Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-κB Inhibition and Nrf2/HO-1 Activation


Abstract
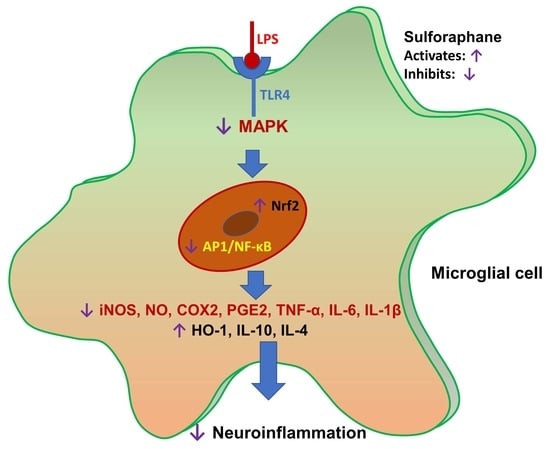
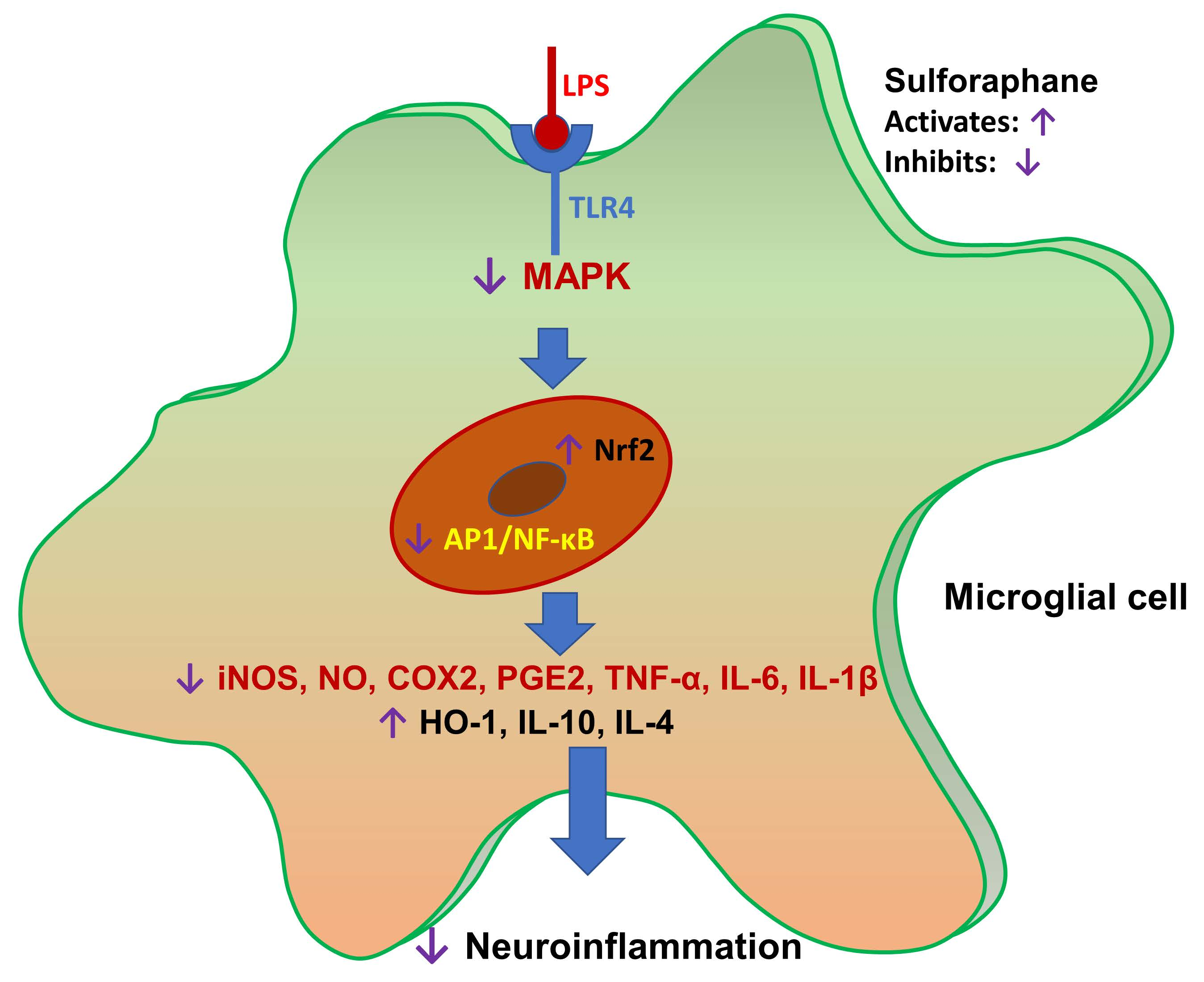
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
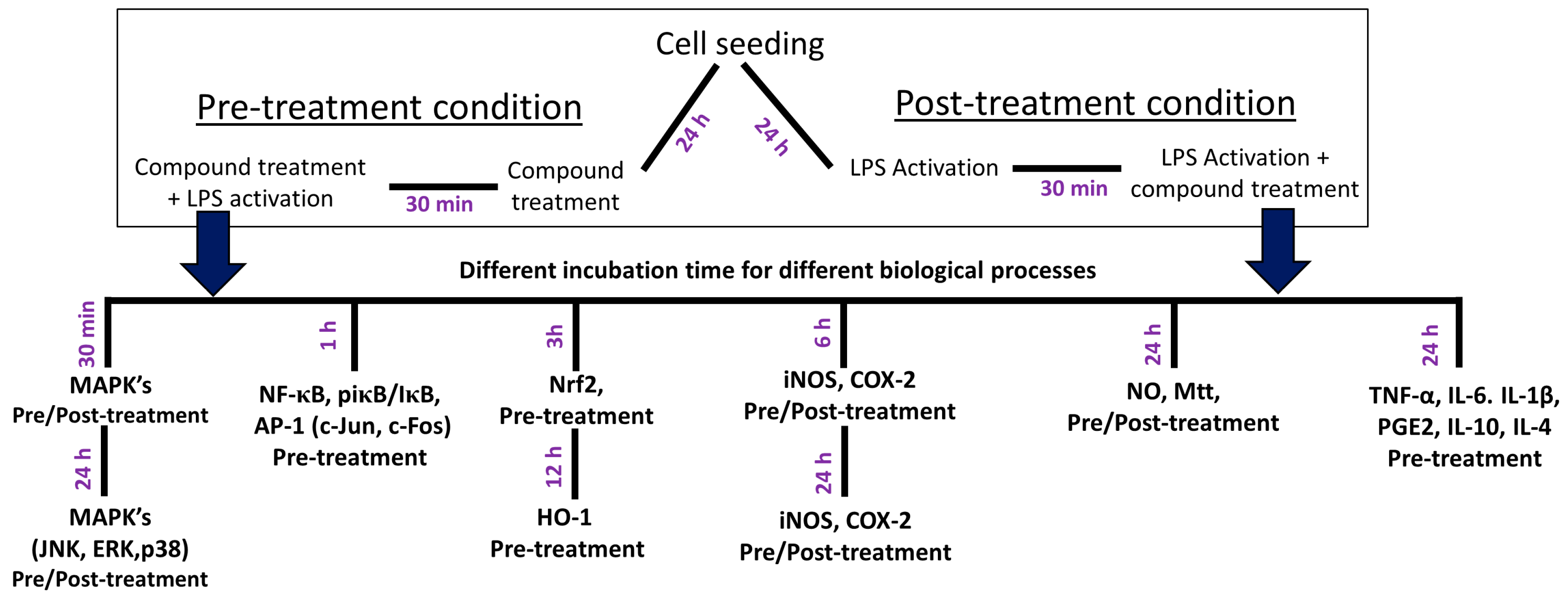
2.3. Cell Treatment and Nitrite and Cell Viability Assays
2.4. Western Blot Analysis
2.5. Measurement of PGE2, TNF-α, IL-1β, IL-6, IL-10, and IL-4 Production
2.6. NF-κB Assay
2.7. Statistical Analysis
3. Results
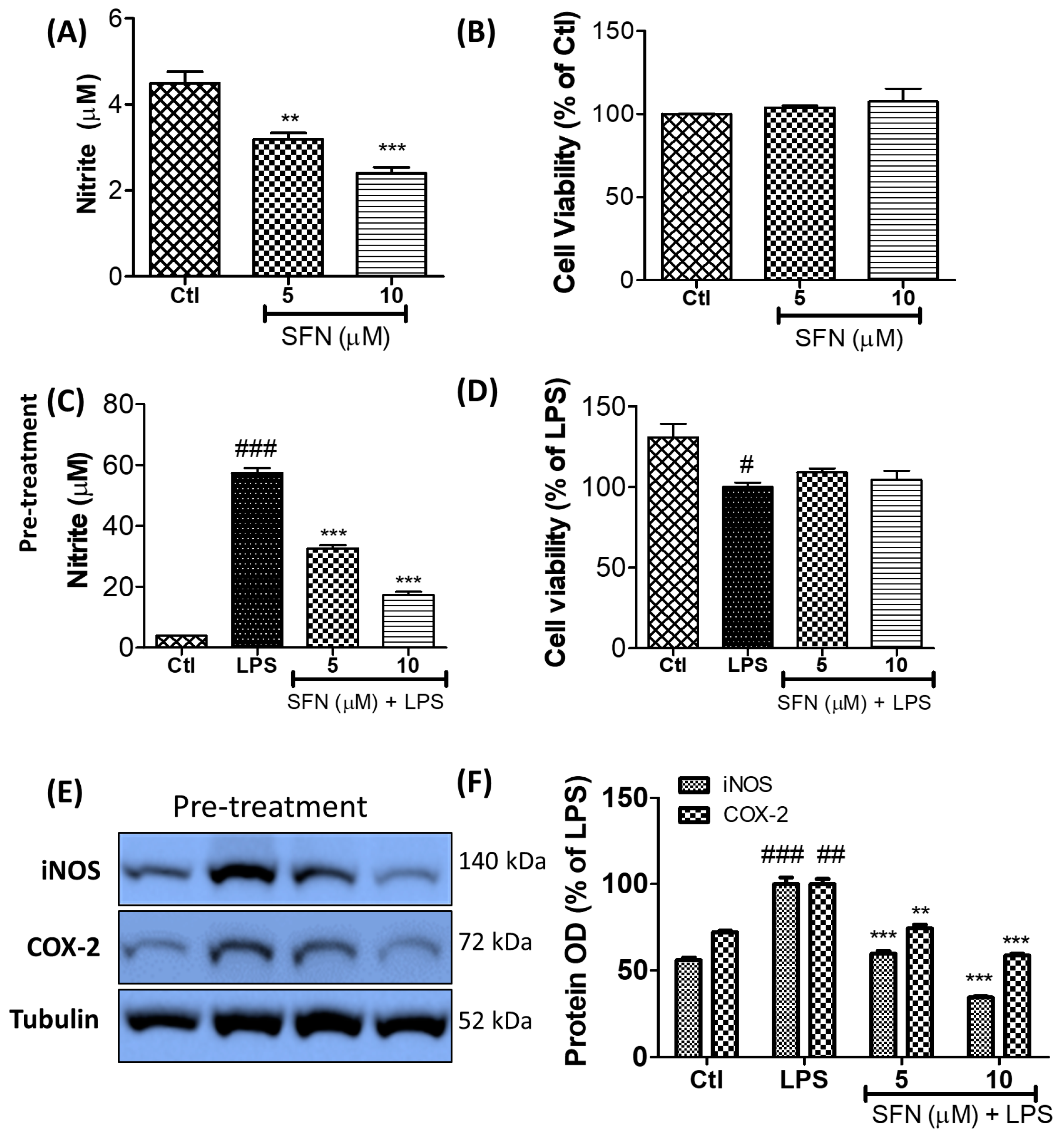
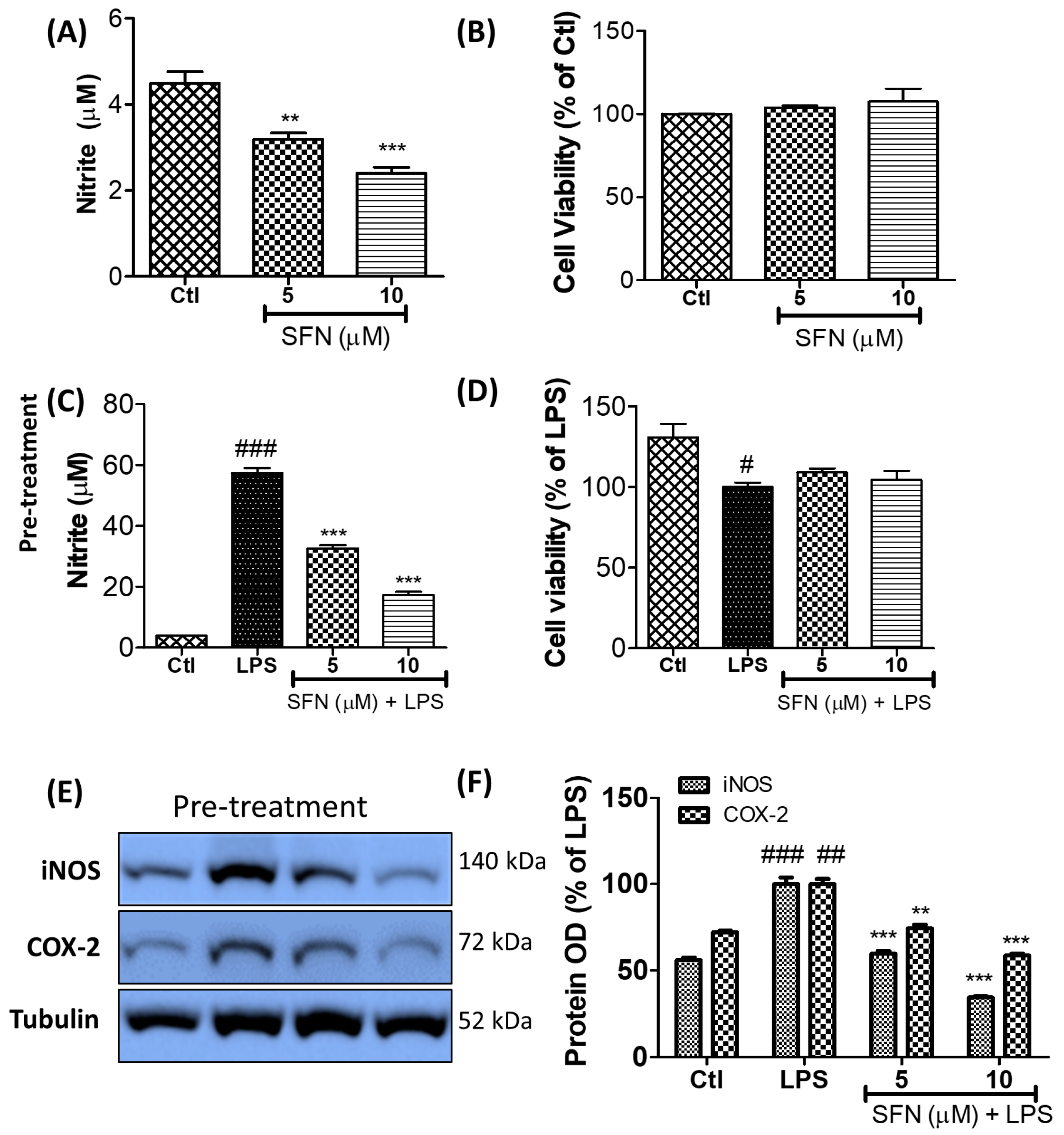
3.1. SFN Inhibited Nitrite Production and iNOS and COX-2 Expression in LPS-Activated Microglial Cells
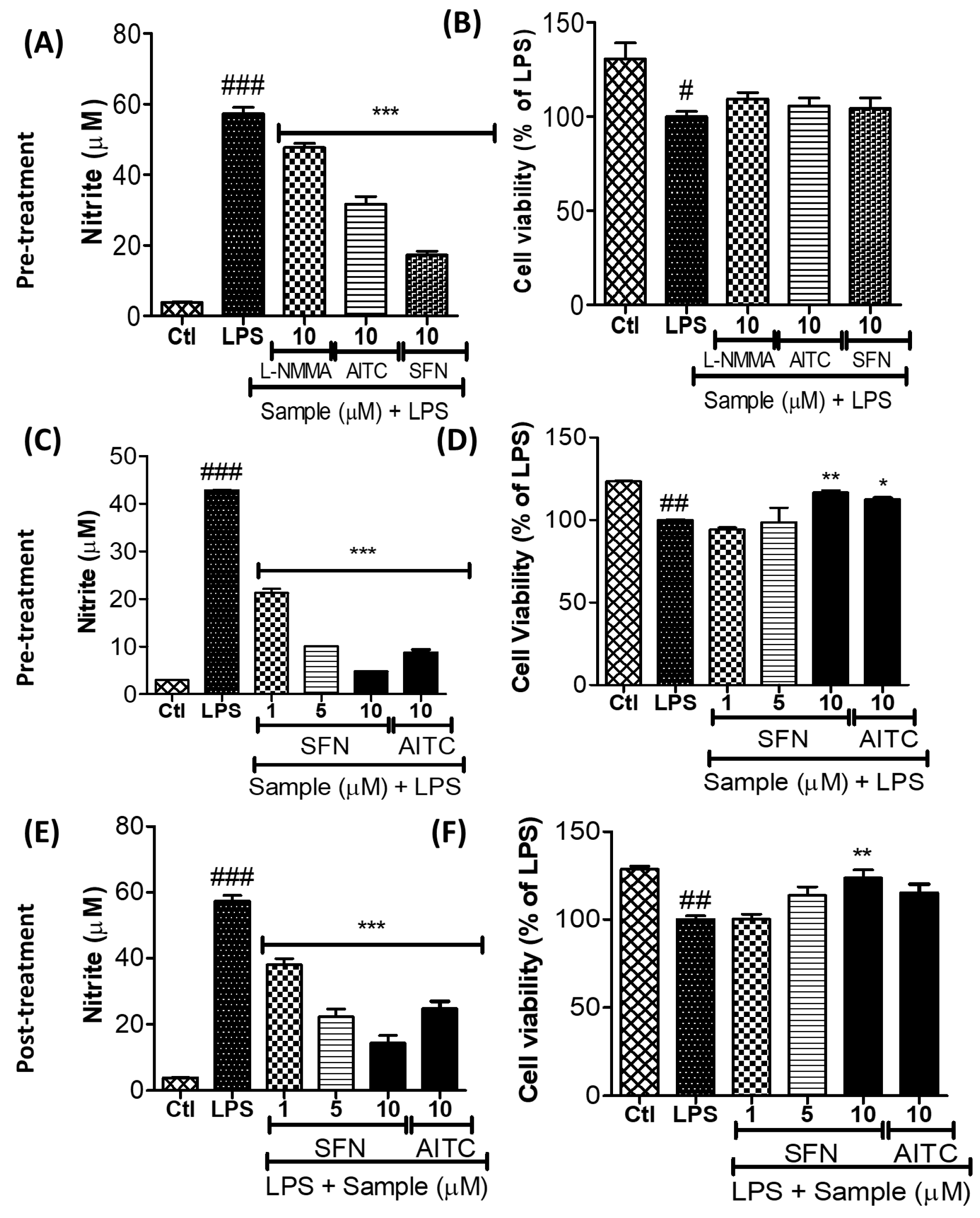
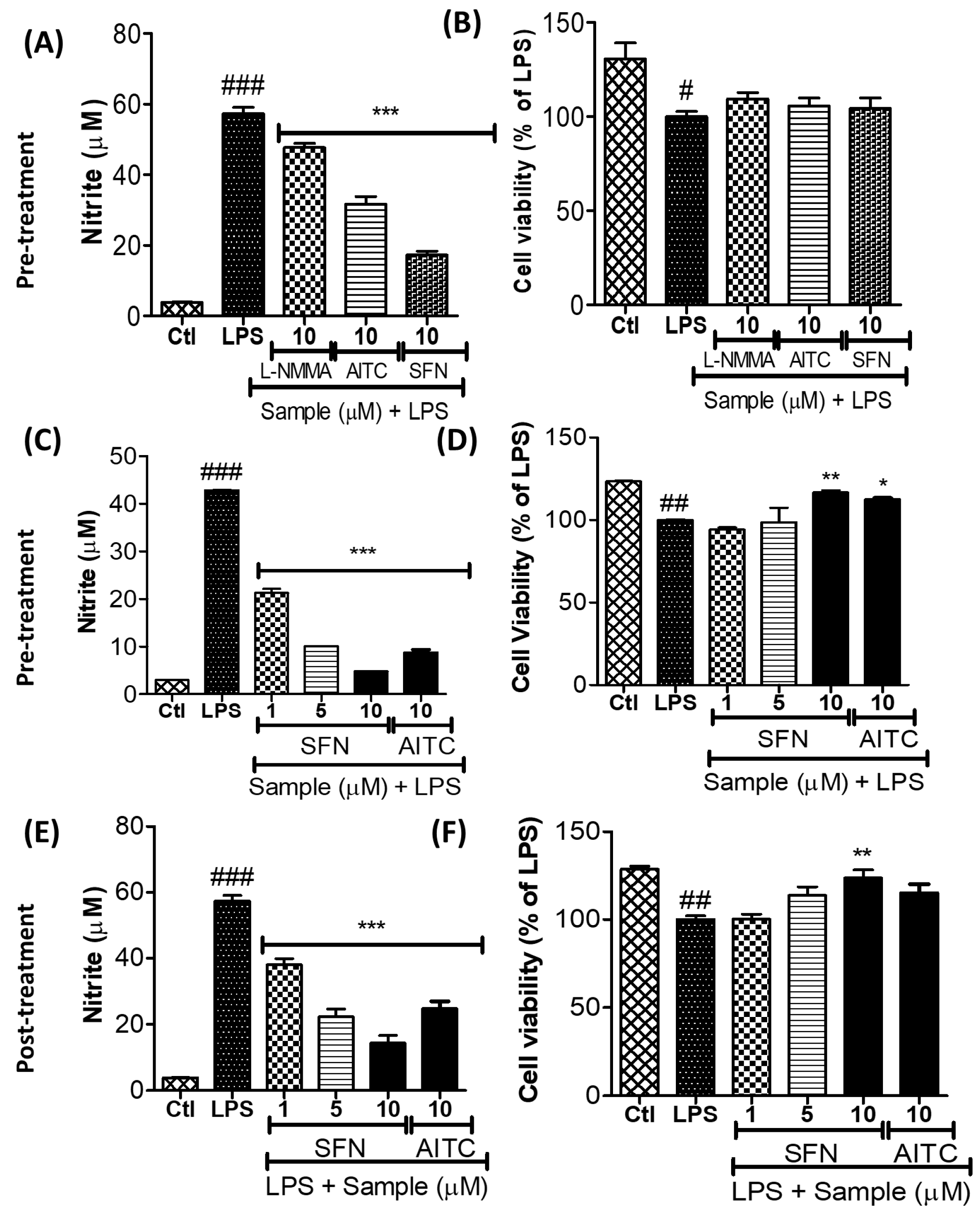
3.2. SFN Inhibited Nitrite Production in LPS-Activated Microglial Cells
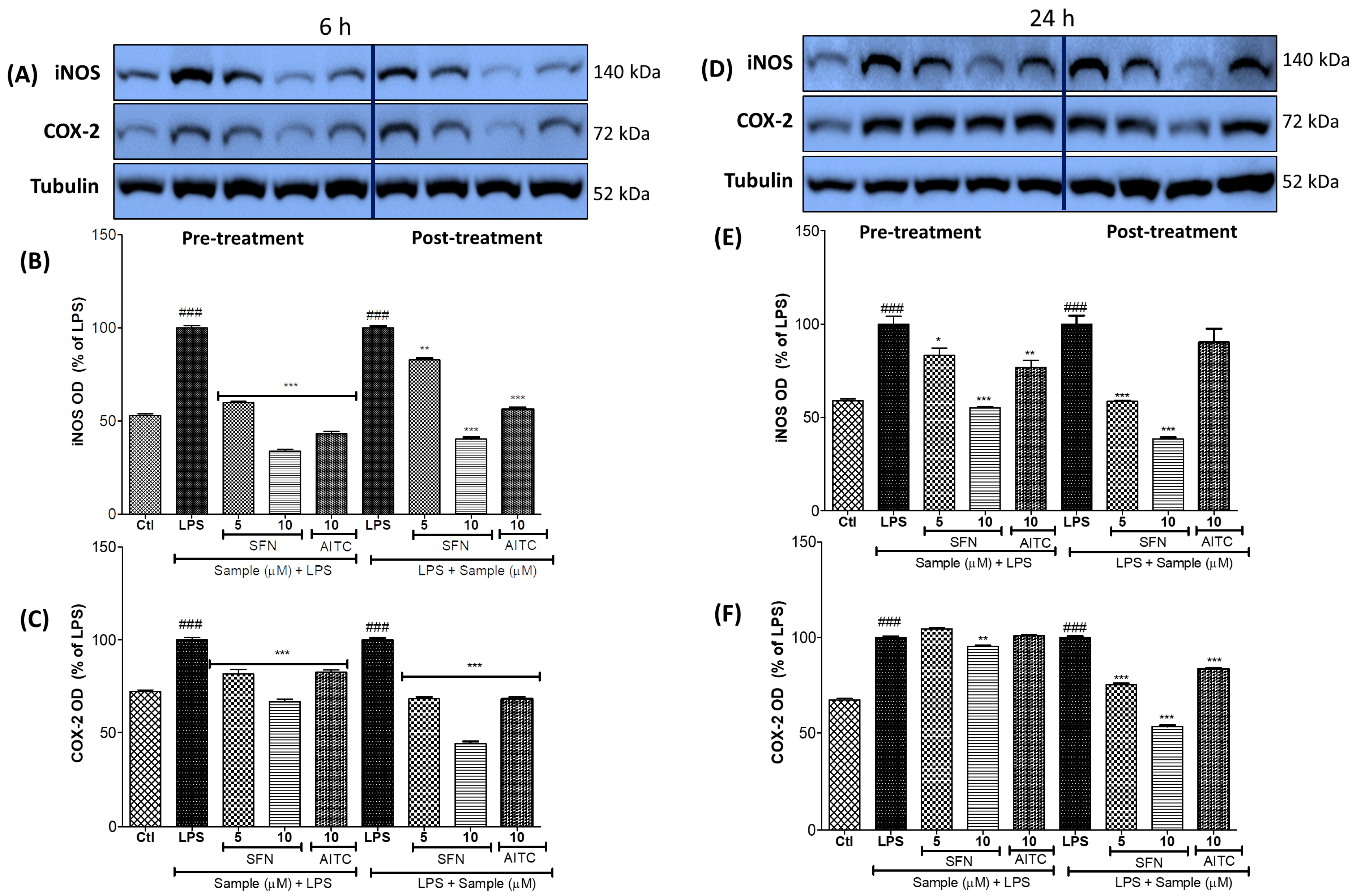
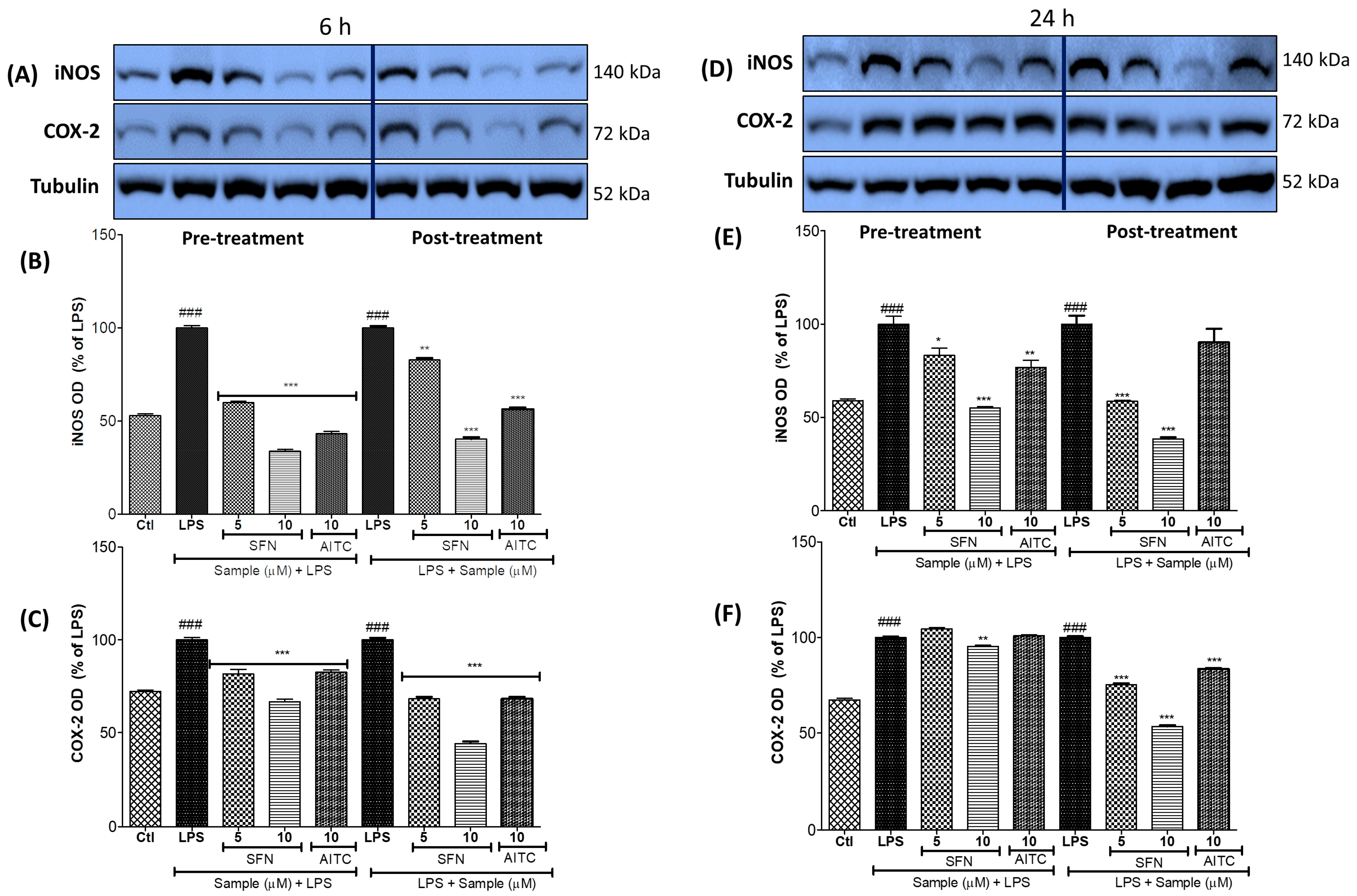
3.3. SFN Significantly Inhibited iNOS and COX-2 Expression, in 6 h and 24 h of LPS Activation, under Both Prophylactic and Therapeutic Strategies
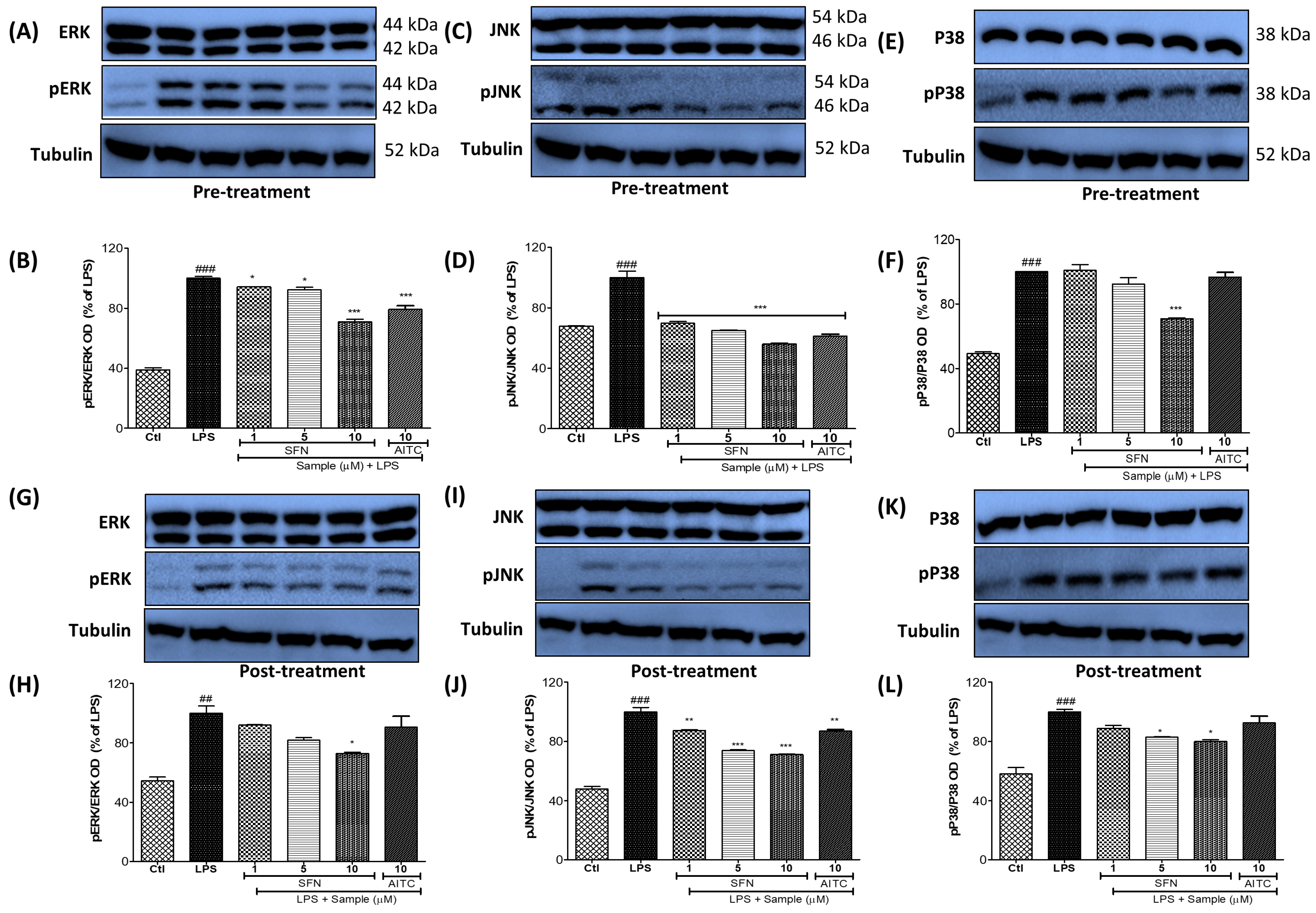
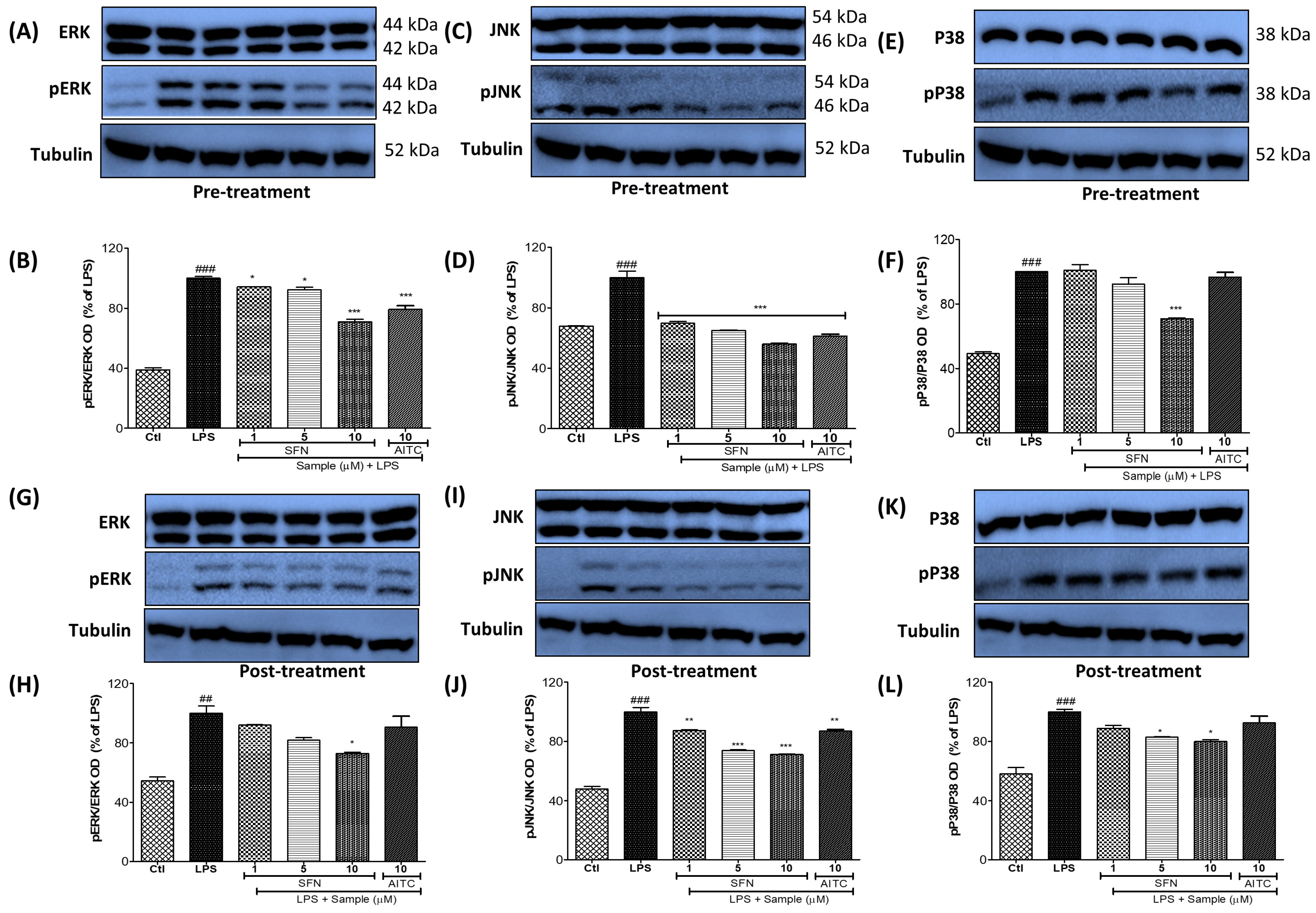
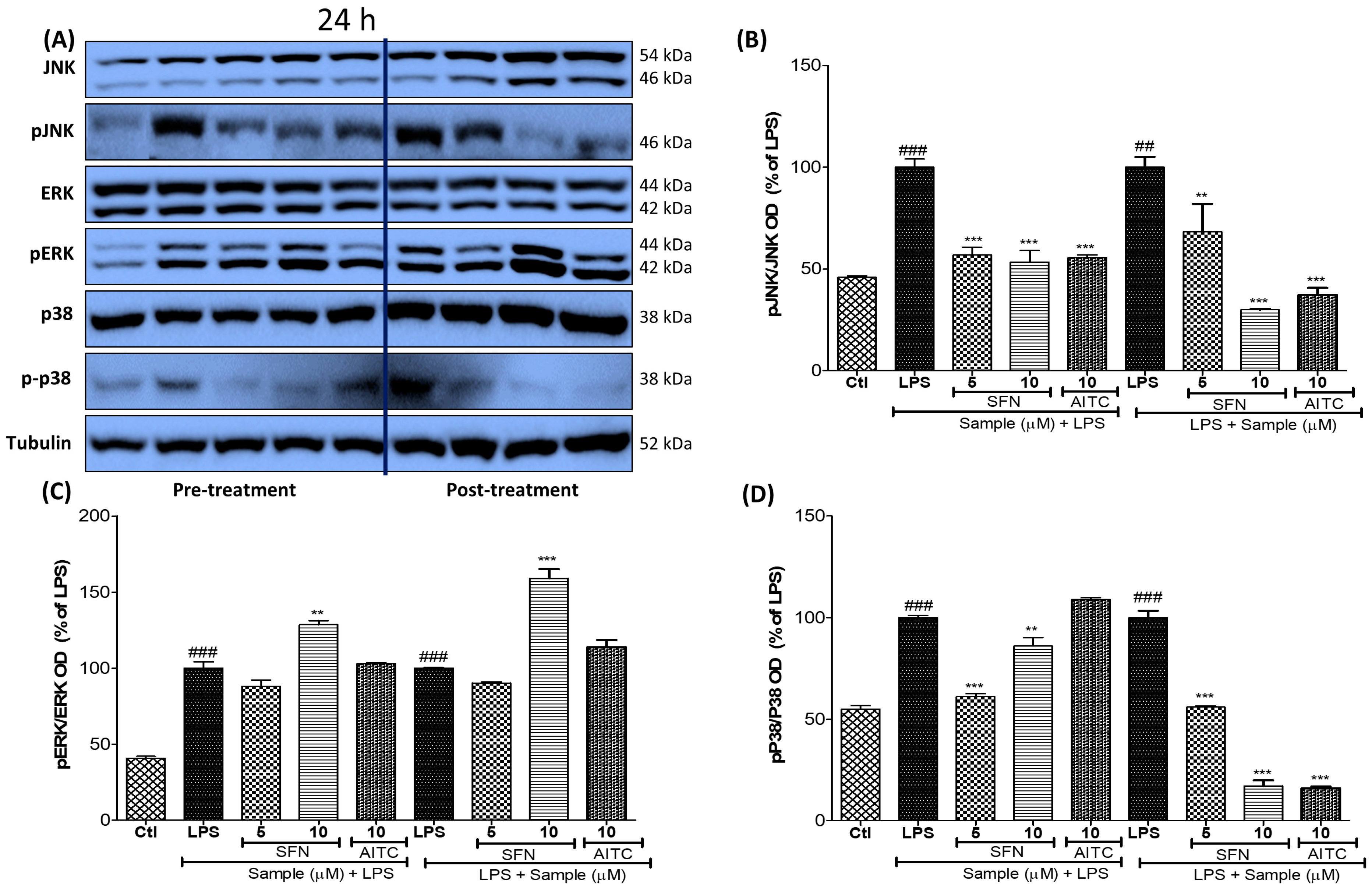
3.4. SFN Pre- and Post-Treatments Significantly Modulated the MAPK Signaling Pathway, Particularly pJNK, in LPS-Activated Microglial Cells
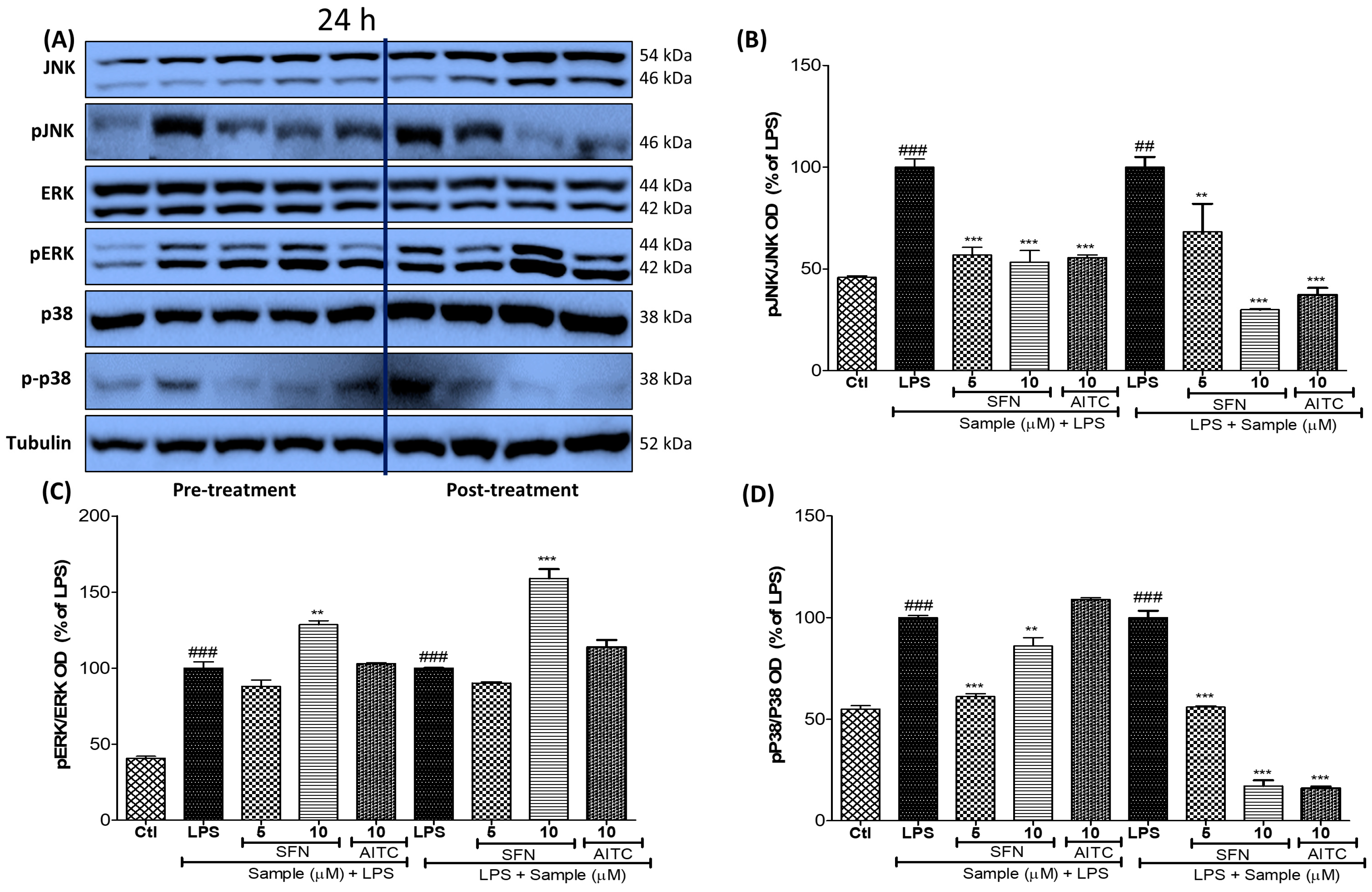
3.5. SFN Pre- and Post-Treatments Significantly Modulated the MAPK Signaling Pathway, Particularly pJNK and p38, in 24 h LPS-Activated Microglial Cells
