Biochemical Differences in Cerebrospinal Fluid between Secondary Progressive and Relapsing–Remitting Multiple Sclerosis


Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Subjects
2.3. Sample Collection
2.4. Metabolite Extraction
2.5. Mass Spectrometry Analysis
2.6. Quantification
2.7. Metabolite Identification
2.8. Statistical Analysis
3. Results
3.1. Participant Demographics
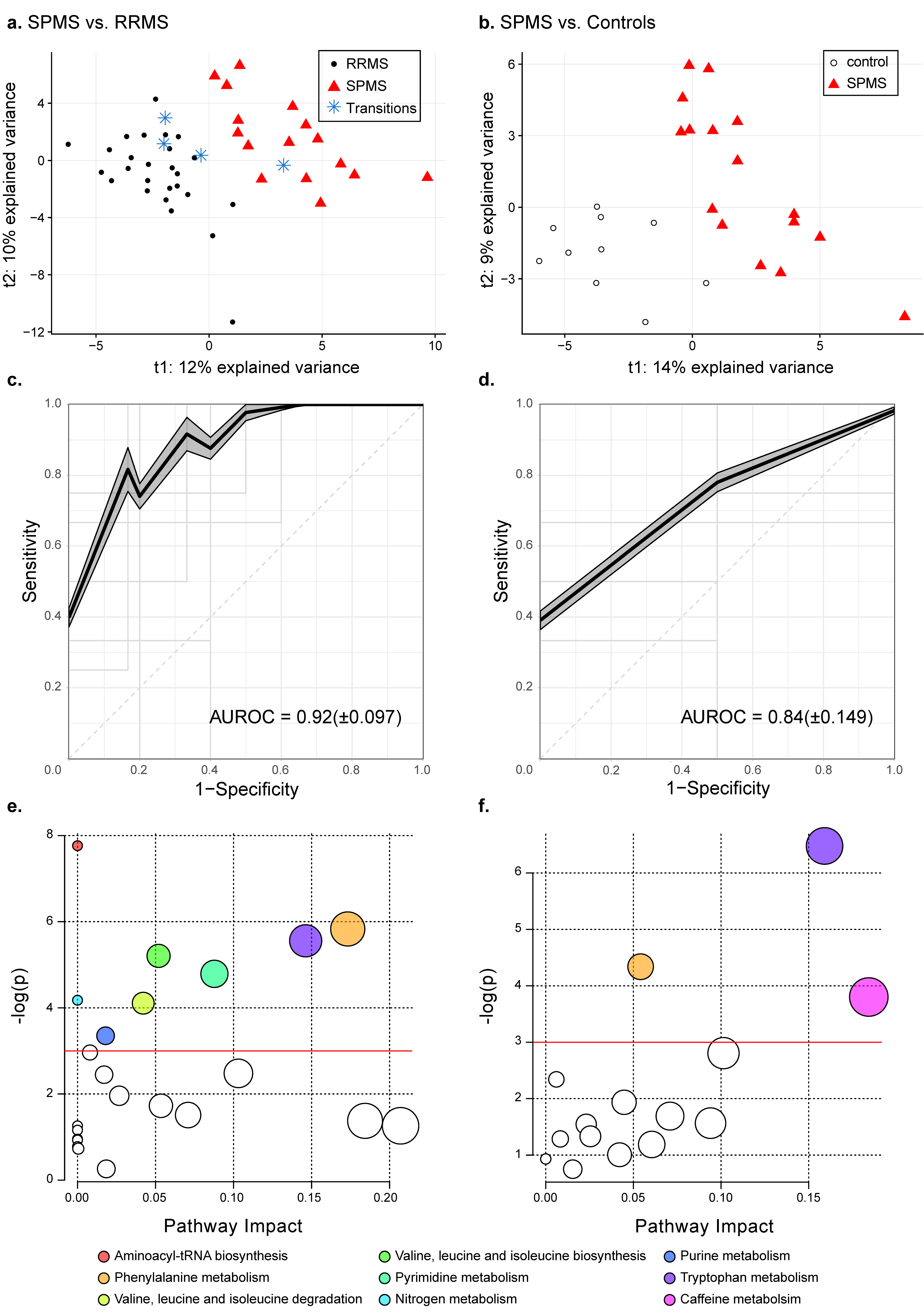
3.2. The CSF Metabolome Could Distinguish SPMS Patients from RRMS and Controls
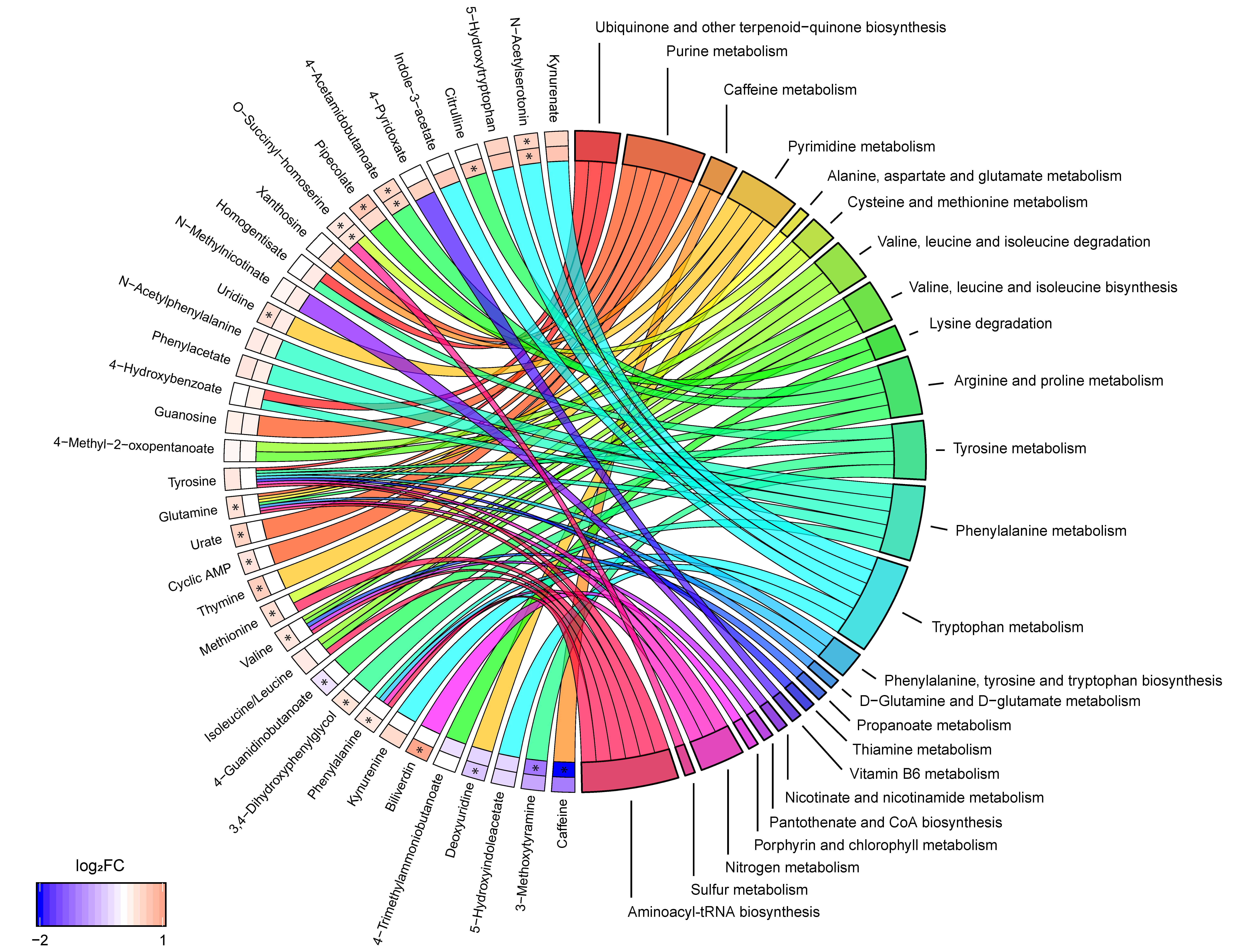
3.3. Phenylalanine and Tryptophan Metabolisms Were Altered in SPMS Compared with RRMS Patients
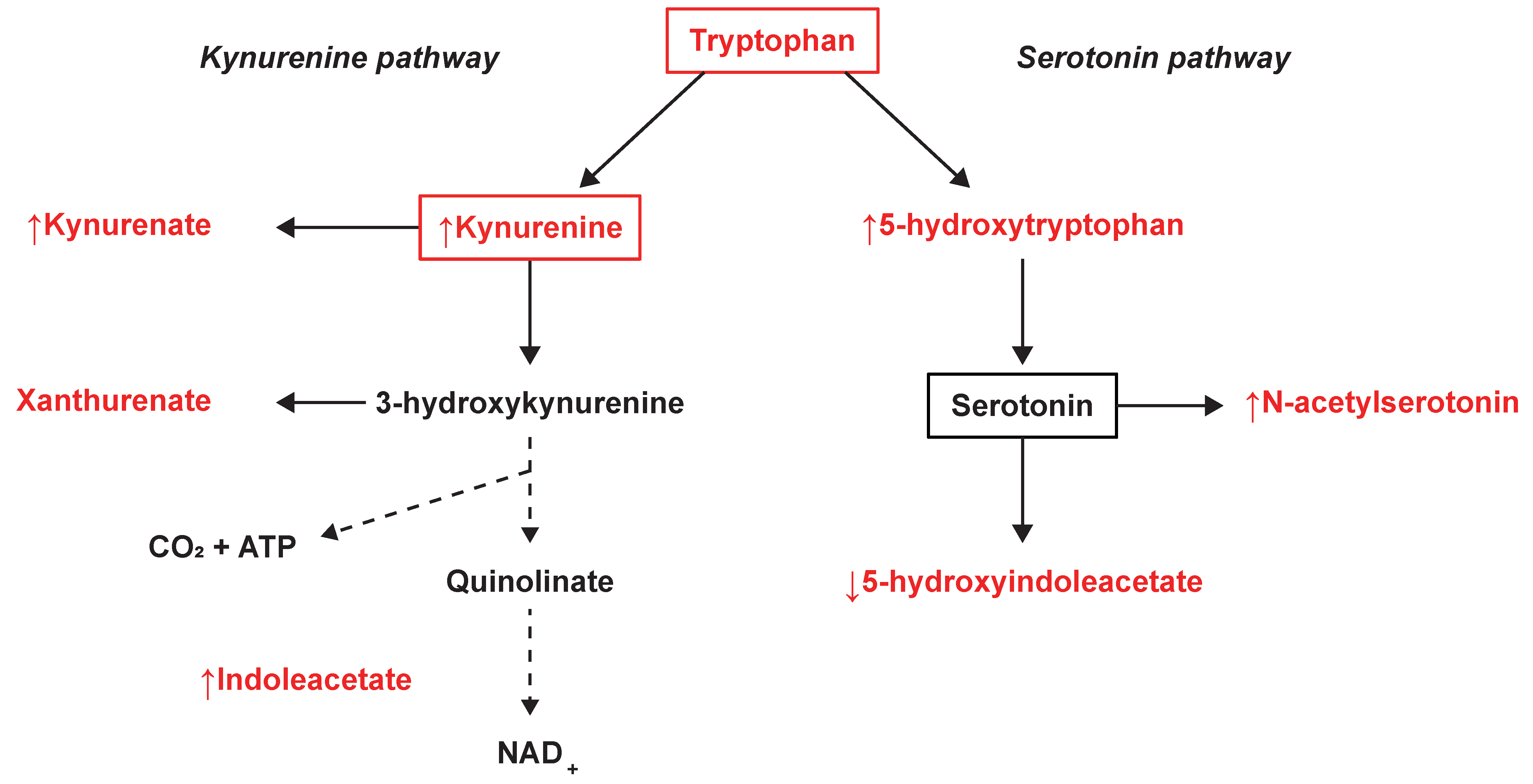
3.4. Tryptophan Metabolism Were Altered in SPMS Compared with Controls
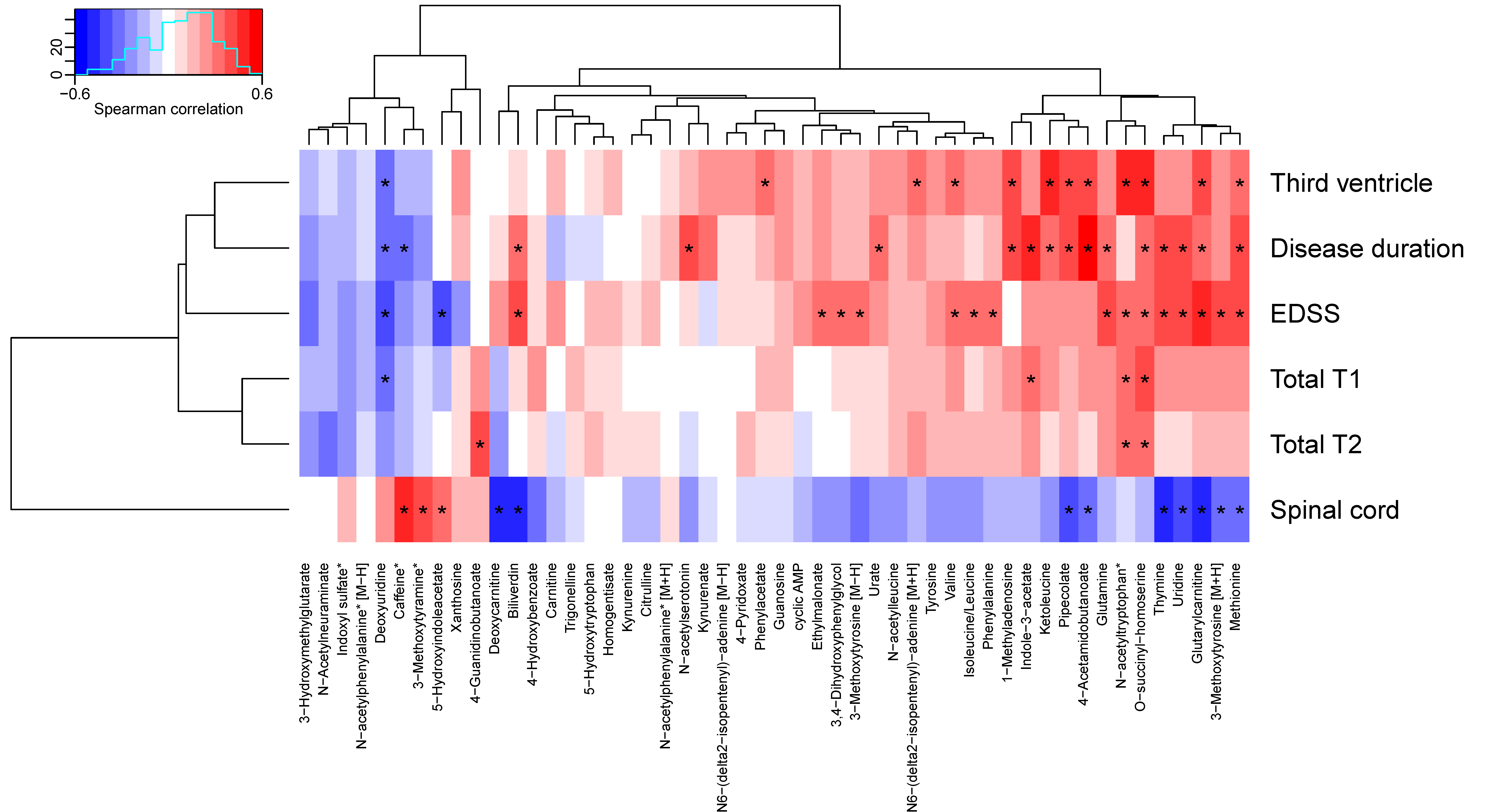
3.5. Metabolites Linked to Pyrimidine and Tryptophan Metabolisms Were Associated with Clinical Measurements in MS Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of Multiple Sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Weinshenker, B.G. Disease Modifying Therapies for Relapsing Multiple Sclerosis. BMJ 2016, 354, 3518. [Google Scholar] [CrossRef] [PubMed]
- Ontaneda, D.; Fox, R.J.; Chataway, J. Clinical Trials in Progressive Multiple Sclerosis: Lessons Learned and Future Perspectives. Lancet Neurol. 2015, 14, 208–223. [Google Scholar] [CrossRef]
- Mulero, P.; Midaglia, L.; Montalban, X. Ocrelizumab: A New Milestone in Multiple Sclerosis Therapy. Ther. Adv. Neurol. Disord. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Calabresi, P.A. Metabolomics in Multiple Sclerosis. Mult. Scler. 2016, 22, 451–460. [Google Scholar] [CrossRef]
- Stoessel, D.; Stellmann, J.-P.; Willing, A.; Behrens, B.; Rosenkranz, S.C.; Hodecker, S.C.; Stürner, K.H.; Reinhardt, S.; Fleischer, S.; Deuschle, C.; et al. Metabolomic Profiles for Primary Progressive Multiple Sclerosis Stratification and Disease Course Monitoring. Front. Hum. Neurosci. 2018, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Jurynczyk, M.; Probert, F.; Yeo, T.; Tackley, G.; Claridge, T.D.W.; Cavey, A.; Woodhall, M.R.; Arora, S.; Winkler, T.; Schiffer, E.; et al. Metabolomics Reveals Distinct, Antibody-Independent, Molecular Signatures of MS, AQP4-Antibody and MOG-Antibody Disease. Acta Neuropathol. Commun. 2017, 5, 95. [Google Scholar] [CrossRef]
- Bhargava, P.; Fitzgerald, K.C.; Calabresi, P.A.; Mowry, E.M. Metabolic Alterations in Multiple Sclerosis and the Impact of Vitamin D Supplementation. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Poddighe, S.; Murgia, F.; Lorefice, L.; Liggi, S.; Cocco, E.; Marrosu, M.G.; Atzori, L. Metabolomic Analysis Identifies Altered Metabolic Pathways in Multiple Sclerosis. Int. J. Biochem. Cell Biol. 2017, 93, 148–155. [Google Scholar] [CrossRef]
- Villoslada, P.; Alonso, C.; Agirrezabal, I.; Kotelnikova, E.; Zubizarreta, I.; Pulido-Valdeolivas, I.; Saiz, A.; Comabella, M.; Montalban, X.; Villar, L.; et al. Metabolomic Signatures Associated with Disease Severity in Multiple Sclerosis. Neurol Neuroimmunol Neuroinflamm 2017, 4, e321. [Google Scholar] [CrossRef]
