Gli Proteins: Regulation in Development and Cancer

 ,
,
Abstract
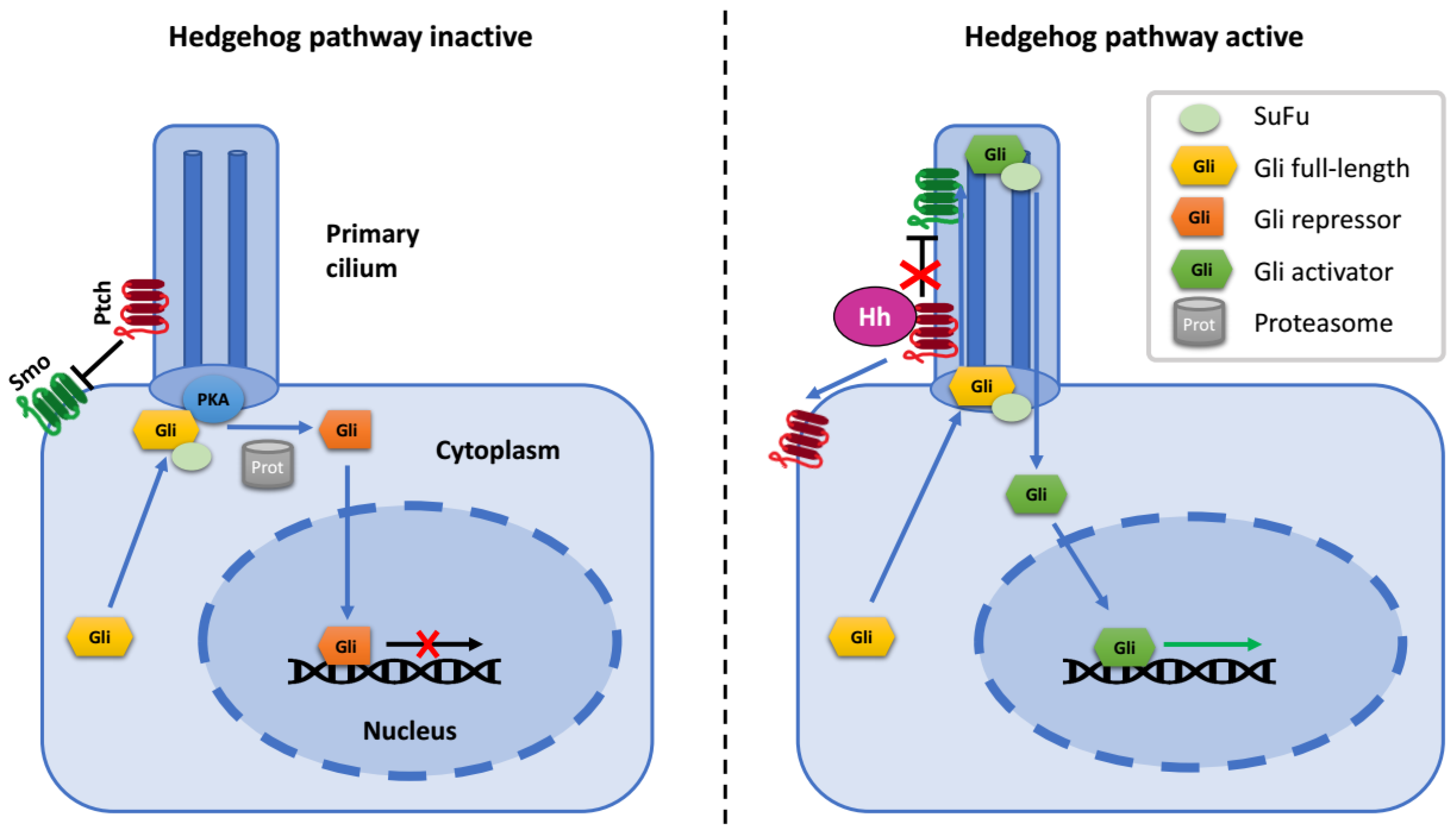
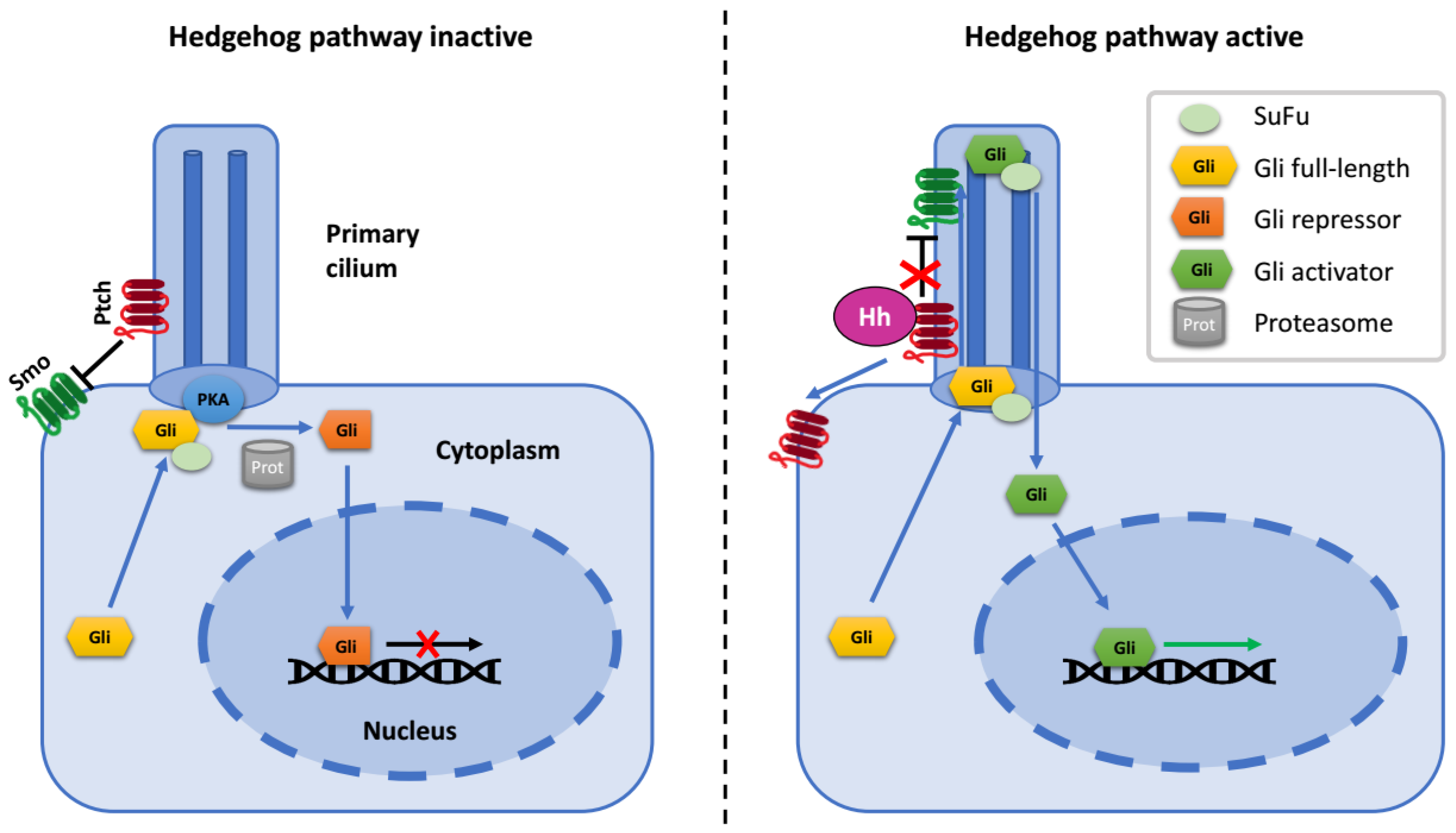
:1. The Hedgehog Signaling Pathway