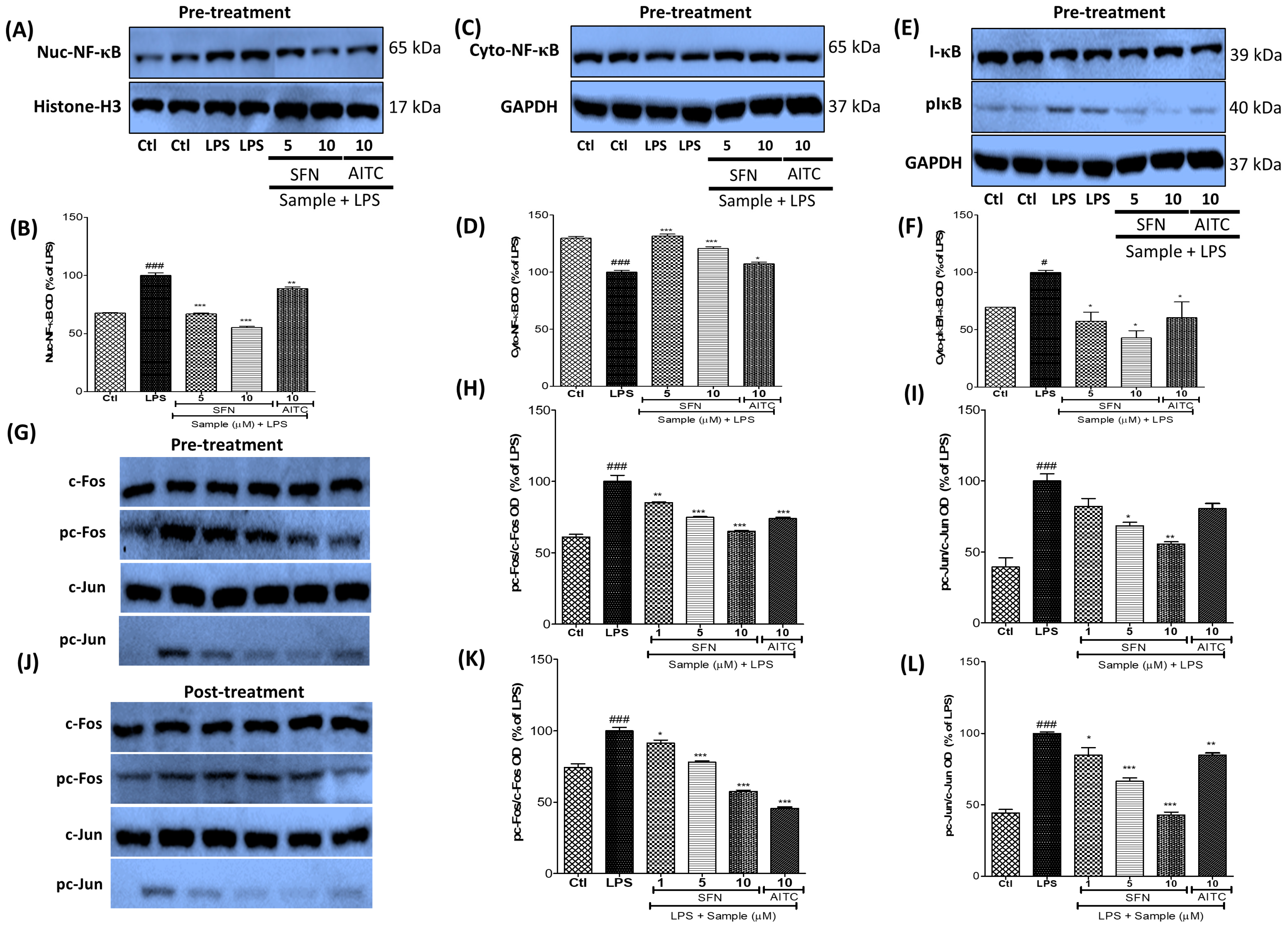
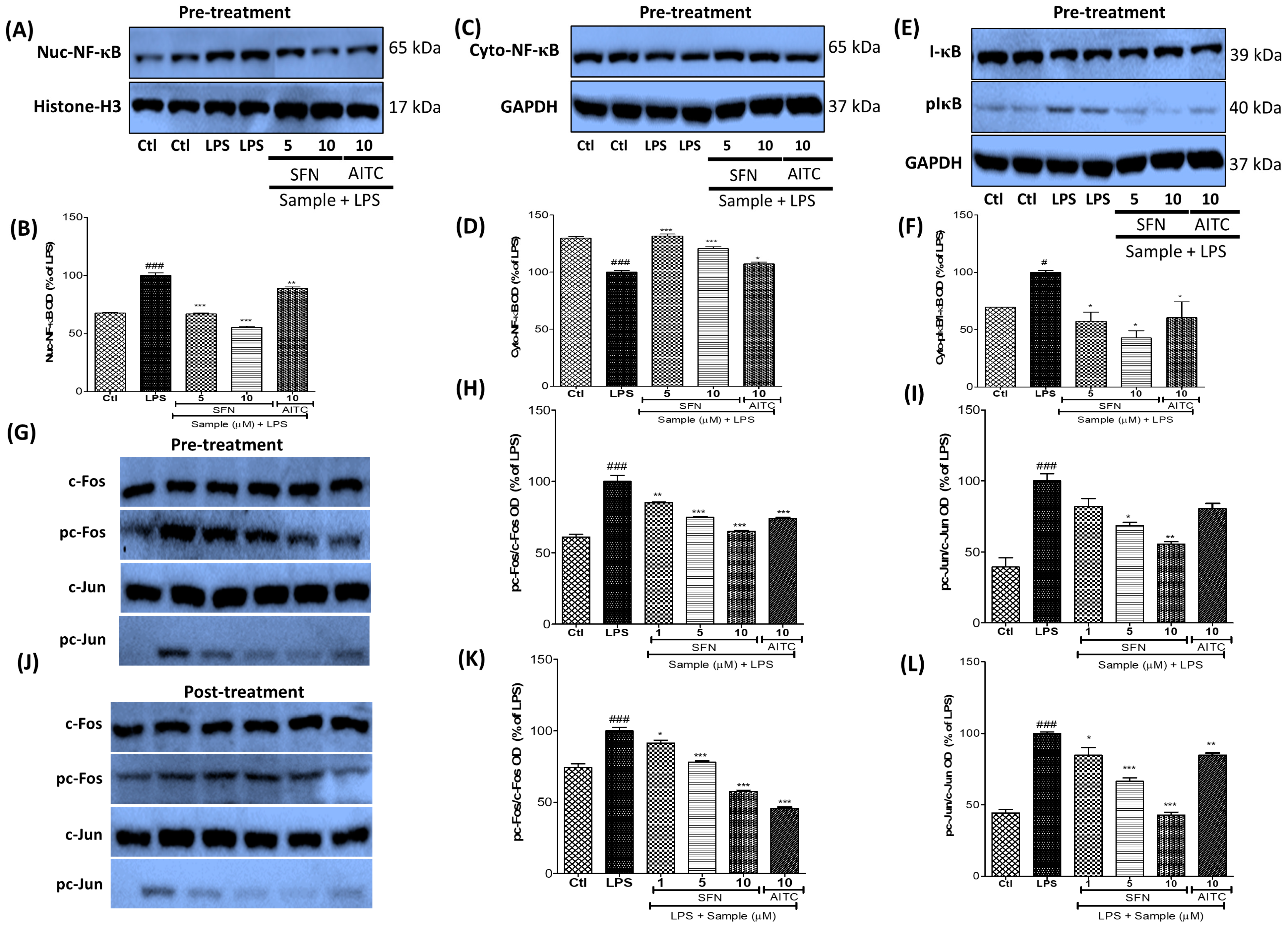
3.6. SFN Treatment Significantly Inhibited NF-κB and AP-1 Signaling in LPS-Activated Microglial Cells
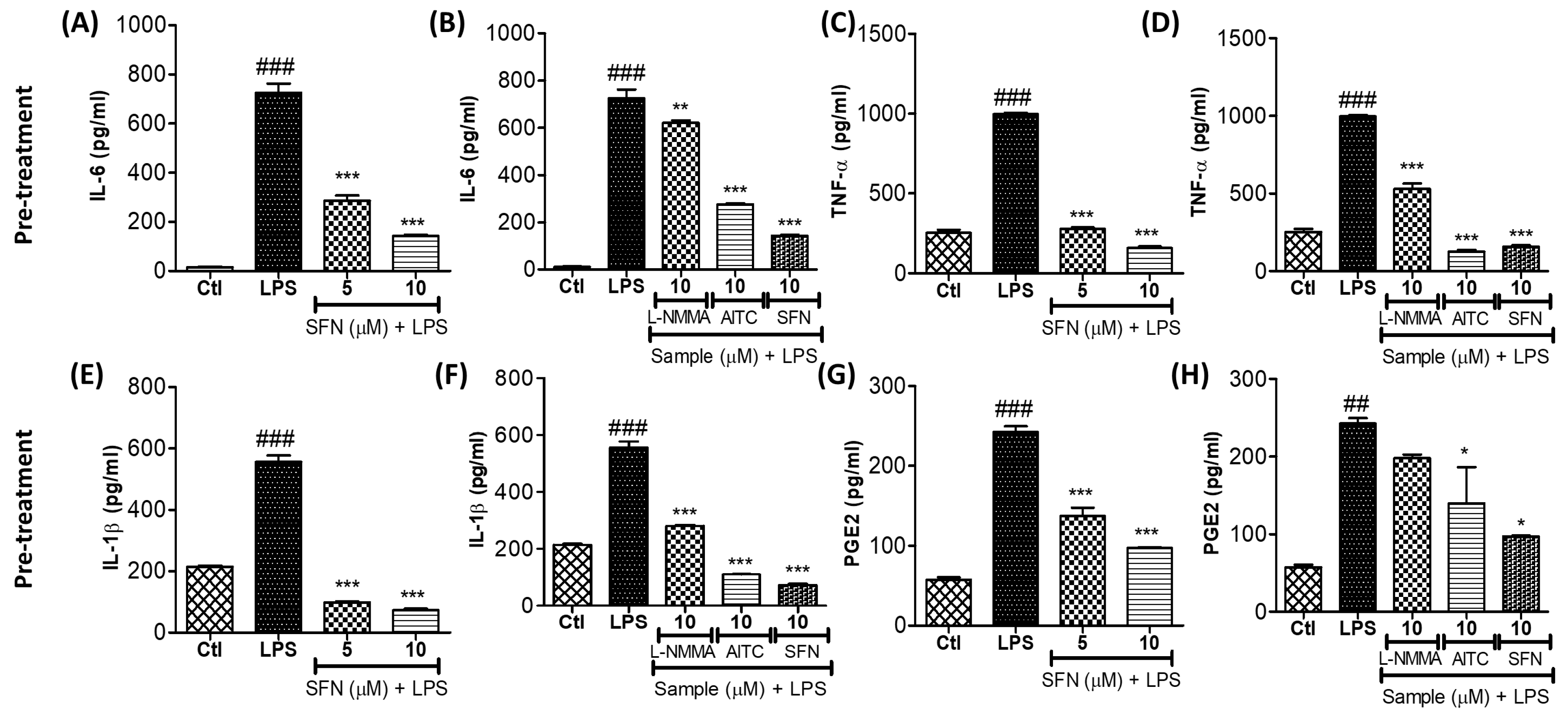
3.7. SFN Treatment Significantly Inhibited the Production of Proinflammatory Cytokines in LPS-Activated Microglial Cells
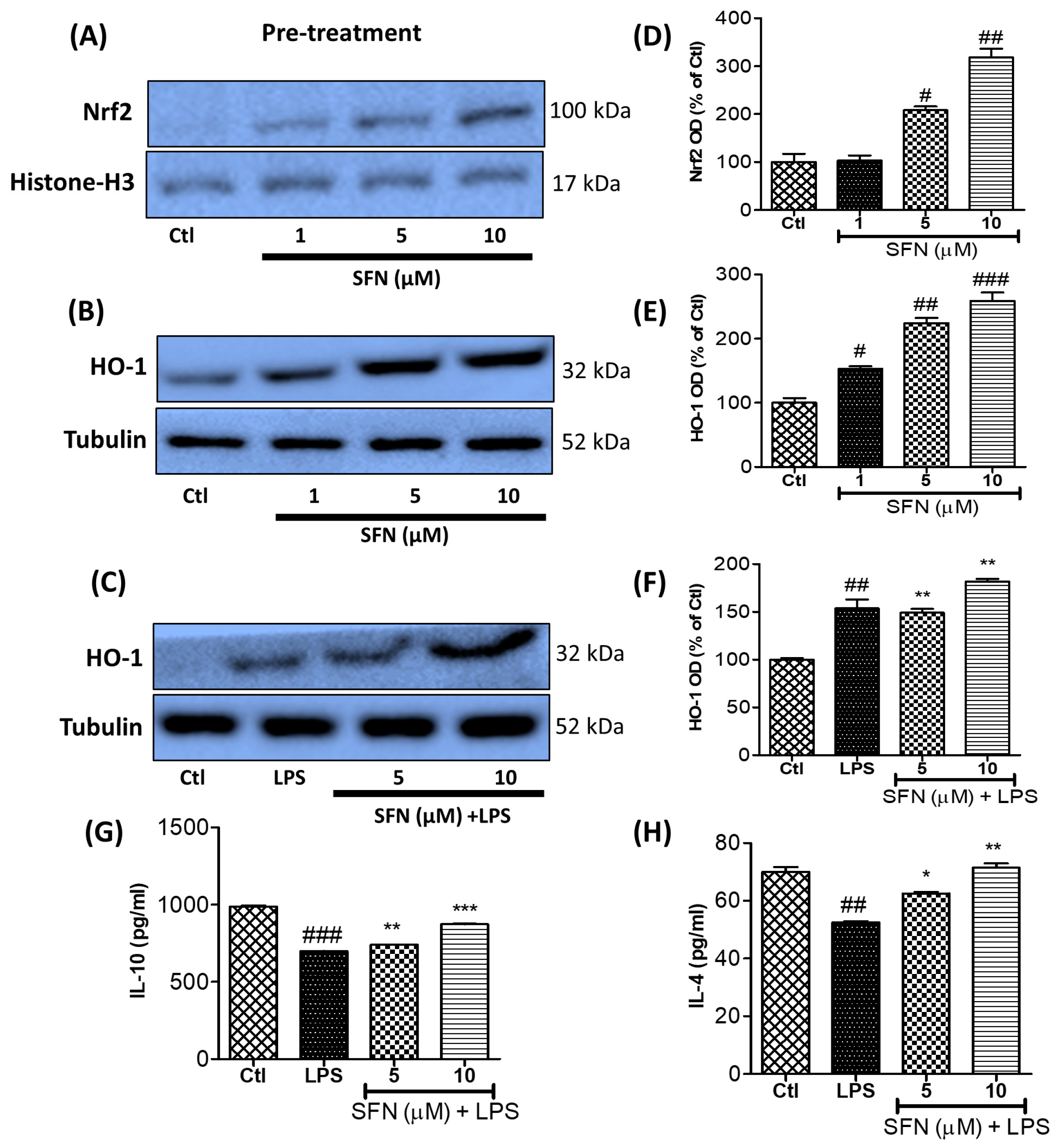
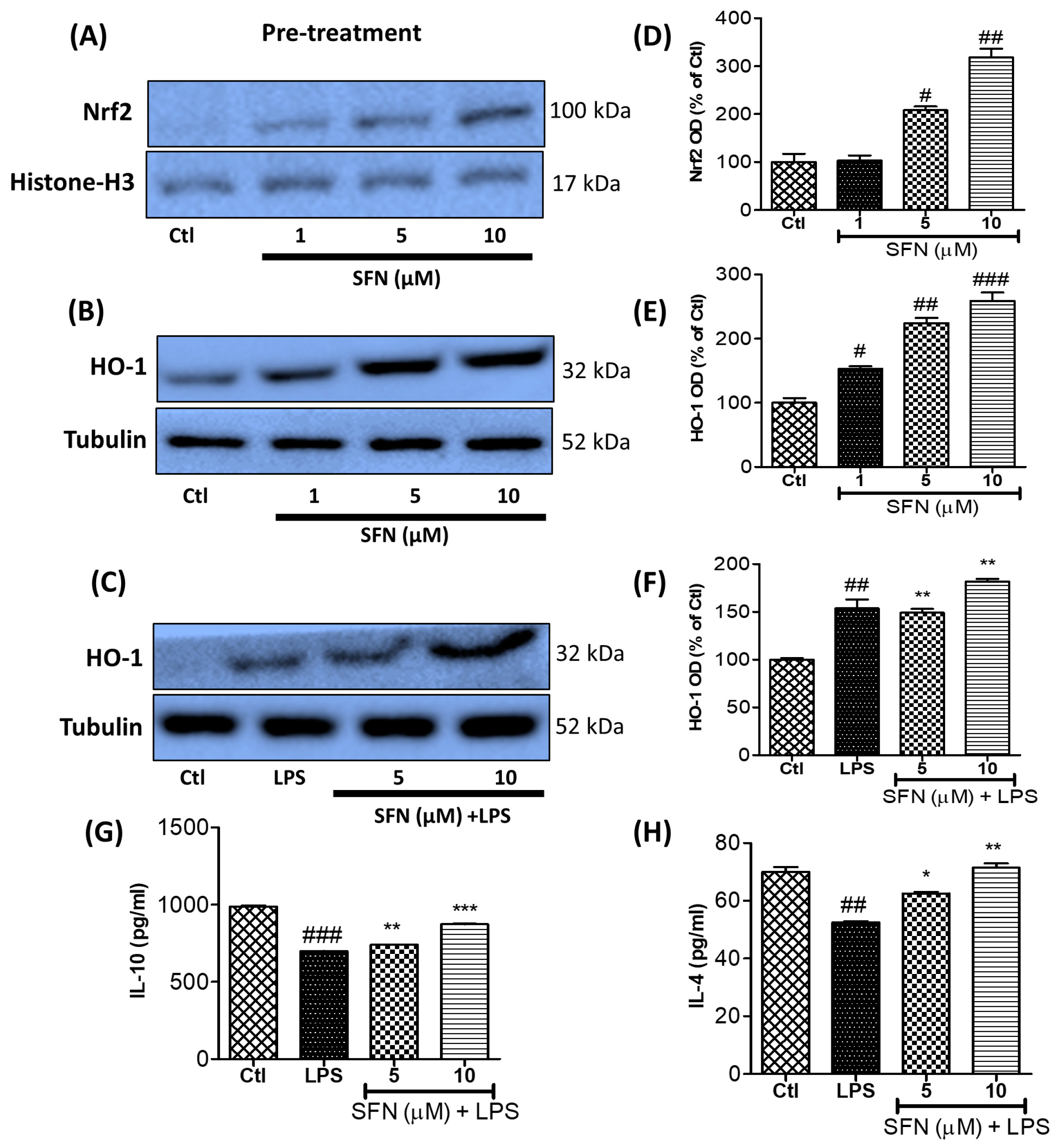
3.8. SFN Treatment Significantly Increased Anti-Inflammatory Protein (Nrf2 and HO-1) Expression and Anti-Inflammatory Cytokine (IL-10 and IL-4) Production in LPS-Activated Microglial Cells
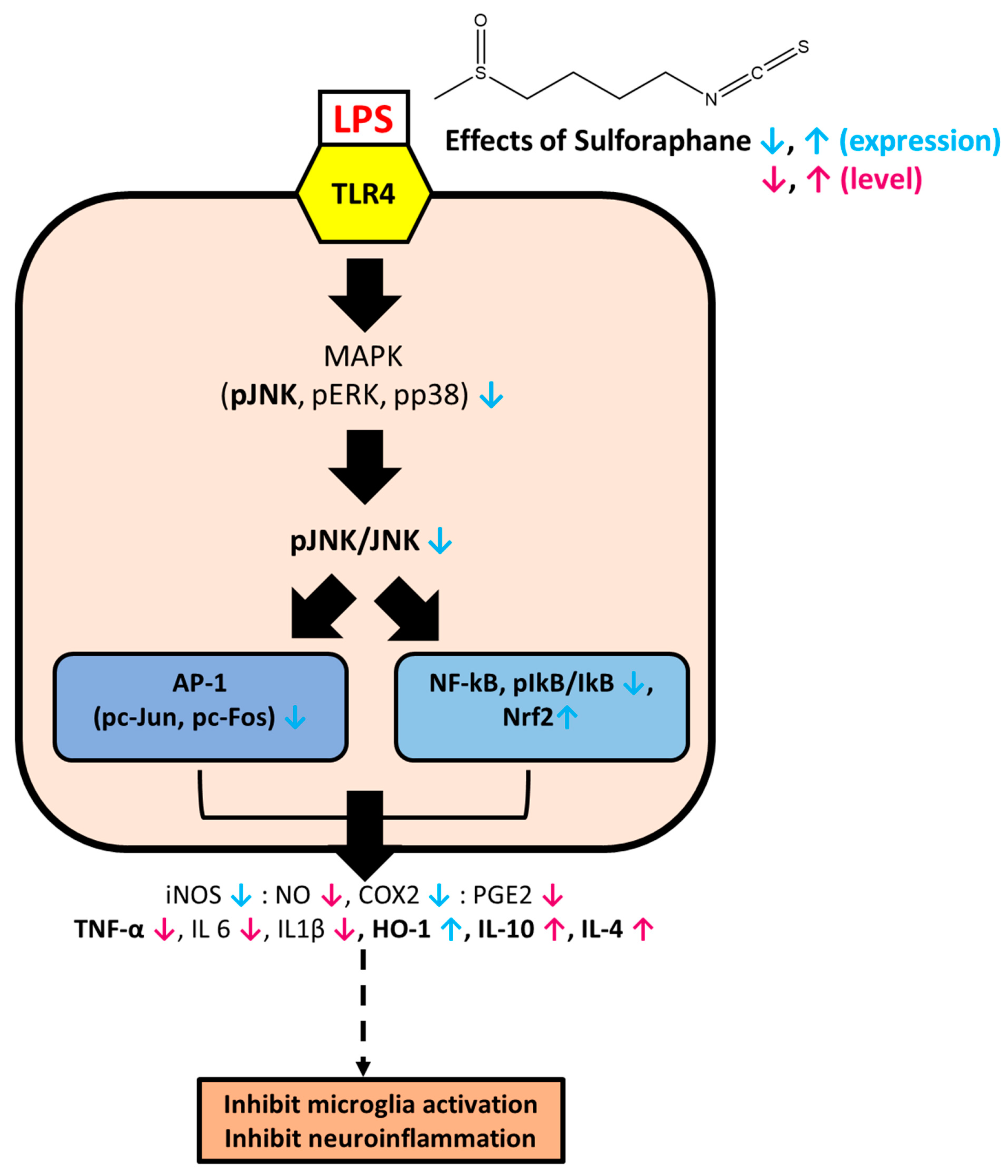
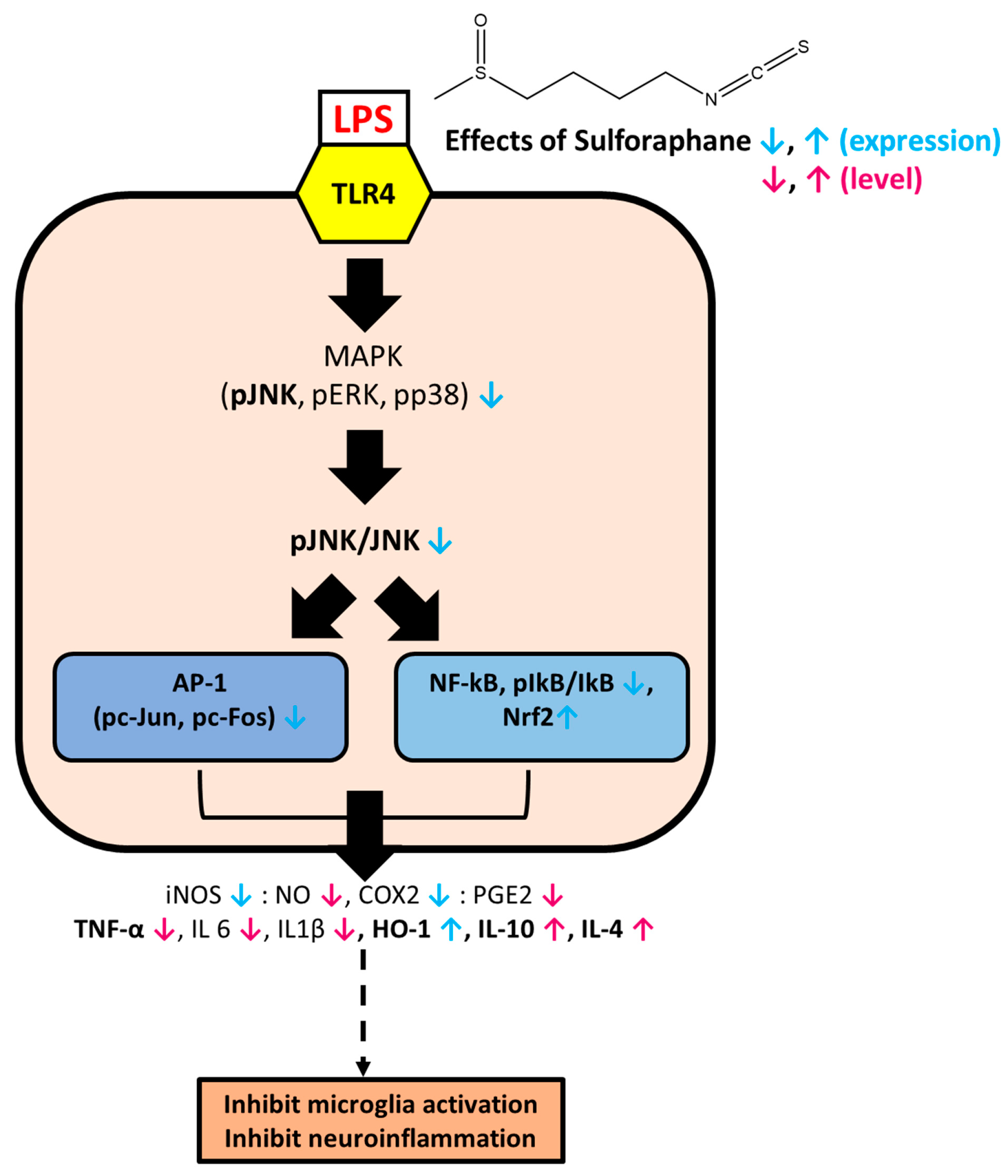
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and other nutrigenomic nrf2 activators: Can the clinician’s expectation be matched by the reality? Oxid. Med. Cell. Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef]
- Kim, J.K.; Park, S.U. Current potential health benefits of sulforaphane. EXCLI J. 2016, 15, 571–577. [Google Scholar]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxid. Med. Cell. Longev. 2013, 2013, 415078. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an nrf2-independent mechanism. J. Leukoc. Biol. 2016, 99, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Eren, E.; Tufekci, K.U.; Isci, K.B.; Tastan, B.; Genc, K.; Genc, S. Sulforaphane inhibits lipopolysaccharide-induced inflammation, cytotoxicity, oxidative stress, and mir-155 