- Lim, C.K.; Bilgin, A.; Lovejoy, D.B.; Tan, V.; Bustamante, S.; Taylor, B.V.; Bessede, A.; Brew, B.J.; Guillemin, G.J. Kynurenine Pathway Metabolomics Predicts and Provides Mechanistic Insight into Multiple Sclerosis Progression. Sci. Rep. 2017, 7, 41473. [Google Scholar] [CrossRef] [PubMed]
- Lutz, N.W.; Cozzone, P.J. Metabolic Profiling in Multiple Sclerosis and Other Disorders by Quantitative Analysis of Cerebrospinal Fluid Using Nuclear Magnetic Resonance Spectroscopy. Curr. Pharm. Biotechnol. 2011, 12, 1016–1025. [Google Scholar] [CrossRef]
- Sinclair, A.J.; Viant, M.R.; Ball, A.K.; Burdon, M.A.; Walker, E.A.; Stewart, P.M.; Rauz, S.; Young, S.P. NMR-Based Metabolomic Analysis of Cerebrospinal Fluid and Serum in Neurological Diseases--a Diagnostic Tool? NMR Biomed. 2010, 23, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Lutz, N.W.; Viola, A.; Malikova, I.; Confort-Gouny, S.; Audoin, B.; Ranjeva, J.-P.; Pelletier, J.; Cozzone, P.J. Inflammatory Multiple-Sclerosis Plaques Generate Characteristic Metabolic Profiles in Cerebrospinal Fluid. PLoS ONE 2007, 2, e595. [Google Scholar] [CrossRef] [PubMed]
- Reinke, S.N.; Broadhurst, D.L.; Sykes, B.D.; Baker, G.B.; Catz, I.; Warren, K.G.; Power, C. Metabolomic Profiling in Multiple Sclerosis: Insights into Biomarkers and Pathogenesis. Mult. Scler. 2014, 20, 1396–1400. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Murgia, F.; Lorefice, L.; Barberini, L.; Poddighe, S.; Frau, J.; Fenu, G.; Coghe, G.; Murru, M.R.; Murru, R.; et al. (1)H-NMR Analysis Provides a Metabolomic Profile of Patients with Multiple Sclerosis. Neurol Neuroimmunol. Neuroinflamm 2016, 3, e185. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-H.; Jeong, I.H.; Hyun, J.-S.; Kong, B.S.; Kim, H.J.; Park, S.J. Metabolomic Profiling of CSF in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorder by Nuclear Magnetic Resonance. PLoS ONE 2017, 12, e0181758. [Google Scholar] [CrossRef]
- Herman, S.; Khoonsari, P.E.; Tolf, A.; Steinmetz, J.; Zetterberg, H.; Åkerfeldt, T.; Jakobsson, P.-J.; Larsson, A.; Spjuth, O.; Burman, J.; et al. Integration of Magnetic Resonance Imaging and Protein and Metabolite CSF Measurements to Enable Early Diagnosis of Secondary Progressive Multiple Sclerosis. Theranostics 2018, 8, 4477–4490. [Google Scholar] [CrossRef]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic Criteria for Multiple Sclerosis: 2010 Revisions to the McDonald Criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef]
- Burman, J.; Zetterberg, H.; Fransson, M.; Loskog, A.S.; Raininko, R.; Fagius, J. Assessing Tissue Damage in Multiple Sclerosis: A Biomarker Approach. Acta Neurol. Scand. 2014, 130, 81–89. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Petzold, A.; Bennett, J.L.; Berven, F.S.; Brundin, L.; Comabella, M.; Franciotta, D.; Frederiksen, J.L.; Fleming, J.O.; Furlan, R.; et al. A Consensus Protocol for the Standardization of Cerebrospinal Fluid Collection and Biobanking. Neurology 2009, 73, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Studies in Classification, Data Analysis, and Knowledge Organization; Springer: Berlin/Heidelberg, Germany, 2008; pp. 319–326. [Google Scholar]
