Cellular and Molecular Therapeutic Targets in Inflammatory Bowel Disease—Focusing on Intestinal Barrier Function


Abstract
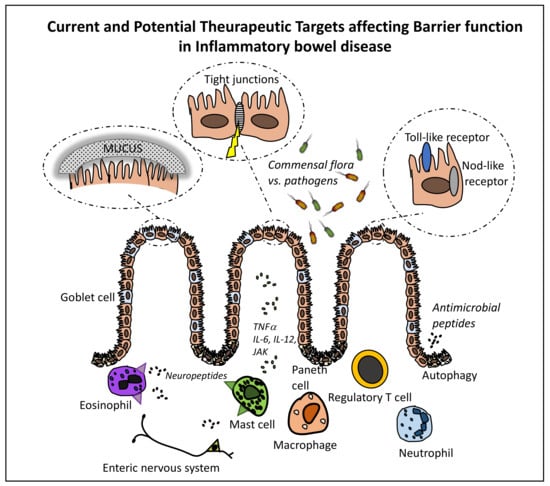
{kind=link}
{kind=link}
{kind=link}
1. Introduction
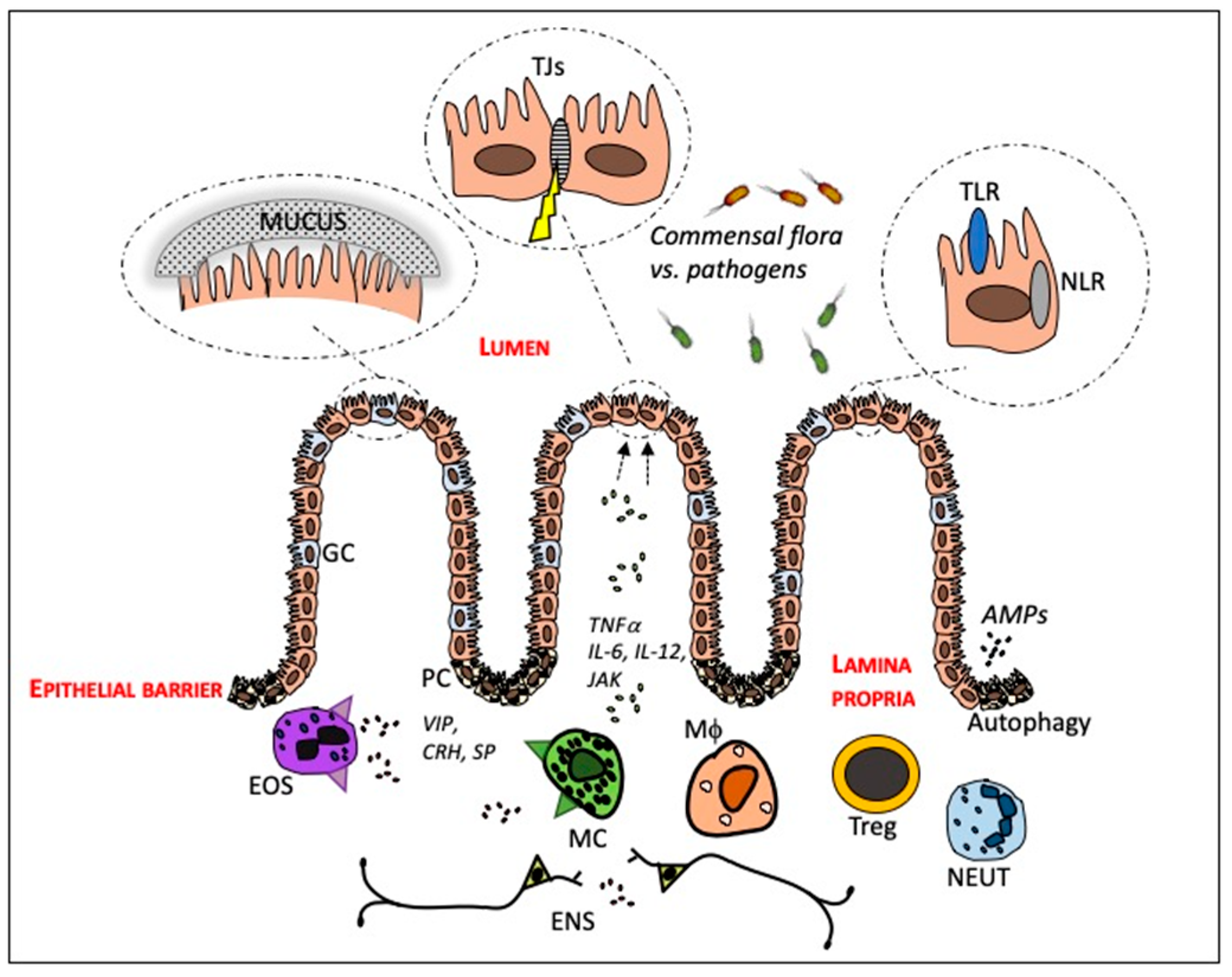
2. The Intestinal Mucosa—In Health and in IBD
2.1. The Crosstalk between the Intestinal Epithelium and Gut Microbiota
2.2. Innate and Adaptive Immune Cells
2.2.1. Paneth Cells
2.2.2. Neutrophils
2.2.3. T Regulatory Cells
2.2.4. Macrophages
2.2.5. Mast Cells
2.2.6. Eosinophils
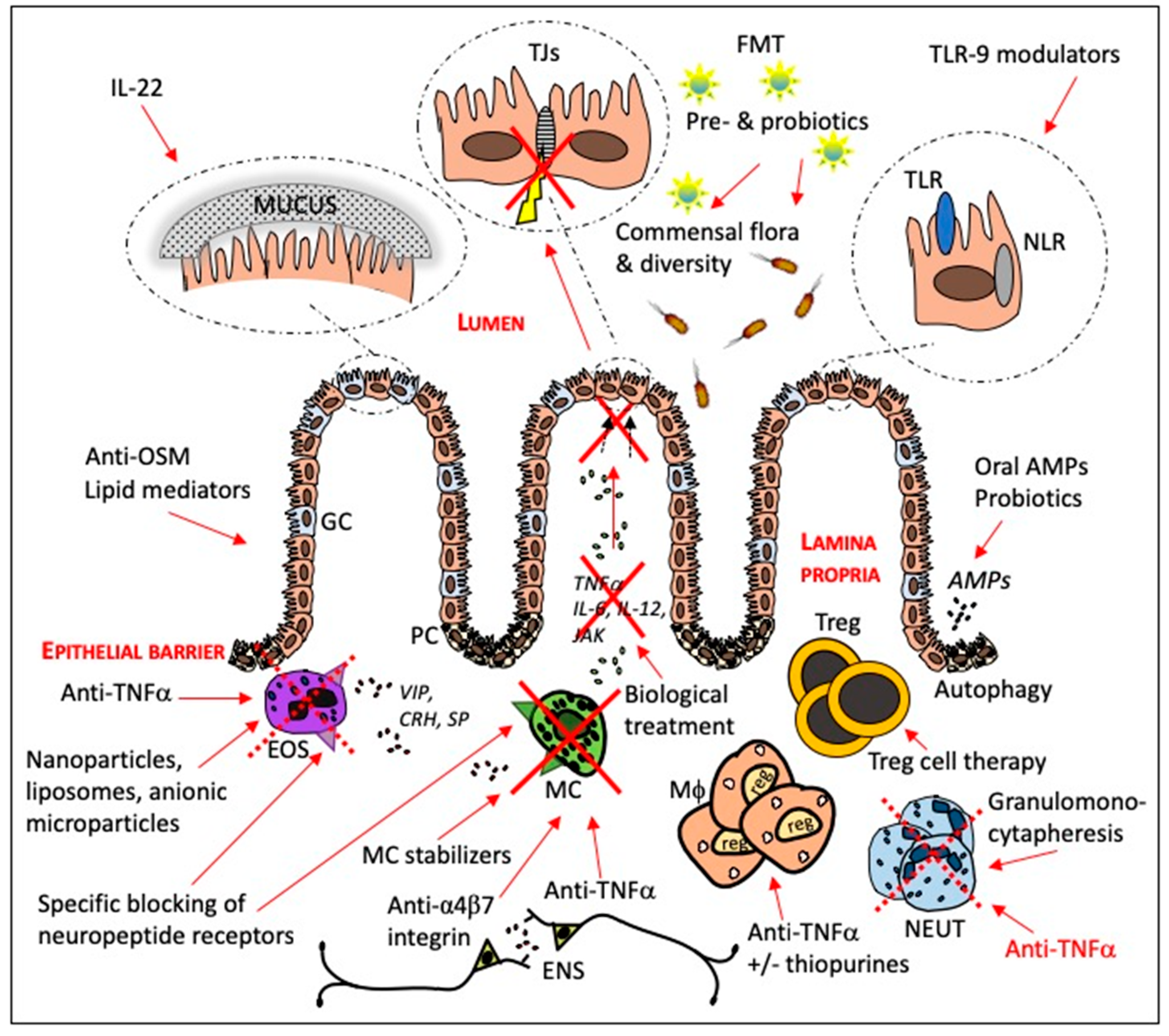
3. Current and Potential Targets for IBD Therapy
3.1. Targeting Pro-Inflammatory Pathways
3.1.1. Antibodies against Anti-TNFα
3.1.2. Targeting the Pro-inflammatory Cytokine IL-22
3.1.3. Anti-IL-6 Treatment
3.1.4. Lipid Mediators as a Therapeutic Approach in IBD
3.2. Manipulation of the Intestinal Microbiota
3.3. Neutrophils as Targets
3.4. The Use of Treg Cell Therapy
3.5. Macrophages as Therapeutic Targets
3.6. Mast Cells as Therapeutic Targets
3.7. Eosinophils as Therapeutic Targets
4. Conclusions
Funding
Conflicts of Interest
References
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Schoultz, I.; Söderholm, J.D.; McKay, D.M. Is metabolic stress a common denominator in inflammatory bowel disease? Inflamm. Bowel Dis. 2011, 17, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Michielan, A.; D’Incà, R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediat. Inflamm. 2015, 2015, 628157. [Google Scholar] [CrossRef] [PubMed]
- Neut, C.; Bulois, P.; Desreumaux, P.; Membré, J.-M.; Lederman, E.; Gambiez, L.; Cortot, A.; Quandalle, P.; van Kruiningen, H.; Colombel, J.-F. Changes in the bacterial flora of the neoterminal ileum after ileocolonic resection for Crohn’s disease. Am. J. Gastroenterol. 2002, 97, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory Bowel Disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; Devinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm. Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef]
- Natividad, J.M.M.; Verdu, E.F. Modulation of intestinal barrier by intestinal microbiota: Pathological and therapeutic implications. Pharmacol. Res. 2013, 69, 42–51. [Google Scholar] [CrossRef]
- Hayes, C.L.; Dong, J.; Galipeau, H.J.; Jury, J.; McCarville, J.; Huang, X.; Wang, X.-Y.; Naidoo, A.; Anbazhagan, A.N.; Libertucci, J.; et al. Commensal microbiota induces colonic barrier structure and functions that contribute to homeostasis. Sci. Rep. 2018, 8, 14184. [Google Scholar] [CrossRef]
- Cornick, S.; Tawiah, A.; Chadee, K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers 2015, 3, e982426. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.F.; Fisher, R.E.; Everett, M.L.; Thomas, A.D.; Bollinger, R.R.; Parker, W. Comparative anatomy and phylogenetic distribution of the mammalian cecal appendix. J. Evol. Biol. 2009, 22, 1984–1999. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Ho, S.B. Intestinal goblet cells and mucins in health and disease: Recent insights and progress. Curr. Gastroenterol. Rep. 2010, 12, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef]
- Bull, M.J.; Plummer, N.T. Part 1: The Human Gut Microbiome in Health and Disease. Integr. Med. (Encinitas) 2014, 13, 17–22. [Google Scholar] [PubMed]
- Schäffler, H.; Herlemann, D.P.R.; Alberts, C.; Kaschitzki, A.; Bodammer, P.; Bannert, K.; Köller, T.; Warnke, P.; Kreikemeyer, B.; Lamprecht, G. Mucosa-attached bacterial community in Crohn’s disease coheres with the clinical disease activity index. Environ. Microbiol. Rep. 2016, 8, 614–621. [Google Scholar] [CrossRef]
- Shawki, A.; McCole, D.F. Mechanisms of Intestinal Epithelial Barrier Dysfunction by Adherent-Invasive Escherichia coli. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 41–50. [Google Scholar] [CrossRef]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef]
- Willing, B.P.; Dicksved, J.; Halfvarson, J.; Andersson, A.F.; Lucio, M.; Zheng, Z.; Järnerot, G.; Tysk, C.; Jansson, J.K.; Engstrand, L. A Pyrosequencing Study in Twins Shows That Gastrointestinal Microbial Profiles Vary with Inflammatory Bowel Disease Phenotypes. Gastroenterology 2010, 139, 1844–1854.e1. [Google Scholar] [CrossRef]
- Kyo, K.; Muto, T.; Nagawa, H.; Lathrop, G.M.; Nakamura, Y. Associations of distinct variants of the intestinal mucin gene MUC3A with ulcerative colitis and Crohn’s disease. J. Hum. Genet. 2001, 46, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Seksik, P.; Rigottier-Gois, L.; Gramet, G.; Sutren, M.; Pochart, P.; Marteau, P.; Jian, R.; Doré, J. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 2003, 52, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermudez-Humaran, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Bach Knudsen, K.E.; Lærke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Gundelund Nielsen, D.S.; Theil, P.K.; Purup, S.; Hald, S.; Schioldan, A.G.; et al. Impact of Diet-Modulated Butyrate Production on Intestinal Barrier Function and Inflammation. Nutrients 2018, 10, 1499. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; He, Z.; Chen, W.; Holzman, I.R.; Lin, J. Effects of butyrate on intestinal barrier function in a Caco-2 cell monolayer model of intestinal barrier. Pediatr. Res. 2007, 61, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.Y.; Inohara, N.; Nuñez, G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 2017, 10, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef]