expression and switches to mox phenotype through activating extracellular signal-regulated kinase 1/2-nuclear factor erythroid 2-related factor 2/antioxidant response element pathway in murine microglial cells. Front. Immunol. 2018, 9, 36. [Google Scholar] [PubMed]
- Kwon, J.S.; Joung, H.; Kim, Y.S.; Shim, Y.S.; Ahn, Y.; Jeong, M.H.; Kee, H.J. Sulforaphane inhibits restenosis by suppressing inflammation and the proliferation of vascular smooth muscle cells. Atherosclerosis 2012, 225, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. Tnf and map kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef]
- Eurlings, I.M.; Reynaert, N.L.; van de Wetering, C.; Aesif, S.W.; Mercken, E.M.; de Cabo, R.; van der Velden, J.L.; Janssen-Heininger, Y.M.; Wouters, E.F.; Dentener, M.A. Involvement of c-jun n-terminal kinase in tnf-alpha-driven remodeling. Am. J. Respir. Cell. Mol. Biol. 2017, 56, 393–401. [Google Scholar] [CrossRef]
- Besirli, C.G.; Wagner, E.F.; Johnson, E.M., Jr. The limited role of nh2-terminal c-jun phosphorylation in neuronal apoptosis: Identification of the nuclear pore complex as a potential target of the jnk pathway. J. Cell Biol. 2005, 170, 401–411. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. Jnk signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Subedi, L.; Gaire, B.P.; Do, M.H.; Lee, T.H.; Kim, S.Y. Anti-neuroinflammatory and neuroprotective effects of the lindera neesiana fruit in vitro. Phytomedicine 2016, 23, 872–881. [Google Scholar] [CrossRef]