- Kenar, E.; Franken, H.; Forcisi, S.; Wörmann, K.; Häring, H.-U.; Lehmann, R.; Schmitt-Kopplin, P.; Zell, A.; Kohlbacher, O. Automated Label-Free Quantification of Metabolites from Liquid Chromatography-Mass Spectrometry Data. Mol. Cell. Proteomics 2014, 13, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Weisser, H.; Nahnsen, S.; Grossmann, J.; Nilse, L.; Quandt, A.; Brauer, H.; Sturm, M.; Kenar, E.; Kohlbacher, O.; Aebersold, R.; et al. An Automated Pipeline for High-Throughput Label-Free Quantitative Proteomics. J. Proteome Res. 2013, 12, 1628–1644. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: http://www.R-project.org (accessed on 12 December 2018).
- Herman, S.; Emami Khoonsari, P.; Aftab, O.; Krishnan, S.; Strömbom, E.; Larsson, R.; Hammerling, U.; Spjuth, O.; Kultima, K.; Gustafsson, M. Mass Spectrometry Based Metabolomics for in Vitro Systems Pharmacology: Pitfalls, Challenges, and Computational Solutions. Metabolomics 2017, 13, 79. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Falahati, F.; Ferreira, D.; Soininen, H.; Mecocci, P.; Vellas, B.; Tsolaki, M.; Kłoszewska, I.; Lovestone, S.; Eriksdotter, M.; Wahlund, L.-O.; et al. The Effect of Age Correction on Multivariate Classification in Alzheimer’s Disease, with a Focus on the Characteristics of Incorrectly and Correctly Classified Subjects. Brain Topogr. 2016, 29, 296–307. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An Open-Source Package for R and S to Analyze and Compare ROC Curves. BMC Bioinformatics 2011, 12, 77. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making Metabolomics More Meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Rajda, C.; Majláth, Z.; Pukoli, D.; Vécsei, L. Kynurenines and Multiple Sclerosis: The Dialogue between the Immune System and the Central Nervous System. Int. J. Mol. Sci. 2015, 16, 18270–18282. [Google Scholar] [CrossRef] [PubMed]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent Advances and New Questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Nourbakhsh, B.; Bhargava, P.; Tremlett, H.; Hart, J.; Graves, J.; Waubant, E. Altered Tryptophan Metabolism Is Associated with Pediatric Multiple Sclerosis Risk and Course. Ann. Clin. Transl. Neurol. 2018, 5, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine Metabolism in Multiple Sclerosis. Acta Neurol. Scand. 2005, 112, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Rejdak, K.; Bartosik-Psujek, H.; Dobosz, B.; Kocki, T.; Grieb, P.; Giovannoni, G.; Turski, W.A.; Stelmasiak, Z. Decreased Level of Kynurenic Acid in Cerebrospinal Fluid of Relapsing-Onset Multiple Sclerosis Patients. Neurosci. Lett. 2002, 331, 63–65. [Google Scholar] [CrossRef]
- Rejdak, K.; Petzold, A.; Kocki, T.; Kurzepa, J.; Grieb, P.; Turski, W.A.; Stelmasiak, Z. Astrocytic Activation in Relation to Inflammatory Markers during Clinical Exacerbation of Relapsing-Remitting Multiple Sclerosis. J. Neural Transm. 2007, 114, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Markianos, M.; Koutsis, G.; Evangelopoulos, M.-E.; Mandellos, D.; Karahalios, G.; Sfagos, C. Relationship of CSF Neurotransmitter Metabolite Levels to Disease Severity and Disability in Multiple Sclerosis. J. Neurochem. 2009, 108, 158–164. [Google Scholar] [CrossRef]