- Sansonetti, P.J. The innate signaling of dangers and the dangers of innate signaling. Nat. Immunol. 2006, 7, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.M.; Rossi, O.; Meijerink, M.; van Baarlen, P. Epithelial crosstalk at the microbiota-mucosal interface. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4607–4614. [Google Scholar] [CrossRef]
- Boyapati, R.K.; Rossi, A.G.; Satsangi, J.; Ho, G.-T. Gut mucosal DAMPs in IBD: From mechanisms to therapeutic implications. Mucosal Immunol. 2016, 9, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Al Nabhani, Z.; Dietrich, G.; Hugot, J.-P.; Barreau, F. Nod2: The intestinal gate keeper. PLoS Pathog. 2017, 13, e1006177. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Zeng, B.; Zhi, F.-C. Regulation of human enteric α-defensins by NOD2 in the Paneth cell lineage. Eur. J. Cell Biol. 2015, 94, 60–66. [Google Scholar] [CrossRef]
- Wallace, K.L.; Zheng, L.-B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef]
- Török, H.-P.; Glas, J.; Tonenchi, L.; Bruennler, G.; Folwaczny, M.; Folwaczny, C. Crohn’s disease is associated with a toll-like receptor-9 polymorphism. Gastroenterology 2004, 127, 365–366. [Google Scholar] [CrossRef]
- Saruta, M.; Targan, S.R.; Mei, L.; Ippoliti, A.F.; Taylor, K.D.; Rotter, J.I. High-frequency haplotypes in the X chromosome locus TLR8 are associated with both CD and UC in females. Inflamm. Bowel Dis. 2009, 15, 321–327. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, M.; De Koning, B.A.E.; De Bruijn, A.C.J.M.; Velcich, A.; Meijerink, J.P.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Mitoma, H.; Harashima, S.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-alpha: Structure, function and interaction with anti-TNF agents. Rheumatology (Oxford) 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Reimund, J.M.; Wittersheim, C.; Dumont, S.; Muller, C.D.; Kenney, J.S.; Baumann, R.; Poindron, P.; Duclos, B. Increased production of tumour necrosis factor-alpha interleukin-1 beta, and interleukin-6 by morphologically normal intestinal biopsies from patients with Crohn’s disease. Gut 1996, 39, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.-D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Garcia-Carbonell, R.; Wong, J.; Kim, J.Y.; Close, L.A.; Boland, B.S.; Wong, T.L.; Harris, P.A.; Ho, S.B.; Das, S.; Ernst, P.B.; et al. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc. Natl. Acad. Sci. USA 2018, 115, E9192–E9200. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Lichtenstein, G.R. Comprehensive review: Antitumor necrosis factor agents in inflammatory bowel disease and factors implicated in treatment response. Therap. Adv. Gastroenterol. 2013, 6, 269–293. [Google Scholar] [CrossRef]
- Li, L.-J.; Gong, C.; Zhao, M.-H.; Feng, B.-S. Role of interleukin-22 in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 18177–18188. [Google Scholar] [CrossRef]
- Ngo, V.L.; Abo, H.; Maxim, E.; Harusato, A.; Geem, D.; Medina-Contreras, O.; Merlin, D.; Gewirtz, A.T.; Nusrat, A.; Denning, T.L. A cytokine network involving IL-36γ, IL-23, and IL-22 promotes antimicrobial defense and recovery from intestinal barrier damage. Proc. Natl. Acad. Sci. USA 2018, 115, E5076–E5085. [Google Scholar] [CrossRef]
- Stoll, M.; Corneliussen, B.; Costello, C.M.; Waetzig, G.H.; Mellgard, B.; Koch, W.A.; Rosenstiel, P.; Albrecht, M.; Croucher, P.J.P.; Seegert, D.; et al. Genetic variation in DLG5 is associated with inflammatory bowel disease. Nat. Genet. 2004, 36, 476–480. [Google Scholar] [CrossRef]
- Peltekova, V.D.; Wintle, R.F.; Rubin, L.A.; Amos, C.I.; Huang, Q.; Gu, X.; Newman, B.; Van Oene, M.; Cescon, D.; Greenberg, G.; et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat. Genet. 2004, 36, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.-E.; Guan, R.; Song, Y.-T. The association of DLG5 polymorphisms with inflammatory bowel disease: A meta-analysis of 25 studies. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2324–2337. [Google Scholar] [PubMed]
- Girardin, M.; Dionne, S.; Goyette, P.; Rioux, J.; Bitton, A.; Elimrani, I.; Charlebois, P.; Qureshi, I.; Levy, E.; Seidman, E.G. Expression and functional analysis of intestinal organic cation/L-carnitine transporter (OCTN) in Crohn’s disease. J. Crohns Colitis 2012, 6, 189–197. [Google Scholar] [CrossRef]
- McCole, D.F. IBD candidate genes and intestinal barrier regulation. Inflamm. Bowel Dis. 2014, 20, 1829–1849. [Google Scholar] [CrossRef] [PubMed]
- Danoy, P.; Pryce, K.; Hadler, J.; Bradbury, L.A.; Farrar, C.; Pointon, J.; Australo-Anglo-American Spondyloarthritis Consortium; Ward, M.; Weisman, M.; Reveille, J.D.; et al. Association of variants at 1q32 and STAT3 with ankylosing spondylitis suggests genetic overlap with Crohn’s disease. PLoS Genet. 2010, 6, e1001195. [Google Scholar] [CrossRef] [PubMed]