- Subedi, L.; Kwon, O.W.; Pak, C.; Lee, G.; Lee, K.; Kim, H.; Kim, S.Y. N,N-disubstituted azines attenuate lps-mediated neuroinflammation in microglia and neuronal apoptosis via inhibiting mapk signaling pathways. BMC Neurosci. 2017, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, T.; Mao, L.; Zhang, F. Sulforaphane protects against brain diseases: Roles of cytoprotective enzymes. Austin J. Cerebrovasc. Dis. Stroke 2017, 4, 1054. [Google Scholar]
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y. Amelioration of alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Masci, A.; Mattioli, R.; Costantino, P.; Baima, S.; Morelli, G.; Punzi, P.; Giordano, C.; Pinto, A.; Donini, L.M.; d’Erme, M.; et al. Neuroprotective effect of brassica oleracea sprouts crude juice in a cellular model of alzheimer’s disease. Oxid. Med. Cell. Longev. 2015, 2015, 781938. [Google Scholar] [CrossRef]
- Chen, H.G.; Xie, K.L.; Han, H.Z.; Wang, W.N.; Liu, D.Q.; Wang, G.L.; Yu, Y.H. Heme oxygenase-1 mediates the anti-inflammatory effect of molecular hydrogen in lps-stimulated raw 264.7 macrophages. Int. J. Surg. 2013, 11, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Withers, C.M.; Bartz, R.R.; MacGarvey, N.C.; Fu, P.; Sweeney, T.E.; Welty-Wolf, K.E.; Suliman, H.B. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J. Biol. Chem. 2011, 286, 16374–16385. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Subedi, L.; Shin, D.; Kim, C.S.; Lee, K.R.; Kim, S.Y. A new neolignan derivative, balanophonin isolated from firmiana simplex delays the progress of neuronal cell death by inhibiting microglial activation. Biomol. Ther. 2017, 25, 519–527. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, X.; Zhao, S.; Ma, C.; Cui, J.; Zheng, Y. Sulforaphane protects against cardiovascular disease via nrf2 activation. Oxid. Med. Cell. Longev. 2015, 2015, 407580. [Google Scholar] [CrossRef]
- Luo, J.F.; Shen, X.Y.; Lio, C.K.; Dai, Y.; Cheng, C.S.; Liu, J.X.; Yao, Y.D.; Yu, Y.; Xie, Y.; Luo, P.; et al. Activation of nrf2/ho-1 pathway by nardochinoid c inhibits inflammation and oxidative stress in lipopolysaccharide-stimulated macrophages. Front. Pharmacol. 2018, 9, 911. [Google Scholar] [CrossRef]
- Lee, T.S.; Chau, L.Y. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 2002, 8, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Peri, F.; Calabrese, V. Toll-like receptor 4 (tlr4) modulation by synthetic and natural compounds: An update. J. Med. Chem. 2014, 57, 3612–3622. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.; Wang, W. Toll-like receptor 4 agonist, lipopolysaccharide, increases the expression levels of cytokines and chemokines in human peripheral blood mononuclear cells. Exp. Ther. Med. 2014, 8, 1914–1918. [Google Scholar] [CrossRef] [PubMed]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing nrf2/are/prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Seibert, K.; Masferrer, J.L. Role of inducible cyclooxygenase (cox-2) in inflammation. Receptor 1994, 4, 17–23. [Google Scholar] [PubMed]