- Hesse, S.; Moeller, F.; Petroff, D.; Lobsien, D.; Luthardt, J.; Regenthal, R.; Becker, G.-A.; Patt, M.; Thomae, E.; Seese, A.; et al. Altered Serotonin Transporter Availability in Patients with Multiple Sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 827–835. [Google Scholar] [CrossRef]
- Malinova, T.S.; Dijkstra, C.D.; de Vries, H.E. Serotonin: A Mediator of the Gut-Brain Axis in Multiple Sclerosis. Mult. Scler. 2018, 24, 1144–1150. [Google Scholar] [CrossRef]
- Vesterinen, H.M.; Connick, P.; Irvine, C.M.J.; Sena, E.S.; Egan, K.J.; Carmichael, G.G.; Tariq, A.; Pavitt, S.; Chataway, J.; Macleod, M.R.; et al. Drug Repurposing: A Systematic Approach to Evaluate Candidate Oral Neuroprotective Interventions for Secondary Progressive Multiple Sclerosis. PLoS ONE 2015, 10, e0117705. [Google Scholar] [CrossRef]
- Chataway, J.; Chandran, S.; Miller, D.; Connick, P.; Giovannoni, G.; Pavitt, S.; Stallard, N.; Hawkins, C.; Sharrack, B.; Plantone, D. The Ms-Smart Trial In Secondary Progressive Ms—Current Update. J. Neurol. Neurosurg. Psychiatry 2016, 87, e1. [Google Scholar] [CrossRef]
- Arreola, R.; Becerril-Villanueva, E.; Cruz-Fuentes, C.; Velasco-Velázquez, M.A.; Garcés-Alvarez, M.E.; Hurtado-Alvarado, G.; Quintero-Fabian, S.; Pavón, L. Immunomodulatory Effects Mediated by Serotonin. J. Immunol. Res. 2015, 2015, 354957. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s Metabolites in Exercise, Inflammation, and Mental Health. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, E.L.; Tam, R.; Zhao, Y.; Vavasour, I.M.; Li, D.K.B.; Oger, J.; Freedman, M.S.; Kolind, S.H.; Traboulsee, A.L. Progressive Multiple Sclerosis Exhibits Decreasing Glutamate and Glutamine over Two Years. Mult. Scler. 2016, 22, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Fitzgerald, K.C.; Venkata, S.L.V.; Smith, M.D.; Kornberg, M.D.; Mowry, E.M.; Haughey, N.J.; Calabresi, P.A. Dimethyl Fumarate Treatment Induces Lipid Metabolism Alterations That Are Linked to Immunological Changes. Ann. Clin. Transl. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Vincenzetti, S.; Polzonetti, V.; Micozzi, D.; Pucciarelli, S. Enzymology of Pyrimidine Metabolism and Neurodegeneration. Curr. Med. Chem. 2016, 23, 1408–1431. [Google Scholar] [CrossRef]
- Kori, M.; Aydın, B.; Unal, S.; Arga, K.Y.; Kazan, D. Metabolic Biomarkers and Neurodegeneration: A Pathway Enrichment Analysis of Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. OMICS 2016, 20, 645–661. [Google Scholar] [CrossRef]
- Bar-Or, A.; Pachner, A.; Menguy-Vacheron, F.; Kaplan, J.; Wiendl, H. Teriflunomide and Its Mechanism of Action in Multiple Sclerosis. Drugs 2014, 74, 659–674. [Google Scholar] [CrossRef]
- Kasarełło, K.; Cudnoch-Jędrzejewska, A.; Członkowski, A.; Mirowska-Guzel, D. Mechanism of Action of Three Newly Registered Drugs for Multiple Sclerosis Treatment. Pharmacol. Rep. 2017, 69, 702–708. [Google Scholar] [CrossRef]
- Huynh, J.L.; Garg, P.; Thin, T.H.; Yoo, S.; Dutta, R.; Trapp, B.D.; Haroutunian, V.; Zhu, J.; Donovan, M.J.; Sharp, A.J.; et al. Epigenome-Wide Differences in Pathology-Free Regions of Multiple Sclerosis-Affected Brains. Nat. Neurosci. 2014, 17, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kular, L.; Liu, Y.; Ruhrmann, S.; Zheleznyakova, G.; Marabita, F.; Gomez-Cabrero, D.; James, T.; Ewing, E.; Lindén, M.; Górnikiewicz, B.; et al. DNA Methylation as a Mediator of HLA-DRB1*15:01 and a Protective Variant in Multiple Sclerosis. Nat. Commun. 2018, 9, 2397. [Google Scholar] [CrossRef] [PubMed]