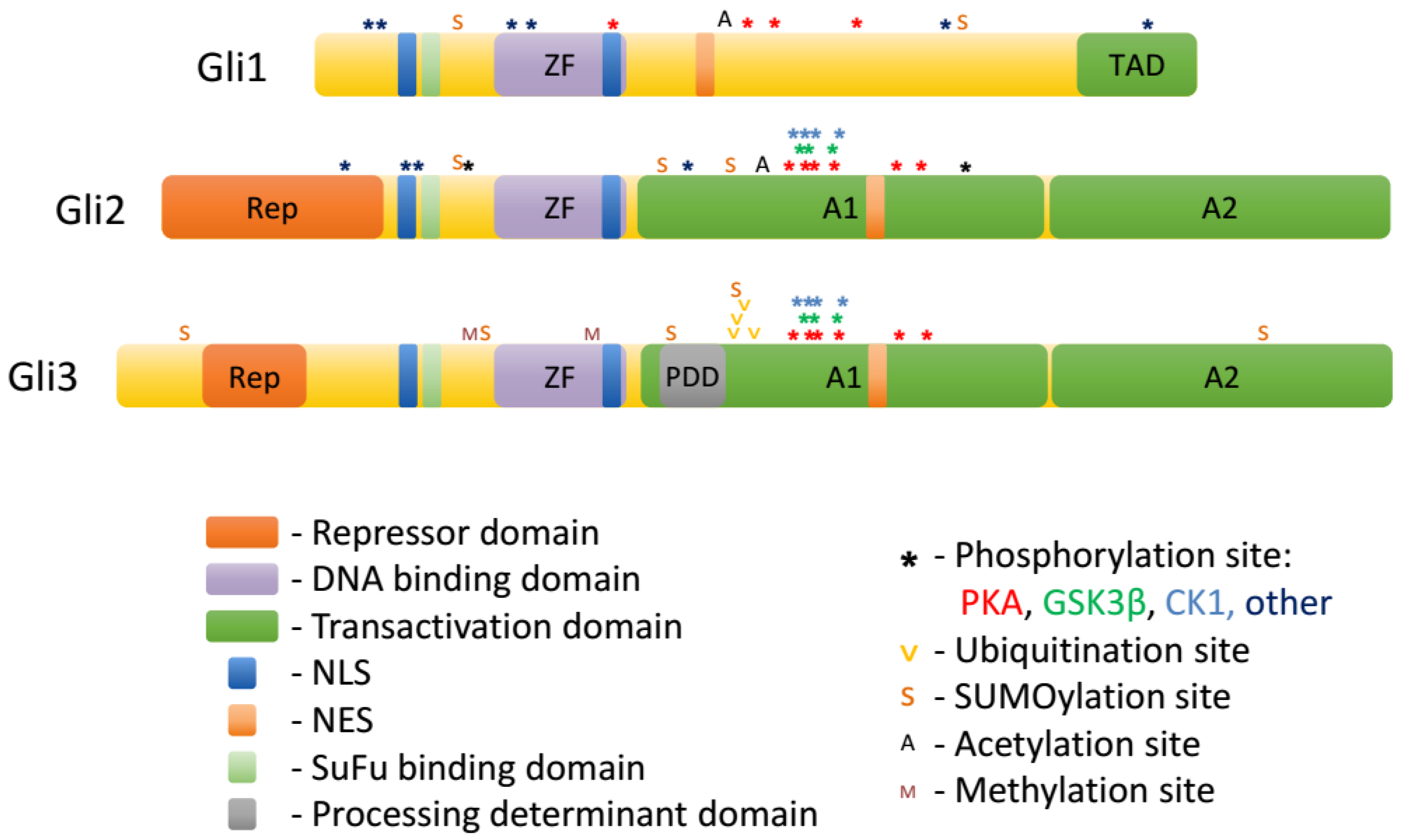
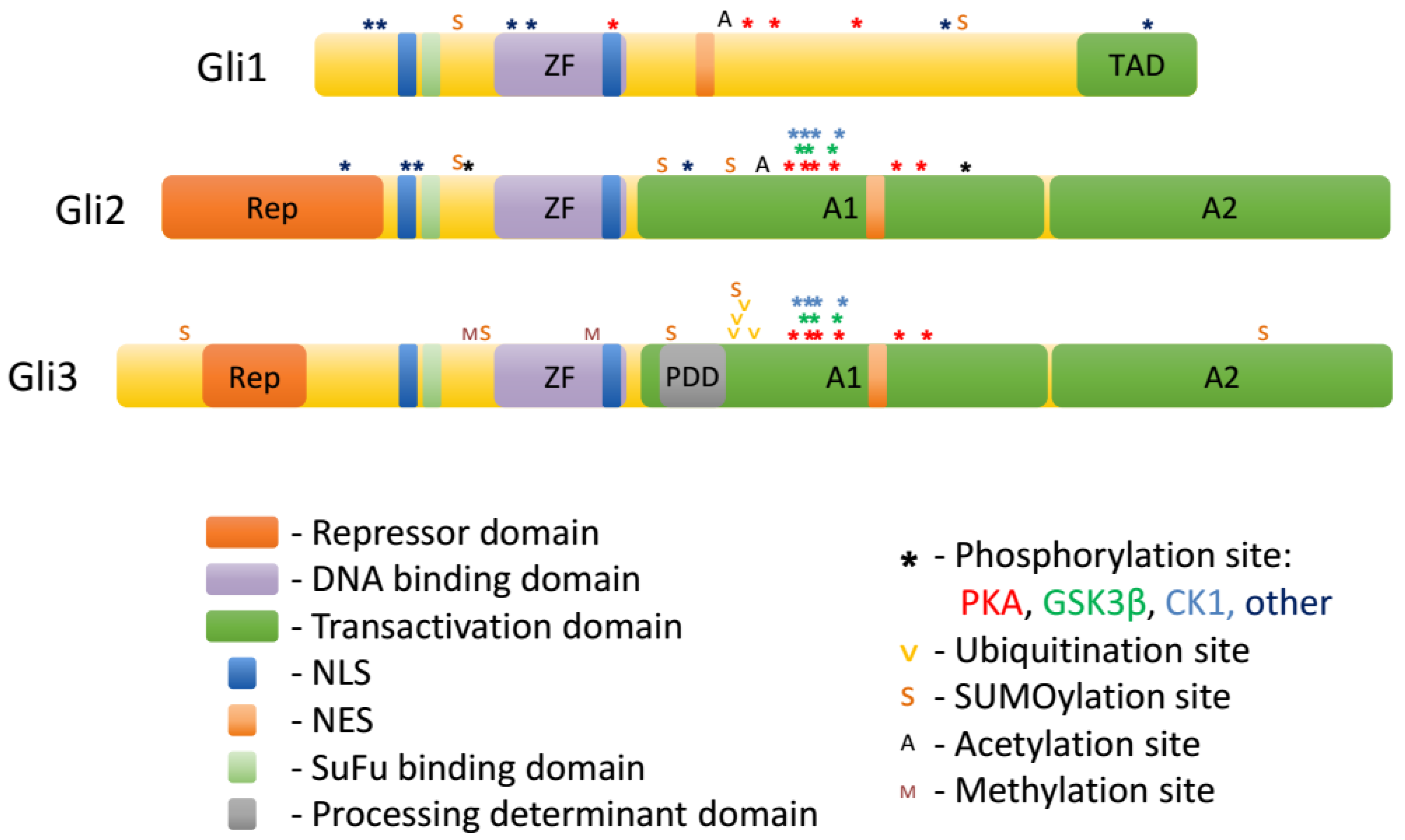
2. Gli Proteins
3. Regulation of Gli Proteins by Posttranslational Modifications
