to +15 | from −20 to +20 |
| Adjusted batch prediction | NO | No | No | No | No | No | No | Yes | Yes | No | No | Yes |
| Processing time for a protein | - | Within 20 s | - | - | - | - | - | Within 20 s | Within 5 min | - | - | Within 5 min |
| Species | Datasets | Positive Samples | Negative Samples |
|---|---|---|---|
| H. sapiens | Training | 1351 | 2702 |
| Independent | 54 | 2004 | |
| M. musculus | Training | 414 | 828 |
| Independent | 24 | 679 | |
| E. coli | Training | 1942 | 3884 |
| Independent | 289 | 1381 | |
| M. tuberculosis | Training | 699 | 1398 |
| Independent | 61 | 242 | |
| S. cerevisiae | Training | 961 | 1922 |
| Independent | 90 | 1423 | |
| T. gondii | Training | 282 | 564 |
| Independent | 26 | 261 | |
| S. lycopersicum | Training | 242 | 484 |
| Independent | 33 | 274 | |
| A. capsulatus | Training | 332 | 664 |
| Independent | 50 | 591 | |
| T. aestivum | Training | 113 | 226 |
| Independent | 32 | 309 |
| Encoding Types | Genetic Explanation | References |
|---|---|---|
| AAindex | Based on the AAindex indices database, the encoding scheme of AAindex reveals the biochemical properties of the sequences. | [56,59,62] |
| ACF | The auto correlation function features for surrounding succinylation sequences. | [54] |
| EBGW | Coding based on grouped weight of physicochemical properties of sequences surrounding succinylation sites. | [54] |
| VDWV | Van der Waals volume properties of surrounding succinylation sequences. | [54] |
| WAAC | Position weight amino acid composition of surrounding succinylation sequences. | [54] |
| AAC | The amino acid composition characterizes the specific state of the surrounding succinylation sequences. | [65] |
| CKSAAP | The CKSAAP encoding represents the short sequence motif information in surrounding succinylation sites. | [56,59] |
| PseAAC | The pseudo amino acid composition reflects a vectorized sequence-coupling model of surrounding succinylation sites. | [56,61,70] |
| SF | The predicted structural feature reflects the structural properties of protein in surrounding succinylation sites. | [66] |
| Binary | The position-specific information measured by binary profile for the curated sequences. | [59,62,65] |
| PSSM | The PSSM exposes the evolutionary information from the sequences. | [57] |
| pCKSAAP | The pCKSAAP reflects the sequence patterns and evolutionary information from the query sequences. | [62,65] |
| Methods | Training | Independent | |||
|---|---|---|---|---|---|
| H. sapiens | RF | SVM | RF | SVM | |
| pCKSAAP | 0.856 | 0.838 | 0.695 | 0.691 | |
| CKSAAP | 0.816 | 0.831 | 0.677 | 0.663 | |
| AAindex | 0.739 | 0.728 | 0.759 | 0.755 | |
| Binary | 0.767 | 0.754 | 0.822 | 0.809 | |
| PseAAC | 0.819 | 0.822 | 0.658 | 0.649 | |
| H. capsulatum | pCKSAAP | 0.789 | 0.792 | 0.638 | 0.634 |
| CKSAAP | 0.788 | 0.783 | 0.619 | 0.607 | |
| AAindex | 0.712 | 0.722 | 0.658 | 0.666 | |
| Binary | 0.713 | 0.698 | 0.665 | 0.647 | |
| PseAAC | 0.759 | 0.743 | 0.612 | 0.614 | |
| M. musculus | pCKSAAP | 0.801 | 0.788 | 0.637 | 0.634 |
| CKSAAP | 0.777 | 0.767 | 0.646 | 0.651 | |
| AAindex | 0.648 | 0.655 | 0.679 | 0.672 | |
| Binary | 0.639 | 0.641 | 0.677 | 0.659 | |
| PseAAC | 0.711 | 0.722 | 0.609 | 0.611 | |
| E. coli | pCKSAAP | 0.769 | 0.761 | 0.679 | 0.684 |
| CKSAAP | 0.773 | 0.782 | 0.646 | 0.631 | |
| AAindex | 0.719 | 0.721 | 0.633 | 0.619 | |
| Binary | 0.689 | 0.674 | 0.619 | 0.607 | |
| PseAAC | 0.733 | 0.734 | 0.608 | 0.603 | |
| M. tuberculosis | pCKSAAP | 0.708 | 0.712 | 0.688 | 0.679 |
| CKSAAP | 0.689 | 0.675 | 0.664 | 0.671 | |
| AAindex | 0.667 | 0.658 | 0.656 | 0.655 | |
| Binary | 0.629 | 0.617 | 0.639 | 0.634 | |
| PseAAC | 0.643 | 0.634 | 0.629 | 0.617 | |
| S. cerevisiae | pCKSAAP | 0.882 | 0.869 | 0.776 | 0.772 |
| CKSAAP | 0.879 | 0.863 | 0.752 | 0.744 | |
| AAindex | 0.742 | 0.733 | 0.759 | 0.749 | |
| Binary | 0.741 | 0.745 | 0.798 | 0.787 | |
| PseAAC | 0.790 | 0.768 | 0.699 | 0.675 | |
| T. gondii | pCKSAAP | 0.834 | 0.836 | 0.657 | 0.666 |
| CKSAAP | 0.826 | 0.822 | 0.655 | 0.638 | |
| AAindex | 0.726 | 718 | 0.663 | 0.647 | |
| Binary | 0.744 | 0.745 | 0.679 | 0.671 | |
| PseAAC | 0.801 | 0.788 | 0.678 | 0.664 | |
| S. lycopersicum | pCKSAAP | 0.842 | 0.836 | 0.649 | 0.642 |
| CKSAAP | 0.833 | 0.824 | 0.648 | 0.637 | |
| AAindex | 0.753 | 0.765 | 0.644 | 0.629 | |
| Binary | 0.729 | 0.722 | 0.637 | 0.631 | |
| PseAAC | 0.801 | 0.783 | 0.678 | 0.658 | |
| T. aestivum | pCKSAAP | 0.822 | 0.826 | 0.649 | 0.654 |
| CKSAAP | 0.821 | 0.811 | 0.638 | 0.634 | |
| AAindex | 0.736 | 0.734 | 0.604 | 0.611 | |
| Binary | 0.726 | 0.719 | 0.612 | 0.596 | |
| PseAAC | 0.778 | 0.769 | 0.632 | 0.628 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, M.M.; Khatun, M.S.; Kurata, H. Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites. Cells 2019, 8, 95. https://doi.org/10.3390/cells8020095
Hasan MM, Khatun MS, Kurata H. Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites. Cells. 2019; 8(2):95. https://doi.org/10.3390/cells8020095
Chicago/Turabian StyleHasan, Md. Mehedi, Mst. Shamima Khatun, and Hiroyuki Kurata. 2019. "Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites" Cells 8, no. 2: 95. https://doi.org/10.3390/cells8020095
APA StyleHasan, M. M., Khatun, M. S., & Kurata, H. (2019). Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites. Cells, 8(2), 95. https://doi.org/10.3390/cells8020095