- Qi, T.; Xu, F.; Yan, X.; Li, S.; Li, H. Sulforaphane exerts anti-inflammatory effects against lipopolysaccharide-induced acute lung injury in mice through the nrf2/are pathway. Int. J. Mol. Med. 2016, 37, 182–188. [Google Scholar] [CrossRef]
- Kaminska, B. Mapk signalling pathways as molecular targets for anti-inflammatory therapy--from molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta 2005, 1754, 253–262. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, J.; Fang, L.; Li, X.; Zhao, Y.; Shi, W.; An, L. Neuroprotective effects of sulforaphane on cholinergic neurons in mice with alzheimer’s disease-like lesions. Int. J. Mol. Sci. 2014, 15, 14396–14410. [Google Scholar] [CrossRef]
- Sun, J.; Nan, G. The mitogen-activated protein kinase (mapk) signaling pathway as a discovery target in stroke. J. Mol. Neurosci. 2016, 59, 90–98. [Google Scholar] [CrossRef]
- Jeong, Y.H.; Hyun, J.W.; Kim Van Le, T.; Kim, D.H.; Kim, H.S. Kalopanaxsaponin a exerts anti-inflammatory effects in lipopolysaccharide-stimulated microglia via inhibition of jnk and nf-kappab/ap-1 pathways. Biomol. Ther. 2013, 21, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Venkatesan, R.; Kim, S.Y. Neuroprotective and anti-inflammatory activities of allyl isothiocyanate through attenuation of jnk/nf-kappab/tnf-alpha signaling. Int. J. Mol. Sci. 2017, 18. [Google Scholar]