- Zheleznyakova, G.Y.; Piket, E.; Marabita, F.; Pahlevan Kakhki, M.; Ewing, E.; Ruhrmann, S.; Needhamsen, M.; Jagodic, M.; Kular, L. Epigenetic Research in Multiple Sclerosis: Progress, Challenges, and Opportunities. Physiol. Genomics 2017, 49, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.K.; Freeman, E.; Arning, E.; Wasek, B.; Clements, R.; Sheppard, C.; Blake, P.; Bottiglieri, T.; McDonough, J. Dysregulation of Methionine Metabolism in Multiple Sclerosis. Neurochem. Int. 2018, 112, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.K.; Li, S.; Arning, E.; Alkhayer, K.; Clements, R.; Sarcyk, Z.; Dassanayake, R.S.; Brasch, N.E.; Freeman, E.J.; Bottiglieri, T.; et al. Changes in Methionine Metabolism and Histone H3 Trimethylation Are Linked to Mitochondrial Defects in Multiple Sclerosis. J. Neurosci. 2015, 35, 15170–15186. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Kastenmüller, G.; Petersen, A.K.; Bell, J.T.; Psatha, M.; Tsai, P.-C.; Gieger, C.; Schulz, H.; Erte, I.; John, S.; et al. Metabolomic Markers Reveal Novel Pathways of Ageing and Early Development in Human Populations. Int. J. Epidemiol. 2013, 42, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Van der Goot, A.T.; Nollen, E.A.A. Tryptophan Metabolism: Entering the Field of Aging and Age-Related Pathologies. Trends Mol. Med. 2013, 19, 336–344. [Google Scholar] [CrossRef]
- García-Campos, M.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Pathway Analysis: State of the Art. Front. Physiol. 2015, 6, 383. [Google Scholar] [CrossRef]
- Creek, D.J.; Dunn, W.B.; Fiehn, O.; Griffin, J.L.; Hall, R.D.; Lei, Z.; Mistrik, R.; Neumann, S.; Schymanski, E.L.; Sumner, L.W.; et al. Metabolite Identification: Are You Sure? And How Do Your Peers Gauge Your Confidence? Metabolomics 2014, 10, 350–353. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | Controls | RRMS | SPMS |
| n | 10 | 30 | 16 |
| On treatment, n | 0 | 15 | 1 |
| Age *, mean (±SD) | 39 (±13.1) | 39 (±10.6) | 58 (±9.3) |
| Female/Male | 6/4 | 21/9 | 10/6 |
| EDSS *, median(range) | n/a | 2.0 (0–7.5) | 5.5 (3.0–7.5) |
| Disease duration *, median (range) | n/a | 92 (0.5–364) | 283 (109–538) |
| Follow up | RRMS | SPMS | |
| n | 27 | 13 | |
| ΔEDSS, median (range) | 0.0 (−3.5–3.0) | 1.5 (0–4.0) | |
| Time interval in months, mean (±SD) | 67.6 (±15.4) | 54.8 (±18.6) | |
| Transitioned, n | 4 | n/a | |
| Deceased, n | 1 | 0 |
| Metabolite | KEGG | VIP Mean (95% CI) | log2 FC SP-RR | p-Value | FDR | CV | Validation Level |
|---|---|---|---|---|---|---|---|
| Thymine | C00178 | 2.01 (1.95, 2.07) | 0.49 | 5.0 × 10−5 | 1.8 × 10-3 | 7.9% | 1 |
| Glutarylcarnitine | - | 1.84 (1.79, 1.90) | 0.48 | 2.4 × 10−4 | 4.4 × 10-3 | 9.3% | 2 |
| Biliverdin | C00500 | 1.78 (1.72, 1.83) | 0.90 | 1.6 × 10−3 | 0.011 | 24.7% | 1 |
| Pipecolate | C00408 | 1.77 (1.70, 1.83) | 0.56 | 1.9 × 10−3 | 0.011 | 7.5% | 1 |
| Uridine | C00299 | 1.76 (1.70, 1.82) | 0.33 | 9.4 × 10−4 | 0.011 | 7.4% | 2 |
| 4-Acetamidobutanoate | C02946 | 1.72 (1.67, 1.78) | 0.40 | 2.1 × 10−3 | 0.011 | 9.2% | 2 |