- Satsangi, J.; Parkes, M.; Louis, E.; Hashimoto, L.; Kato, N.; Welsh, K.; Terwilliger, J.D.; Lathrop, G.M.; Bell, J.I.; Jewell, D.P. Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat. Genet. 1996, 14, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Tysk, C.; Riedesel, H.; Lindberg, E.; Panzini, B.; Podolsky, D.; Järnerot, G. Colonic glycoproteins in monozygotic twins with inflammatory bowel disease. Gastroenterology 1991, 100, 419–423. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef]
- UK IBD Genetics Consortium; Barrett, J.C.; Lee, J.C.; Lees, C.W.; Prescott, N.J.; Anderson, C.A.; Phillips, A.; Wesley, E.; Parnell, K.; Zhang, H.; et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet. 2009, 41, 1330–1334. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.-H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.S.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Muise, A.M.; Walters, T.D.; Glowacka, W.K.; Griffiths, A.M.; Ngan, B.-Y.; Lan, H.; Xu, W.; Silverberg, M.S.; Rotin, D. Polymorphisms in E-cadherin (CDH1) result in a mis-localised cytoplasmic protein that is associated with Crohn’s disease. Gut 2009, 58, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, M.S.; Cho, J.H.; Rioux, J.D.; McGovern, D.P.B.; Wu, J.; Annese, V.; Achkar, J.-P.; Goyette, P.; Scott, R.; Xu, W.; et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat. Genet. 2009, 41, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, V.; Nakata, T.; Desch, A.N.; Lévesque, C.; Boroughs, A.; Guzman, G.; Cao, Z.; Creasey, E.; Yao, J.; Boucher, G.; et al. C1orf106 is a colitis risk gene that regulates stability of epithelial adherens junctions. Science 2018, 359, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Görtz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Philip Schumm, L.; Sharma, Y.; Anderson, C.A.; et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef]
- Pothoven, K.L.; Schleimer, R.P. The barrier hypothesis and Oncostatin M: Restoration of epithelial barrier function as a novel therapeutic strategy for the treatment of type 2 inflammatory disease. Tissue Barriers 2017, 5, e1341367. [Google Scholar] [CrossRef]
- Dubuquoy, L.; Jansson, E.A.; Deeb, S.; Rakotobe, S.; Karoui, M.; Colombel, J.-F.; Auwerx, J.; Pettersson, S.; Desreumaux, P. Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology 2003, 124, 1265–1276. [Google Scholar] [CrossRef]
- Da Silva, S.; Keita, Å.V.; Mohlin, S.; Påhlman, S.; Theodorou, V.; Påhlman, I.; Mattson, J.P.; Söderholm, J.D. A Novel Topical PPARγ Agonist Induces PPARγ Activity in Ulcerative Colitis Mucosa and Prevents and Reverses Inflammation in Induced Colitis Models. Inflamm. Bowel Dis. 2018, 24, 792–805. [Google Scholar] [CrossRef]
- Onnie, C.M.; Fisher, S.A.; Pattni, R.; Sanderson, J.; Forbes, A.; Lewis, C.M.; Mathew, C.G. Associations of allelic variants of the multidrug resistance gene (ABCB1 or MDR1) and inflammatory bowel disease and their effects on disease behavior: A case-control and meta-analysis study. Inflamm. Bowel Dis. 2006, 12, 263–271. [Google Scholar] [CrossRef]
- Wang, S.-L.; Shao, B.-Z.; Zhao, S.-B.; Fang, J.; Gu, L.; Miao, C.-Y.; Li, Z.-S.; Bai, Y. Impact of Paneth Cell Autophagy on Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 693. [Google Scholar] [CrossRef]
- Iida, T.; Onodera, K.; Nakase, H. Role of autophagy in the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2017, 23, 1944–1953. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Thachil, E.; Hugot, J.-P.; Arbeille, B.; Paris, R.; Grodet, A.; Peuchmaur, M.; Codogno, P.; Barreau, F.; Ogier-Denis, E.; Berrebi, D.; et al. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn’s disease. Gastroenterology 2012, 142, 1097–1099.e4. [Google Scholar] [CrossRef]
- Hosomi, S.; Kaser, A.; Blumberg, R.S. Role of endoplasmic reticulum stress and autophagy as interlinking pathways in the pathogenesis of inflammatory bowel disease. Curr. Opin. Gastroenterol. 2015, 31, 81–88. [Google Scholar] [CrossRef]
- Rosales, C.; Demaurex, N.; Lowell, C.A.; Uribe-Querol, E. Neutrophils: Their Role in Innate and Adaptive Immunity. J. Immunol. Res. 2016, 2016, 1469780. [Google Scholar] [CrossRef]
- Zhou, G.X.; Liu, Z.J. Potential roles of neutrophils in regulating intestinal mucosal inflammation of inflammatory bowel disease. J. Dig. Dis. 2017, 18, 495–503. [Google Scholar] [CrossRef]
- Tanaka, T.; Okanobu, H.; Yoshimi, S.; Murakami, E.; Kogame, A.; Imagawa, H.; Numata, Y.; Kuga, Y.; Moriya, T.; Ohya, T.; et al. In patients with ulcerative colitis, adsorptive depletion of granulocytes and monocytes impacts mucosal level of neutrophils and clinically is most effective in steroid naïve patients. Dig. Liver Dis. 2008, 40, 731–736. [Google Scholar] [CrossRef]
- Muthas, D.; Reznichenko, A.; Balendran, C.A.; Böttcher, G.; Clausen, I.G.; Kärrman Mårdh, C.; Ottosson, T.; Uddin, M.; MacDonald, T.T.; Danese, S.; et al. Neutrophils in ulcerative colitis: A review of selected biomarkers and their potential therapeutic implications. Scand. J. Gastroenterol. 2017, 52, 125–135. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Mayne, C.G.; Williams, C.B. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 1772–1788. [Google Scholar] [CrossRef]
- Saleh, M.; Elson, C.O. Experimental inflammatory bowel disease: Insights into the host-microbiota dialog. Immunity 2011, 34, 293–302. [Google Scholar] [CrossRef]