3.1. Phosphorylation
3.2. Ubiquitination
3.3. Sumoylation
3.4. Acetylation
3.5. O-GlcNAcylation
3.6. Methylation
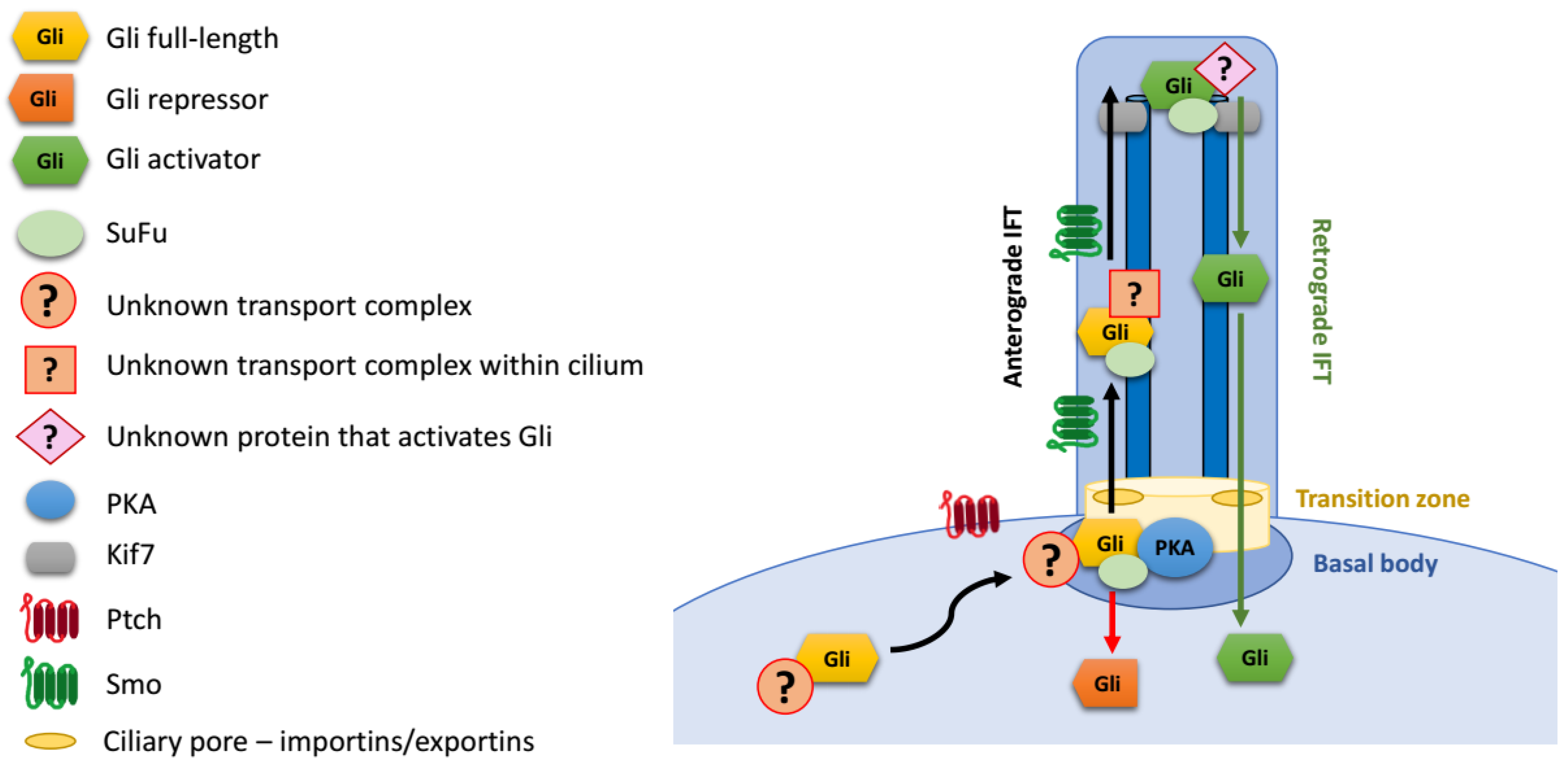
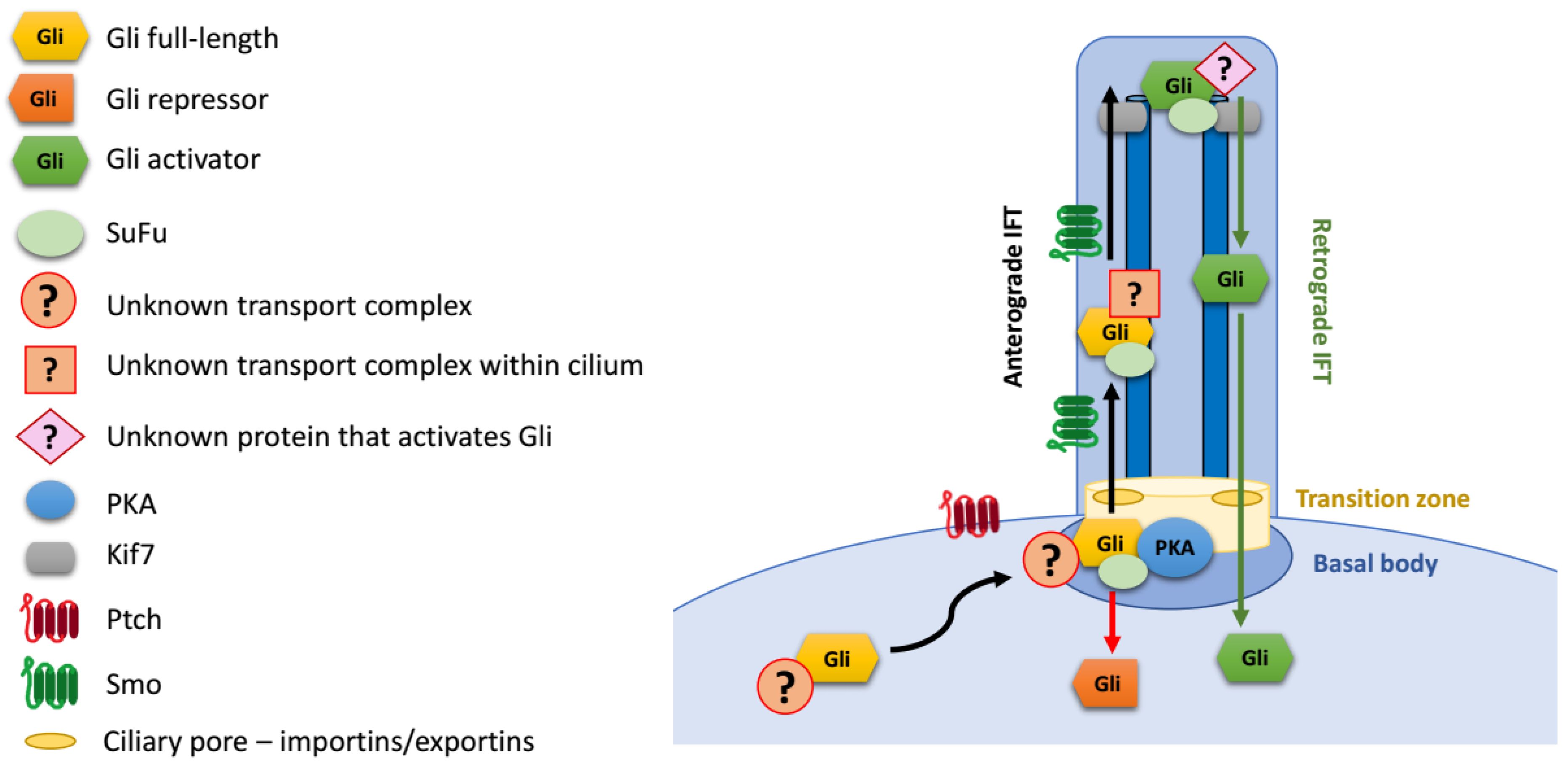
4. Regulation of Gli Proteins at Primary Cilia
Gli Protein Transport to the Primary Cilium
5. Nuclear Transport of Gli Proteins
6. Mechanisms of Transcriptional Activation by Gli Proteins
7. Mechanisms of Transcriptional Repression by Gli Proteins
8. Gli Proteins in Cancer
9. Gli Proteins in Development and Tissue Regeneration
10. Summary
- How do the different posttranslational modifications of Gli proteins interact with one another? Which modifications coexist, and which are mutually exclusive?