| Deoxyuridine | C00526 | 1.67 (1.62, 1.72) | −0.50 | 1.4 × 10−3 | 0.011 | 13.8% | 1 |
| Ethylmalonate | - | 1.63 (1.56, 1.70) | 0.46 | 9.7 × 10−3 | 0.030 | 7.1% | 2 |
| Valine | C00183 | 1.61 (1.56, 1.66) | 0.25 | 4.2 × 10−3 | 0.020 | 4.5% | 2 |
| O-Succinyl-homoserine | C01118 | 1.57 (1.52, 1.62) | 0.24 | 6.4 × 10−3 | 0.021 | 14.3% | 1 |
| Methionine | C00073 | 1.56 (1.51, 1.62) | 0.32 | 6.0 × 10−3 | 0.021 | 3.4% | 2 |
| Glutamine | C00064 | 1.51 (1.46, 1.56) | 0.31 | 6.3 × 10−3 | 0.021 | 22.3% | 2 |
| 3-Methoxytyrosine [M + H] | - | 1.38 (1.31, 1.45) | 0.80 | 0.075 | 0.088 | 6.8% | 2 |
| Phenylacetate | C07086 | 1.38 (1.32, 1.43) | 0.26 | 0.031 | 0.052 | 8.7% | 2 |
| N-Acetylleucine | C02710 | 1.37 (1.33, 1.42) | 0.24 | 0.016 | 0.038 | 5.8% | 1 |
| Phenylalanine | C00079 | 1.35 (1.30, 1.39) | 0.23 | 0.021 | 0.043 | 4.6% | 2 |
| 1-Methyladenosine | C02494 | 1.34 (1.28, 1.39) | 0.26 | 0.017 | 0.040 | 11.5% | 2 |
| Urate | C00366 | 1.25 (1.20, 1.31) | 0.44 | 0.013 | 0.037 | 8.6% | 2 |
| Caffeine * | C07481 | 1.25 (1.18, 1.32) | −1.12 | 0.078 | 0.088 | 24.4% | 2 |
| Ketoleucine | C00233 | 1.25 (1.19, 1.31) | 0.09 | 0.034 | 0.052 | 7.6% | 2 |
| Tyrosine | C00082 | 1.25 (1.21, 1.29) | 0.25 | 0.034 | 0.052 | 5.2% | 2 |
| N6-(delta2-isopentenyl)-adenine | C04083 | 1.23 (1.17, 1.29) | 0.29 | 0.028 | 0.049 | 5.4% | 1 |
| N-Acetylphenylalanine * [M + H] | C03519 | 1.23 (1.18, 1.27) | 0.21 | 0.044 | 0.061 | 6.9% | 1 |
| 3-Methoxytyramine * | C05587 | 1.20 (1.14, 1.26) | −0.75 | 0.038 | 0.054 | 26.7% | 1 |
| Cyclic AMP | C00575 | 1.20 (1.10, 1.31) | 0.28 | 0.020 | 0.043 | 12.0% | 1 |
| N-Acetylserotonin | C00978 | 1.20 (1.15, 1.25) | 0.44 | 0.023 | 0.045 | 16.8% | 1 |
| 3,4-Dihydroxyphenylglycol | C05576 | 1.18 (1.13, 1.24) | 0.28 | 0.015 | 0.038 | 15.9% | 1 |
| Guanosine | C00387 | 1.17 (1.10, 1.25) | 0.16 | 0.035 | 0.052 | 7.8% | 2 |
| Kynurenine | C00328 | 1.12 (1.06, 1.18) | 0.37 | 0.048 | 0.063 | 7.9% | 2 |
| Isoleucine/Leucine | C00407 | 1.10 (1.05, 1.15) | 0.21 | 0.078 | 0.088 | 6.5% | 2 |
| Kynurenate | C01717 | 1.09 (1.02, 1.16) | 0.43 | 0.050 | 0.064 | 9.1% | 1 |
| 5-Hydroxytryptophan | C00643 | 1.09 (1.02, 1.15) | 0.45 | 0.055 | 0.068 | 15.8% | 1 |
| 3-Methoxytyrosine [M − H] | - | 1.08 (1.05, 1.12) | 0.65 | 0.146 | 0.150 | 12.0% | 1 |
| 4-Guanidinobutanoate | C01035 | 1.06 (1.01, 1.12) | −0.24 | 0.028 | 0.049 | 7.9% | 1 |
| 5-Hydroxyindoleacetate | C05635 | 1.06 (0.99, 1.12) | −0.35 | 0.083 | 0.091 | 11.7% | 1 |
| Trigonelline | C01004 | 1.05 (0.97, 1.12) | 0.09 | 0.157 | 0.157 | 11.3% | 1 |
| 3-Hydroxymethylglutarate | C03761 | 1.01 (0.94, 1.08) | −0.25 | 0.107 | 0.113 | 13.3% | 1 |
| Pathway | Coverage | p-Value | FDR | Impact |
|---|---|---|---|---|
| Aminoacyl-tRNA biosynthesis | 6/56 | 4.2 × 10−4 | 0.034 | 0 |
| Phenylalanine metabolism | 4/45 | 2.9 × 10−3 | 0.103 | 0.173 |
| Tryptophan metabolism | 5/79 | 3.9 × 10−3 | 0.103 | 0.146 |
| Valine, leucine & isoleucine biosynthesis | 3/27 | 5.5 × 10−3 | 0.110 | 0.052 |
| Pyrimidine metabolism | 4/60 | 8.3 × 10−3 | 0.133 | 0.088 |
| Nitrogen metabolism | 3/39 | 0.015 | 0.188 | 0 |
| Valine, leucine & isoleucine degradation | 3/40 | 0.016 | 0.188 | 0.042 |