- Maul, J.; Loddenkemper, C.; Mundt, P.; Berg, E.; Giese, T.; Stallmach, A.; Zeitz, M.; Duchmann, R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 2005, 128, 1868–1878. [Google Scholar] [CrossRef]
- Takahashi, M.; Nakamura, K.; Honda, K.; Kitamura, Y.; Mizutani, T.; Araki, Y.; Kabemura, T.; Chijiiwa, Y.; Harada, N.; Nawata, H. An inverse correlation of human peripheral blood regulatory T cell frequency with the disease activity of ulcerative colitis. Dig. Dis. Sci. 2006, 51, 677–686. [Google Scholar] [CrossRef]
- Saruta, M.; Yu, Q.T.; Fleshner, P.R.; Mantel, P.-Y.; Schmidt-Weber, C.B.; Banham, A.H.; Papadakis, K.A. Characterization of FOXP3+CD4+ regulatory T cells in Crohn’s disease. Clin. Immunol. 2007, 125, 281–290. [Google Scholar] [CrossRef]
- Okabe, Y.; Medzhitov, R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 2014, 157, 832–844. [Google Scholar] [CrossRef]
- Gabanyi, I.; Muller, P.A.; Feighery, L.; Oliveira, T.Y.; Costa-Pinto, F.A.; Mucida, D. Neuro-immune Interactions Drive Tissue Programming in Intestinal Macrophages. Cell 2016, 164, 378–391. [Google Scholar] [CrossRef]
- Man, A.L.; Gicheva, N.; Regoli, M.; Rowley, G.; De Cunto, G.; Wellner, N.; Bassity, E.; Gulisano, M.; Bertelli, E.; Nicoletti, C. CX3CR1+ Cell-Mediated Salmonella Exclusion Protects the Intestinal Mucosa during the Initial Stage of Infection. J. Immunol. 2017, 198, 335–343. [Google Scholar] [CrossRef]
- Rivollier, A.; He, J.; Kole, A.; Valatas, V.; Kelsall, B.L. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J. Exp. Med. 2012, 209, 139–155. [Google Scholar] [CrossRef]
- Hadis, U.; Wahl, B.; Schulz, O.; Hardtke-Wolenski, M.; Schippers, A.; Wagner, N.; Müller, W.; Sparwasser, T.; Förster, R.; Pabst, O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 2011, 34, 237–246. [Google Scholar] [CrossRef]
- Al-Ghadban, S.; Kaissi, S.; Homaidan, F.R.; Naim, H.Y.; El-Sabban, M.E. Cross-talk between intestinal epithelial cells and immune cells in inflammatory bowel disease. Sci. Rep. 2016, 6, 29783. [Google Scholar] [CrossRef]
- Smythies, L.E.; Sellers, M.; Clements, R.H.; Mosteller-Barnum, M.; Meng, G.; Benjamin, W.H.; Orenstein, J.M.; Smith, P.D. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Investig. 2005, 115, 66–75. [Google Scholar] [CrossRef]
- Takada, Y.; Hisamatsu, T.; Kamada, N.; Kitazume, M.T.; Honda, H.; Oshima, Y.; Saito, R.; Takayama, T.; Kobayashi, T.; Chinen, H.; et al. Monocyte chemoattractant protein-1 contributes to gut homeostasis and intestinal inflammation by composition of IL-10-producing regulatory macrophage subset. J. Immunol. 2010, 184, 2671–2676. [Google Scholar] [CrossRef]
- Zigmond, E.; Varol, C.; Farache, J.; Elmaliah, E.; Satpathy, A.T.; Friedlander, G.; Mack, M.; Shpigel, N.; Boneca, I.G.; Murphy, K.M.; et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity 2012, 37, 1076–1090. [Google Scholar] [CrossRef]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef]
- De Winter, B.Y.; van den Wijngaard, R.M.; de Jonge, W.J. Intestinal mast cells in gut inflammation and motility disturbances. Biochim. Biophys. Acta 2012, 1822, 66–73. [Google Scholar] [CrossRef]
- Stead, R.H. Innervation of mucosal immune cells in the gastrointestinal tract. Reg. Immunol. 1992, 4, 91–99. [Google Scholar]
- Bienenstock, J.; Tomioka, M.; Matsuda, H.; Stead, R.H.; Quinonez, G.; Simon, G.T.; Coughlin, M.D.; Denburg, J.A. The role of mast cells in inflammatory processes: Evidence for nerve/mast cell interactions. Int. Arch. Allergy Appl. Immunol. 1987, 82, 238–243. [Google Scholar] [CrossRef]
- He, S.-H. Key role of mast cells and their major secretory products in inflammatory bowel disease. World J. Gastroenterol. 2004, 10, 309–318. [Google Scholar] [CrossRef]
- Santos, J.; Yang, P.C.; Söderholm, J.D.; Benjamin, M.; Perdue, M.H. Role of mast cells in chronic stress induced colonic epithelial barrier dysfunction in the rat. Gut 2001, 48, 630–636. [Google Scholar] [CrossRef]
- Keita, A.V.; Carlsson, A.H.; Cigéhn, M.; Ericson, A.-C.; McKay, D.M.; Söderholm, J.D. Vasoactive intestinal polypeptide regulates barrier function via mast cells in human intestinal follicle-associated epithelium and during stress in rats. Neurogastroenterol. Motil. 2013, 25, e406–e417. [Google Scholar] [CrossRef]
- Keita, A.V.; Söderholm, J.D.; Ericson, A.-C. Stress-induced barrier disruption of rat follicle-associated epithelium involves corticotropin-releasing hormone, acetylcholine, substance P, and mast cells. Neurogastroenterol. Motil. 2010, 770–778, e221–e222. [Google Scholar] [CrossRef]
- Casado-Bedmar, M.; Heil, S.D.S.; Myrelid, P.; Söderholm, J.D.; Keita, Å.V. Upregulation of intestinal mucosal mast cells expressing VPAC1 in close proximity to vasoactive intestinal polypeptide in inflammatory bowel disease and murine colitis. Neurogastroenterol. Motil. 2018, e13503. [Google Scholar] [CrossRef]
- Wallon, C.; Persborn, M.; Jönsson, M.; Wang, A.; Phan, V.; Lampinen, M.; Vicario, M.; Santos, J.; Sherman, P.M.; Carlson, M.; et al. Eosinophils express muscarinic receptors and corticotropin-releasing factor to disrupt the mucosal barrier in ulcerative colitis. Gastroenterology 2011, 140, 1597–1607. [Google Scholar] [CrossRef]