- Bohush, A.; Niewiadomska, G.; Filipek, A. Role of mitogen activated protein kinase signaling in parkinson’s disease. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of mapk signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nan, G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target (review). Int. J. Mol. Med. 2017, 39, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.N.; Huang, H.P.; Wang, C.J.; Liu, K.L.; Lii, C.K. Sulforaphane inhibits tnf-alpha-induced adhesion molecule expression through the rho a/rock/nf-kappab signaling pathway. J. Med. Food 2014, 17, 1095–1102. [Google Scholar] [CrossRef]
- Dickinson, S.E.; Melton, T.F.; Olson, E.R.; Zhang, J.; Saboda, K.; Bowden, G.T. Inhibition of activator protein-1 by sulforaphane involves interaction with cysteine in the cfos DNA-binding domain: Implications for chemoprevention of uvb-induced skin cancer. Cancer Res. 2009, 69, 7103–7110. [Google Scholar] [CrossRef]
- Karin, M. The regulation of ap-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef]
- Monje, P.; Hernandez-Losa, J.; Lyons, R.J.; Castellone, M.D.; Gutkind, J.S. Regulation of the transcriptional activity of c-fos by erk. A novel role for the prolyl isomerase pin1. J. Biol. Chem. 2005, 280, 35081–35084. [Google Scholar] [CrossRef]
- Ishikawa, T.; Morris, P.L. Interleukin-1beta signals through a c-Jun N-terminal kinase-dependent inducible nitric oxide synthase and nitric oxide production pathway in Sertoli epithelial cells. Endocrinology 2006, 147, 5424–5430. [Google Scholar] [CrossRef]