| Purine metabolism | 4/92 | 0.035 | 0.350 | 0.018 |
| Metabolite | KEGG | VIP Mean (95% CI) | log2 FC SP-C | p-Value | FDR | CV | Validation Level |
|---|---|---|---|---|---|---|---|
| Caffeine * | C07481 | 1.84 (1.80, 1.88) | −1.97 | 4.3 × 10−3 | 0.033 | 24.4% | 2 |
| Citrulline | C00327 | 1.83 (1.76, 1.90) | 0.55 | 5.4 × 10−3 | 0.033 | 14.2% | 1 |
| 1-Methyladenosine | C02494 | 1.80 (1.77, 1.84) | 0.40 | 1.9 × 10−3 | 0.033 | 11.5% | 2 |
| 3-Methoxytyramine * | C05587 | 1.79 (1.72, 1.85) | −1.16 | 0.012 | 0.049 | 26.7% | 1 |
| 4-Acetamidobutanoate | C02946 | 1.69 (1.64, 1.74) | 0.43 | 6.1×10−3 | 0.033 | 9.2% | 2 |
| N-Acetylserotonin | C00978 | 1.65 (1.57, 1.73) | 0.59 | 6.2 × 10−3 | 0.033 | 16.8% | 1 |
| O-Succinyl-homoserine | C01118 | 1.64 (1.60, 1.69) | 0.28 | 4.7 × 10−3 | 0.033 | 14.3% | 1 |
| N6- (delta2-isopentenyl)-adenine [M + H] | C04083 | 1.64 (1.59, 1.69) | 0.36 | 9.8 × 10−3 | 0.045 | 5.4% | 1 |
| Trigonelline | C01004 | 1.59 (1.51, 1.66) | 0.20 | 0.021 | 0.067 | 11.3% | 1 |
| 5-Hydroxytryptophan | C00643 | 1.47 (1.42, 1.52) | 0.57 | 0.016 | 0.057 | 15.8% | 1 |
| Kynurenate | C01717 | 1.37 (1.32, 1.43) | 0.60 | 0.039 | 0.113 | 9.1% | 1 |
| N-Acetylneuraminate | C00270 | 1.37 (1.29, 1.45) | −0.27 | 0.062 | 0.117 | 7.0% | 2 |
| N6- (delta2-isopentenyl)-adenine [M − H] | C04083 | 1.32 (1.26, 1.38) | 0.29 | 0.075 | 0.126 | 8.3% | 1 |
| N-Acetylphenylalanine * [M + H] | C03519 | 1.32 (1.26, 1.37) | 0.23 | 0.054 | 0.116 | 6.9% | 1 |
| Deoxyuridine | C00526 | 1.31 (1.24, 1.37) | −0.37 | 0.050 | 0.114 | 13.8% | 1 |
| Homogentisate | C00544 | 1.28 (1.21, 1.36) | 0.21 | 0.050 | 0.114 | 18.6% | 1 |
| 5-Hydroxyindoleacetate | C05635 | 1.26 (1.20, 1.32) | −0.38 | 0.101 | 0.135 | 11.7% | 1 |
| Pipecolate | C00408 | 1.24 (1.16, 1.31) | 0.37 | 0.042 | 0.113 | 7.5% | 1 |
| N-Acetylleucine | C02710 | 1.19 (1.13, 1.26) | 0.15 | 0.145 | 0.178 | 5.8% | 1 |
| Indole-3-acetate | C00954 | 1.19 (1.13, 1.24) | 0.54 | 0.066 | 0.117 | 12.2% | 2 |
| Uridine | C00299 | 1.17 (1.10, 1.24) | 0.19 | 0.087 | 0.132 | 7.4% | 2 |
| Indoxyl sulfate * | C08481 | 1.17 (1.09, 1.25) | −0.35 | 0.244 | 0.252 | 24.0% | 2 |
| N-Acetyltryptophan * | C03137 | 1.12 (1.06, 1.18) | 0.35 | 0.058 | 0.116 | 4.3% | 1 |
| Deoxycarnitine | C01181 | 1.10 (0.99, 1.20) | −0.27 | 0.214 | 0.228 | 6.8% | 1 |
| Xanthosine | C01762 | 1.09 (1.02, 1.16) | 0.26 | 0.096 | 0.134 | 8.8% | 1 |
| Phenylacetate | C07086 | 1.08 (1.00, 1.16) | 0.18 | 0.202 | 0.222 | 8.7% | 2 |
| Ketoleucine | C00233 | 1.07 (1.02, 1.11) | 0.07 | 0.083 | 0.132 | 7.6% | 2 |
| Carnitine | C00318 | 1.07 (0.97, 1.16) | −0.18 | 0.166 | 0.189 | 5.8% | 2 |
| Guanosine | C00387 | 1.06 (0.99, 1.13) | 0.13 | 0.162 | 0.189 | 7.8% | 2 |
| N-Acetylphenylalanine * [M − H] | C03519 | 1.03 (0.96, 1.11) | 0.13 | 0.260 | 0.260 | 9.3% | 1 |
| 4-Pyridoxate | C00847 | 1.02 (0.97, 1.06) | 0.48 | 0.094 | 0.134 | 12.2% | 1 |
| 4-Hydroxybenzoate | C00156 | 1.02 (0.94, 1.09) | 0.17 | 0.113 | 0.145 | 11.5% | 1 |
| Pathway | Coverage | p-Value | FDR | Impact |
|---|---|---|---|---|
| Tryptophan metabolism | 5/79 | 1.5 × 10−3 | 0.123 | 0.159 |
| Phenylalanine metabolism | 3/45 | 0.013 | 0.522 | 0.054 |
| Caffeine metabolism | 2/21 | 0.022 | 0.595 | 0.184 |