- Winterkamp, S.; Raithel, M.; Hahn, E.G. Secretion and tissue content of eosinophil cationic protein in Crohn’s disease. J. Clin. Gastroenterol. 2000, 30, 170–175. [Google Scholar] [CrossRef]
- Rothenberg, M.E.; Mishra, A.; Brandt, E.B.; Hogan, S.P. Gastrointestinal eosinophils. Immunol. Rev. 2001, 179, 139–155. [Google Scholar] [CrossRef]
- Furuta, G.T.; Nieuwenhuis, E.E.S.; Karhausen, J.; Gleich, G.; Blumberg, R.S.; Lee, J.J.; Ackerman, S.J. Eosinophils alter colonic epithelial barrier function: Role for major basic protein. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G890–G897. [Google Scholar] [CrossRef]
- Lampinen, M.; Rönnblom, A.; Amin, K.; Kristjansson, G.; Rorsman, F.; Sangfelt, P.; Säfsten, B.; Wagner, M.; Wanders, A.; Winqvist, O.; et al. Eosinophil granulocytes are activated during the remission phase of ulcerative colitis. Gut 2005, 54, 1714–1720. [Google Scholar] [CrossRef]
- Zheng, P.-Y.; Feng, B.-S.; Oluwole, C.; Struiksma, S.; Chen, X.; Li, P.; Tang, S.-G.; Yang, P.-C. Psychological stress induces eosinophils to produce corticotrophin releasing hormone in the intestine. Gut 2009, 58, 1473–1479. [Google Scholar] [CrossRef]
- Vergnolle, N. Protease inhibition as new therapeutic strategy for GI diseases. Gut 2016, 65, 1215–1224. [Google Scholar] [CrossRef]
- Neurath, H. Evolution of proteolytic enzymes. Science 1984, 224, 350–357. [Google Scholar] [CrossRef]
- Stack, W.A.; Mann, S.D.; Roy, A.J.; Heath, P.; Sopwith, M.; Freeman, J.; Holmes, G.; Long, R.; Forbes, A.; Kamm, M.A. Randomised controlled trial of CDP571 antibody to tumour necrosis factor-alpha in Crohn’s disease. Lancet 1997, 349, 521–524. [Google Scholar] [CrossRef]
- Targan, S.R.; Hanauer, S.B.; van Deventer, S.J.; Mayer, L.; Present, D.H.; Braakman, T.; DeWoody, K.L.; Schaible, T.F.; Rutgeerts, P.J. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N. Engl. J. Med. 1997, 337, 1029–1035. [Google Scholar] [CrossRef]
- Chudy-Onwugaje, K.O.; Christian, K.E.; Farraye, F.A.; Cross, R.K. A State-of-the-Art Review of New and Emerging Therapies for the Treatment of IBD. Inflamm. Bowel Dis. 2018. [Google Scholar] [CrossRef]
- Holleran, G.; Lopetuso, L.; Petito, V.; Graziani, C.; Ianiro, G.; McNamara, D.; Gasbarrini, A.; Scaldaferri, F. The Innate and Adaptive Immune System as Targets for Biologic Therapies in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2017, 18, 2020. [Google Scholar] [CrossRef]
- Olesen, C.M.; Coskun, M.; Peyrin-Biroulet, L.; Nielsen, O.H. Mechanisms behind efficacy of tumor necrosis factor inhibitors in inflammatory bowel diseases. Pharmacol. Ther. 2016, 159, 110–119. [Google Scholar] [CrossRef]
- Koch, S.; Nusrat, A. The life and death of epithelia during inflammation: Lessons learned from the gut. Annu. Rev. Pathol. 2012, 7, 35–60. [Google Scholar] [CrossRef]
- Yakymenko, O.; Schoultz, I.; Gullberg, E.; Ström, M.; Almer, S.; Wallon, C.; Wang, A.; Keita, Å.V.; Campbell, B.J.; McKay, D.M.; et al. Infliximab restores colonic barrier to adherent-invasive E. coli in Crohn’s disease via effects on epithelial lipid rafts. Scand. J. Gastroenterol. 2018, 53, 677–684. [Google Scholar] [CrossRef]
- Brand, S.; Beigel, F.; Olszak, T.; Zitzmann, K.; Eichhorst, S.T.; Otte, J.-M.; Diepolder, H.; Marquardt, A.; Jagla, W.; Popp, A.; et al. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G827–G838. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Flavell, R.A. Recent advances in IL-22 biology. Int. Immunol. 2011, 23, 159–163. [Google Scholar] [CrossRef]
- Mizoguchi, A. Healing of intestinal inflammation by IL-22. Inflamm. Bowel Dis. 2012, 18, 1777–1784. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef]
- Andoh, A.; Shioya, M.; Nishida, A.; Bamba, S.; Tsujikawa, T.; Kim-Mitsuyama, S.; Fujiyama, Y. Expression of IL-24, an activator of the JAK1/STAT3/SOCS3 cascade, is enhanced in inflammatory bowel disease. J. Immunol. 2009, 183, 687–695. [Google Scholar] [CrossRef]
- Begue, B.; Verdier, J.; Rieux-Laucat, F.; Goulet, O.; Morali, A.; Canioni, D.; Hugot, J.-P.; Daussy, C.; Verkarre, V.; Pigneur, B.; et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am. J. Gastroenterol. 2011, 106, 1544–1555. [Google Scholar] [CrossRef]
- Hyams, J.S.; Fitzgerald, J.E.; Treem, W.R.; Wyzga, N.; Kreutzer, D.L. Relationship of functional and antigenic interleukin 6 to disease activity in inflammatory bowel disease. Gastroenterology 1993, 104, 1285–1292. [Google Scholar] [CrossRef]
- Umehara, Y.; Kudo, M.; Nakaoka, R.; Kawasaki, T.; Shiomi, M. Serum proinflammatory cytokines and adhesion molecules in ulcerative colitis. Hepatogastroenterology 2006, 53, 879–882. [Google Scholar]
- Helbling, R.; Nyddeger, A.; Angelini, F.; Von Scheven Gete, A.; Hofer, M. P03-024—Early onset IBD treated by tocilizumab. Pediatr. Rheumatol. 2013, 11, A222. [Google Scholar] [CrossRef]
- Szeto, M.C.H.; Yalçın, M.D.; Khan, A.; Piotrowicz, A. Successful Use of Tocilizumab in a Patient with Coexisting Rheumatoid Arthritis and Ulcerative Colitis. Case Rep. Immunol. 2016, 2016, 7562123. [Google Scholar] [CrossRef]
- Shipman, L. Rheumatoid arthritis: Tocilizumab and the risk of intestinal perforation. Nat. Rev. Rheumatol. 2016, 12, 499. [Google Scholar] [CrossRef]