- Ventura, J.J.; Cogswell, P.; Flavell, R.A.; Baldwin, A.S., Jr.; Davis, R.J. Jnk potentiates tnf-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004, 18, 2905–2915. [Google Scholar] [CrossRef] [PubMed]
- Neniskyte, U.; Vilalta, A.; Brown, G.C. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014, 588, 2952–2956. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, S.; Ji, H.; Zhang, Z.; Chen, J.; Tan, Y.; Wintergerst, K.; Zheng, Y.; Sun, J.; Cai, L. Broccoli sprout extract prevents diabetic cardiomyopathy via nrf2 activation in db/db t2dm mice. Sci. Rep. 2016, 6, 30252. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subedi, L.; Lee, J.H.; Yumnam, S.; Ji, E.; Kim, S.Y. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-κB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8, 194. https://doi.org/10.3390/cells8020194
Subedi L, Lee JH, Yumnam S, Ji E, Kim SY. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-κB Inhibition and Nrf2/HO-1 Activation. Cells. 2019; 8(2):194. https://doi.org/10.3390/cells8020194
Chicago/Turabian StyleSubedi, Lalita, Jae Hyuk Lee, Silvia Yumnam, Eunhee Ji, and Sun Yeou Kim. 2019. "Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-κB Inhibition and Nrf2/HO-1 Activation" Cells 8, no. 2: 194. https://doi.org/10.3390/cells8020194
APA StyleSubedi, L., Lee, J. H., Yumnam, S., Ji, E., & Kim, S. Y. (2019). Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-κB Inhibition and Nrf2/HO-1 Activation. Cells, 8(2), 194. https://doi.org/10.3390/cells8020194