| Metabolite | Spinal Cord | Third Ventricle | EDSS | Disease Duration | Total T1 | Total T2 |
|---|---|---|---|---|---|---|
| Caffeine * | 0.45 # | −0.12 | −0.21 | −0.33 # | −0.19 | −0.14 |
| 1-Methyladenosine | −0.14 | 0.37 # | 0.04 | 0.44 # | 0.24 | 0.18 |
| 3-Methoxytyramine * | 0.39 # | −0.16 | −0.18 | −0.23 | −0.07 | −0.10 |
| 4-Acetamidobutanoate | −0.33 # | 0.39 # | 0.23 | 0.60 # | 0.23 | 0.10 |
| N-Acetylserotonin | −0.27 | 0.17 | 0.08 | 0.39 # | 0.04 | −0.04 |
| O-Succinyl-homoserine | −0.17 | 0.47 # | 0.30 # | 0.31 # | 0.37 # | 0.33 # |
| N6-(delta2-isopentenyl)-adenine [M + H] | −0.16 | 0.32 # | 0.19 | 0.26 | 0.16 | 0.24 |
| Deoxyuridine | 0.28 | −0.35 # | −0.41 # | −0.34 # | −0.32 # | −0.26 |
| 5-Hydroxyindoleacetate | 0.35 # | 0.03 | −0.38 # | 0.0 | −0.12 | −0.01 |
| Pipecolate | −0.41 # | 0.38 # | 0.21 | 0.42 # | 0.19 | 0.16 |
| Indole-3-acetate | -0.12 | 0.26 | 0.24 | 0.52 # | 0.31 # | 0.27 |
| Uridine | −0.42 | 0.23 | 0.43 # | 0.42 # | 0.27 | 0.11 |
| N-Acetyltryptophan * | −0.09 | 0.45 # | 0.34 # | 0.12 | 0.30 # | 0.31 # |
| Deoxycarnitine | −0.45 # | 0.03 | 0.27 | 0.09 | −0.12 | −0.20 |
| Phenylacetate | −0.08 | 0.33 # | 0.07 | 0.16 | 0.16 | 0.11 |
| Ketoleucine | −0.25 | 0.48 # | 0.24 | 0.34 # | 0.27 | 0.11 |
| Thymine | −0.50 # | 0.28 | 0.42 # | 0.39 # | 0.26 | 0.08 |
| Glutarylcarnitine | −0.44 # | 0.39 # | 0.52 # | 0.29 # | 0.26 | 0.18 |
| Biliverdin | −0.46 # | 0.08 | 0.41 # | 0.32 # | 0.12 | −0.03 |
| Ethylmalonate | −0.27 | 0.26 | 0.31 # | 0.25 | 0.02 | 0.02 |
| Valine | −0.25 | 0.30# | 0.31 # | 0.15 | 0.21 | 0.13 |
| Methionine | −0.35 # | 0.30# | 0.44 # | 0.39 # | 0.21 | 0.16 |
| Glutamine | −0.14 | 0.27 | 0.43 # | 0.31 # | 0.27 | 0.28 |
| 3-Methoxytyrosine [M + H] | −0.33 # | 0.27 | 0.38 # | 0.28 | 0.23 | 0.16 |
| Phenylalanine | −0.17 | 0.23 | 0.35 # | 0.14 | 0.16 | 0.16 |
| Urate | −0.22 | 0.26 | 0.23 | 0.33 # | 0.12 | 0.10 |
| 3,4-Dihydroxyphenylglycol | −0.27 | 0.16 | 0.29 # | 0.19 | 0.11 | 0.01 |
| Isoleucine/Leucine | −0.22 | 0.24 | 0.36 # | 0.1 | 0.12 | 0.15 |
| 3-Methoxytyrosine [M − H] | −0.30 | 0.20 | 0.33 # | 0.14 | 0.09 | 0.11 |
| 4-Guanidinobutanoate | 0.16 | 0.03 | −0.01 | 0.03 | 0.23 | 0.38 # |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herman, S.; Åkerfeldt, T.; Spjuth, O.; Burman, J.; Kultima, K. Biochemical Differences in Cerebrospinal Fluid between Secondary Progressive and Relapsing–Remitting Multiple Sclerosis. Cells 2019, 8, 84. https://doi.org/10.3390/cells8020084
Herman S, Åkerfeldt T, Spjuth O, Burman J, Kultima K. Biochemical Differences in Cerebrospinal Fluid between Secondary Progressive and Relapsing–Remitting Multiple Sclerosis. Cells. 2019; 8(2):84. https://doi.org/10.3390/cells8020084
Chicago/Turabian StyleHerman, Stephanie, Torbjörn Åkerfeldt, Ola Spjuth, Joachim Burman, and Kim Kultima. 2019. "Biochemical Differences in Cerebrospinal Fluid between Secondary Progressive and Relapsing–Remitting Multiple Sclerosis" Cells 8, no. 2: 84. https://doi.org/10.3390/cells8020084
APA StyleHerman, S., Åkerfeldt, T., Spjuth, O., Burman, J., & Kultima, K. (2019). Biochemical Differences in Cerebrospinal Fluid between Secondary Progressive and Relapsing–Remitting Multiple Sclerosis. Cells, 8(2), 84. https://doi.org/10.3390/cells8020084