- Xu, Z.; Bouman-Thio, E.; Comisar, C.; Frederick, B.; Van Hartingsveldt, B.; Marini, J.C.; Davis, H.M.; Zhou, H. Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br. J. Clin. Pharmacol. 2011, 72, 270–281. [Google Scholar] [CrossRef]
- Khanna, R.; Bressler, B.; Levesque, B.G.; Zou, G.; Stitt, L.W.; Greenberg, G.R.; Panaccione, R.; Bitton, A.; Paré, P.; Vermeire, S.; et al. Early combined immunosuppression for the management of Crohn’s disease (REACT): A cluster randomised controlled trial. Lancet 2015, 386, 1825–1834. [Google Scholar] [CrossRef]
- Ungaro, F.; Rubbino, F.; Danese, S.; D’Alessio, S. Actors and Factors in the Resolution of Intestinal Inflammation: Lipid Mediators as a New Approach to Therapy in Inflammatory Bowel Diseases. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front Immunol 2017, 8. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215. [Google Scholar] [CrossRef]
- Headland, S.E.; Norling, L.V. The resolution of inflammation: Principles and challenges. Semin. Immunol. 2015, 27, 149–160. [Google Scholar] [CrossRef]
- Serhan, C.N. Novel lipid mediators and resolution mechanisms in acute inflammation: To resolve or not? Am. J. Pathol. 2010, 177, 1576–1591. [Google Scholar] [CrossRef]
- Skulas-Ray, A.C. Omega-3 fatty acids and inflammation: A perspective on the challenges of evaluating efficacy in clinical research. Prostaglandins Other Lipid Mediat. 2015, 116–117, 104–111. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Simopoulos, A.P. An Increase in the Omega-6/Omega-3 Fatty Acid Ratio Increases the Risk for Obesity. Nutrients 2016, 8, 128. [Google Scholar] [CrossRef]
- Shinohara, M.; Serhan, C.N. Novel Endogenous Proresolving Molecules: Essential Fatty Acid-Derived and Gaseous Mediators in the Resolution of Inflammation. J. Atheroscler. Thromb. 2016, 23, 655–664. [Google Scholar] [CrossRef]
- Pearl, D.S.; Masoodi, M.; Eiden, M.; Brümmer, J.; Gullick, D.; McKeever, T.M.; Whittaker, M.A.; Nitch-Smith, H.; Brown, J.F.; Shute, J.K.; et al. Altered colonic mucosal availability of n-3 and n-6 polyunsaturated fatty acids in ulcerative colitis and the relationship to disease activity. J. Crohns Colitis 2014, 8, 70–79. [Google Scholar] [CrossRef]
- Masoodi, M.; Pearl, D.S.; Eiden, M.; Shute, J.K.; Brown, J.F.; Calder, P.C.; Trebble, T.M. Altered colonic mucosal Polyunsaturated Fatty Acid (PUFA) derived lipid mediators in ulcerative colitis: New insight into relationship with disease activity and pathophysiology. PLoS ONE 2013, 8, e76532. [Google Scholar] [CrossRef]
- Ungaro, F.; Tacconi, C.; Massimino, L.; Corsetto, P.A.; Correale, C.; Fonteyne, P.; Piontini, A.; Garzarelli, V.; Calcaterra, F.; Della Bella, S.; et al. MFSD2A Promotes Endothelial Generation of Inflammation-Resolving Lipid Mediators and Reduces Colitis in Mice. Gastroenterology 2017, 153, 1363–1377.e6. [Google Scholar] [CrossRef]
- McIlroy, J.; Ianiro, G.; Mukhopadhya, I.; Hansen, R.; Hold, G.L. Review article: The gut microbiome in inflammatory bowel disease-avenues for microbial management. Aliment. Pharmacol. Ther. 2018, 47, 26–42. [Google Scholar] [CrossRef]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.-F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef]
- La Fata, G.; Weber, P.; Mohajeri, M.H. Probiotics and the Gut Immune System: Indirect Regulation. Probiotics Antimicrob. Proteins 2018, 10, 11–21. [Google Scholar] [CrossRef]
- Bron, P.A.; Kleerebezem, M.; Brummer, R.-J.; Cani, P.D.; Mercenier, A.; MacDonald, T.T.; Garcia-Ródenas, C.L.; Wells, J.M. Can probiotics modulate human disease by impacting intestinal barrier function? Br. J. Nutr. 2017, 117, 93–107. [Google Scholar] [CrossRef]
- Ghouri, Y.A.; Richards, D.M.; Rahimi, E.F.; Krill, J.T.; Jelinek, K.A.; DuPont, A.W. Systematic review of randomized controlled trials of probiotics, prebiotics, and synbiotics in inflammatory bowel disease. Clin. Exp. Gastroenterol. 2014, 7, 473–487. [Google Scholar]
- Seo, E.; Weibel, S.; Wehkamp, J.; Oelschlaeger, T.A. Construction of recombinant E. coli Nissle 1917 (EcN) strains for the expression and secretion of defensins. Int. J. Med. Microbiol. 2012, 302, 276–287. [Google Scholar] [CrossRef]
- Möndel, M.; Schroeder, B.O.; Zimmermann, K.; Huber, H.; Nuding, S.; Beisner, J.; Fellermann, K.; Stange, E.F.; Wehkamp, J. Probiotic E. coli treatment mediates antimicrobial human beta-defensin synthesis and fecal excretion in humans. Mucosal Immunol. 2009, 2, 166–172. [Google Scholar] [CrossRef]
- Schlee, M.; Wehkamp, J.; Altenhoefer, A.; Oelschlaeger, T.A.; Stange, E.F.; Fellermann, K. Induction of human beta-defensin 2 by the probiotic Escherichia coli Nissle 1917 is mediated through flagellin. Infect. Immun. 2007, 75, 2399–2407. [Google Scholar] [CrossRef]
- Ukena, S.N.; Singh, A.; Dringenberg, U.; Engelhardt, R.; Seidler, U.; Hansen, W.; Bleich, A.; Bruder, D.; Franzke, A.; Rogler, G.; et al. Probiotic Escherichia coli Nissle 1917 inhibits leaky gut by enhancing mucosal integrity. PLoS ONE 2007, 2, e1308. [Google Scholar] [CrossRef]
- O’Toole, P.W.; Marchesi, J.R.; Hill, C. Next-generation probiotics: The spectrum from probiotics to live biotherapeutics. Nat. Microbiol. 2017, 2, 17057. [Google Scholar] [CrossRef]
- Duncan, S.H.; Hold, G.L.; Harmsen, H.J.M.; Stewart, C.S.; Flint, H.J. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 2141–2146. [Google Scholar]
- Carlsson, A.H.; Yakymenko, O.; Olivier, I.; Håkansson, F.; Postma, E.; Keita, A.V.; Söderholm, J.D. Faecalibacterium prausnitzii supernatant improves intestinal barrier function in mice DSS colitis. Scand. J. Gastroenterol. 2013, 48, 1136–1144. [Google Scholar] [CrossRef]
- Rajca, S.; Grondin, V.; Louis, E.; Vernier-Massouille, G.; Grimaud, J.-C.; Bouhnik, Y.; Laharie, D.; Dupas, J.-L.; Pillant, H.; Picon, L.; et al. Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 978–986. [Google Scholar]
- Ganda Mall, J.-P.; Casado-Bedmar, M.; Winberg, M.E.; Brummer, R.J.; Schoultz, I.; Keita, Å.V. A β-Glucan-Based Dietary Fiber Reduces Mast Cell-Induced Hyperpermeability in Ileum from Patients with Crohn’s Disease and Control Subjects. Inflamm. Bowel Dis. 2017, 24, 166–178. [Google Scholar] [CrossRef]
- Hvas, C.L.; Dige, A.; Bendix, M.; Wernlund, P.G.; Christensen, L.A.; Dahlerup, J.F.; Agnholt, J. Casein glycomacropeptide for active distal ulcerative colitis: A randomized pilot study. Eur. J. Clin. Investig. 2016, 46, 555–563. [Google Scholar] [CrossRef]
- Karner, M.; Kocjan, A.; Stein, J.; Schreiber, S.; von Boyen, G.; Uebel, P.; Schmidt, C.; Kupcinskas, L.; Dina, I.; Zuelch, F.; et al. First Multicenter Study of Modified Release Phosphatidylcholine “LT-02” in Ulcerative Colitis: A Randomized, Placebo-Controlled Trial in Mesalazine-Refractory Courses. Am. J. Gastroenterol. 2014, 109, 1041–1051. [Google Scholar] [CrossRef]
- Brandt, L.J.; Aroniadis, O.C.; Mellow, M.; Kanatzar, A.; Kelly, C.; Park, T.; Stollman, N.; Rohlke, F.; Surawicz, C. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am. J. Gastroenterol. 2012, 107, 1079–1087. [Google Scholar] [CrossRef]
- Rossen, N.G.; Fuentes, S.; van der Spek, M.J.; Tijssen, J.G.; Hartman, J.H.A.; Duflou, A.; Löwenberg, M.; van den Brink, G.R.; Mathus-Vliegen, E.M.H.; de Vos, W.M.; et al. Findings from a Randomized Controlled Trial of Fecal Transplantation for Patients with Ulcerative Colitis. Gastroenterology 2015, 149, 110–118.e4. [Google Scholar] [CrossRef]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients with Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109.e6. [Google Scholar] [CrossRef]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Kump, P.; Wurm, P.; Gröchenig, H.P.; Wenzl, H.; Petritsch, W.; Halwachs, B.; Wagner, M.; Stadlbauer, V.; Eherer, A.; Hoffmann, K.M.; et al. The taxonomic composition of the donor intestinal microbiota is a major factor influencing the efficacy of faecal microbiota transplantation in therapy refractory ulcerative colitis. Aliment. Pharmacol. Ther. 2018, 47, 67–77. [Google Scholar] [CrossRef]
- Colman, R.J.; Rubin, D.T. Fecal microbiota transplantation as therapy for inflammatory bowel disease: A systematic review and meta-analysis. J. Crohns Colitis 2014, 8, 1569–1581. [Google Scholar] [CrossRef]
- Imdad, A.; Nicholson, M.R.; Tanner-Smith, E.E.; Zackular, J.P.; Gomez-Duarte, O.G.; Beaulieu, D.B.; Acra, S. Fecal transplantation for treatment of inflammatory bowel disease. Cochrane Database Syst. Rev. 2018, 11, CD012774. [Google Scholar] [CrossRef]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Moltzau-Anderson, J.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients with Clostridium difficile Infection. Gastroenterology 2017, 152, 799–811.e7. [Google Scholar] [CrossRef]
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment. Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef]
- Stange, E.F.; Wehkamp, J. Recent advances in understanding and managing Crohn’s disease. F1000Res 2016, 5, 2896. [Google Scholar] [CrossRef]
- Koeninger, L.; Armbruster, N.; Hu, Z.; Jensen, B.; Stange, E.; Nordkild, P.; Malek, N.; Zender, L.; Wehkamp, J. Oral delivery of Human β-defensin 2 is reversibly increasing microbiome diversity and is effective in the treatment of experimental colitis. J. Crohn’s Colitis 2018, 12, S547. [Google Scholar] [CrossRef]
- Coretti, L.; Natale, A.; Cuomo, M.; Florio, E.; Keller, S.; Lembo, F.; Chiariotti, L.; Pero, R. The Interplay between Defensins and Microbiota in Crohn’s Disease. Mediat. Inflamm. 2017, 2017. [Google Scholar] [CrossRef]
- Tanaka, T.; Okanobu, H.; Kuga, Y.; Yoshifuku, Y.; Fujino, H.; Miwata, T.; Moriya, T.; Nishida, T.; Oya, T. Clinical and endoscopic features of responders and non-responders to adsorptive leucocytapheresis: A report based on 120 patients with active ulcerative colitis. Gastroenterol. Clin. Biol. 2010, 34, 687–695. [Google Scholar] [CrossRef]
- Saniabadi, A.R.; Tanaka, T.; Yamamoto, T.; Kruis, W.; Sacco, R. Granulomonocytapheresis as a cell-dependent treatment option for patients with inflammatory bowel disease: Concepts and clinical features for better therapeutic outcomes. J. Clin. Apher 2018, 34, 51–60. [Google Scholar] [CrossRef]
- Zhang, C.; Shu, W.; Zhou, G.; Lin, J.; Chu, F.; Wu, H.; Liu, Z. Anti-TNF-α Therapy Suppresses Proinflammatory Activities of Mucosal Neutrophils in Inflammatory Bowel Disease. Available online: https://www.hindawi.com/journals/mi/2018/3021863/ (accessed on 26 December 2018).
- Himmel, M.E.; Yao, Y.; Orban, P.C.; Steiner, T.S.; Levings, M.K. Regulatory T-cell therapy for inflammatory bowel disease: More questions than answers. Immunology 2012, 136, 115–122. [Google Scholar] [CrossRef]
- Fantini, M.C.; Monteleone, G. Update on the Therapeutic Efficacy of Tregs in IBD: Thumbs up or Thumbs down? Inflamm. Bowel Dis. 2017, 23, 1682–1688. [Google Scholar] [CrossRef]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hébuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology 2012, 143, 1207–1217.e2. [Google Scholar] [CrossRef]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef]
- Vos, A.C.W.; Wildenberg, M.E.; Arijs, I.; Duijvestein, M.; Verhaar, A.P.; de Hertogh, G.; Vermeire, S.; Rutgeerts, P.; van den Brink, G.R.; Hommes, D.W. Regulatory macrophages induced by infliximab are involved in healing in vivo and in vitro. Inflamm. Bowel Dis. 2012, 18, 401–408. [Google Scholar] [CrossRef]
- Vos, A.C.W.; Wildenberg, M.E.; Duijvestein, M.; Verhaar, A.P.; van den Brink, G.R.; Hommes, D.W. Anti-tumor necrosis factor-α antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology 2011, 140, 221–230. [Google Scholar] [CrossRef]
- Jharap, B.; Seinen, M.L.; de Boer, N.K.H.; van Ginkel, J.R.; Linskens, R.K.; Kneppelhout, J.C.; Mulder, C.J.J.; van Bodegraven, A.A. Thiopurine therapy in inflammatory bowel disease patients: Analyses of two 8-year intercept cohorts. Inflamm. Bowel Dis. 2010, 16, 1541–1549. [Google Scholar] [CrossRef]
- Liu, R.; Tang, A.; Wang, X.; Chen, X.; Zhao, L.; Xiao, Z.; Shen, S. Inhibition of lncRNA NEAT1 suppresses the inflammatory response in IBD by modulating the intestinal epithelial barrier and by exosome-mediated polarization of macrophages. Int. J. Mol. Med. 2018, 42, 2903–2913. [Google Scholar] [CrossRef]
- Marshall, J.K.; Irvine, E.J. Ketotifen treatment of active colitis in patients with 5-aminosalicylate intolerance. Can. J. Gastroenterol. 1998, 12, 273–275. [Google Scholar] [CrossRef]
- Heatley, R.V.; Calcraft, B.J.; Rhodes, J.; Owen, E.; Evans, B.K. Disodium cromoglycate in the treatment of chronic proctitis. Gut 1975, 16, 559–563. [Google Scholar] [CrossRef]
- Chu, H.-Q.; Li, J.; Huang, H.-P.; Hao, W.-D.; Duan, L.-P.; Wei, X.-T. Protective effects of tranilast on oxazolone-induced rat colitis through a mast cell-dependent pathway. Dig. Liver Dis. 2016, 48, 162–171. [Google Scholar] [CrossRef]
- Zhang, T.; Finn, D.F.; Barlow, J.W.; Walsh, J.J. Mast cell stabilisers. Eur. J. Pharmacol. 2016, 778, 158–168. [Google Scholar] [CrossRef]
- Klooker, T.K.; Braak, B.; Koopman, K.E.; Welting, O.; Wouters, M.M.; van der Heide, S.; Schemann, M.; Bischoff, S.C.; van den Wijngaard, R.M.; Boeckxstaens, G.E. The mast cell stabiliser ketotifen decreases visceral hypersensitivity and improves intestinal symptoms in patients with irritable bowel syndrome. Gut 2010, 59, 1213–1221. [Google Scholar] [CrossRef]
- Lobo, B.; Ramos, L.; Martínez, C.; Guilarte, M.; González-Castro, A.M.; Alonso-Cotoner, C.; Pigrau, M.; de Torres, I.; Rodiño-Janeiro, B.K.; Salvo-Romero, E.; et al. Downregulation of mucosal mast cell activation and immune response in diarrhoea-irritable bowel syndrome by oral disodium cromoglycate: A pilot study. United Eur. Gastroenterol. J. 2017, 5, 887–897. [Google Scholar] [CrossRef]
- De Zuani, M.; Dal Secco, C.; Frossi, B. Mast cells at the crossroads of microbiota and IBD. Eur. J. Immunol. 2018, 48, 1929–1937. [Google Scholar] [CrossRef]
- Soler, D.; Chapman, T.; Yang, L.-L.; Wyant, T.; Egan, R.; Fedyk, E.R. The binding specificity and selective antagonism of vedolizumab, an anti-alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases. J. Pharmacol. Exp. Ther. 2009, 330, 864–875. [Google Scholar] [CrossRef]
- Scribano, M.L. Vedolizumab for inflammatory bowel disease: From randomized controlled trials to real-life evidence. World J. Gastroenterol. 2018, 24, 2457–2467. [Google Scholar] [CrossRef]
- Gurish, M.F.; Tao, H.; Abonia, J.P.; Arya, A.; Friend, D.S.; Parker, C.M.; Austen, K.F. Intestinal mast cell progenitors require CD49dbeta7 (alpha4beta7 integrin) for tissue-specific homing. J. Exp. Med. 2001, 194, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Corinaldesi, R.; Stanghellini, V.; Cremon, C.; Gargano, L.; Cogliandro, R.F.; De Giorgio, R.; Bartesaghi, G.; Canovi, B.; Barbara, G. Effect of mesalazine on mucosal immune biomarkers in irritable bowel syndrome: A randomized controlled proof-of-concept study. Aliment. Pharmacol. Ther. 2009, 30, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Wallon, C.; Yang, P.-C.; Keita, A.V.; Ericson, A.-C.; McKay, D.M.; Sherman, P.M.; Perdue, M.H.; Söderholm, J.D. Corticotropin-releasing hormone (CRH) regulates macromolecular permeability via mast cells in normal human colonic biopsies in vitro. Gut 2008, 57, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Al-Haddad, S.; Riddell, R.H. The role of eosinophils in inflammatory bowel disease. Gut 2005, 54, 1674–1675. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Wolters, V.M.; Russell, R.K.; Shakhnovich, V.; Muise, A.M.; Ledder, O.; Ngan, B.; Friesen, C. Anti-TNF, infliximab, and adalimumab can be effective in eosinophilic bowel disease. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 492–497. [Google Scholar] [CrossRef]
- Carlson, M.; Raab, Y.; Peterson, C.; Hällgren, R.; Venge, P. Increased intraluminal release of eosinophil granule proteins EPO, ECP, EPX, and cytokines in ulcerative colitis and proctitis in segmental perfusion. Am. J. Gastroenterol. 1999, 94, 1876–1883. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoultz, I.; Keita, Å.V. Cellular and Molecular Therapeutic Targets in Inflammatory Bowel Disease—Focusing on Intestinal Barrier Function. Cells 2019, 8, 193. https://doi.org/10.3390/cells8020193
Schoultz I, Keita ÅV. Cellular and Molecular Therapeutic Targets in Inflammatory Bowel Disease—Focusing on Intestinal Barrier Function. Cells. 2019; 8(2):193. https://doi.org/10.3390/cells8020193
Chicago/Turabian StyleSchoultz, Ida, and Åsa V. Keita. 2019. "Cellular and Molecular Therapeutic Targets in Inflammatory Bowel Disease—Focusing on Intestinal Barrier Function" Cells 8, no. 2: 193. https://doi.org/10.3390/cells8020193
APA StyleSchoultz, I., & Keita, Å. V. (2019). Cellular and Molecular Therapeutic Targets in Inflammatory Bowel Disease—Focusing on Intestinal Barrier Function. Cells, 8(2), 193. https://doi.org/10.3390/cells8020193