- What are the kinases and phosphorylation sites responsible for the phosphorylation of active nuclear Gli proteins?
- What is the importance of Cul3/Spop for degradation of Gli3R and Gli2?
- What are the proteins that regulate the accumulation of Gli proteins at the base of the primary cilium prior to crossing the ciliary barrier?
- What are the proteins that participate in nuclear and/or ciliary export of Gli proteins?
- What happens to Gli proteins at the tip of the primary cilium that makes them active?
- In what circumstances do the different transcriptional co-activators and co-repressors interact with nuclear Gli proteins? Which of these co-activators and co-repressors play roles in regulation of specific target genes?
- What is the functional significance of mutations in Gli proteins that occur in cancer?
Supplementary Materials
Funding
Conflicts of Interest
References
- Krauss, S.; Concordet, J.P.; Ingham, P.W. A functionally conserved homolog of the Drosophila segment polarity gene hh is expressed in tissues with polarizing activity in zebrafish embryos. Cell 1993, 75, 1431–1444. [Google Scholar] [CrossRef]
- Mohler, J. Requirements for hedgehog, a segmental polarity gene, in patterning larval and adult cuticle of Drosophila. Genetics 1988, 120, 1061–1072. [Google Scholar] [PubMed]
- Lettice, L.A.; Heaney, S.J.H.; Purdie, L.A.; Li, L.; de Beer, P.; Oostra, B.A.; Goode, D.; Elgar, G.; Hill, R.E.; de Graaff, E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003, 12, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog Signal Transduction Network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.K.; Beachy, P.A. Novel lipid modifications of secreted protein signals. Annu. Rev. Biochem. 2004, 73, 891–923. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Small, S. Morphogen rules: Design principles of gradient-mediated embryo patterning. Development 2015, 142, 3996–4009. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Yang, J.-J.; Tao, H.; Li, J. Hedgehog signaling pathway as key player in liver fibrosis: New insights and perspectives. Expert Opin. Ther. Targets 2014, 18, 1011–1021. [Google Scholar] [CrossRef]
- Pathi, S.; Pagan-Westphal, S.; Baker, D.P.; Garber, E.A.; Rayhorn, P.; Bumcrot, D.; Tabin, C.J.; Blake Pepinsky, R.; Williams, K.P. Comparative biological responses to human Sonic, Indian, and Desert hedgehog. Mech. Dev. 2001, 106, 107–117. [Google Scholar] [CrossRef]
- Rash, B.G.; Grove, E.A. Patterning the Dorsal Telencephalon: A Role for Sonic Hedgehog? J. Neurosci. 2007, 27, 11595–11603. [Google Scholar] [CrossRef]
- Riddle, R.D.; Johnson, R.L.; Laufer, E.; Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993, 75, 1401–1416. [Google Scholar] [CrossRef]
- Wechsler-Reya, R.J.; Scott, M.P. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999, 22, 103–114. [Google Scholar] [CrossRef]
- Arensdorf, A.M.; Marada, S.; Ogden, S.K. Smoothened Regulation: A Tale of Two Signals. Trends Pharmacol. Sci. 2016, 37, 62–72. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hui, C.-C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 1990, 10, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Ruppert, J.M.; Bigner, S.H.; Vogelstein, B. The GLI gene is a member of the Kruppel family of zinc finger proteins. Nature 1988, 332, 371–374. [Google Scholar] [CrossRef]
- Chen, M.-H.; Wilson, C.W.; Li, Y.-J.; Law, K.K.L.; Lu, C.-S.; Gacayan, R.; Zhang, X.; Hui, C.; Chuang, P.-T. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu–Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef]
- Rohatgi, R.; Scott, M.P. Patching the gaps in Hedgehog signalling. Nat. Cell Biol. 2007, 9, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Haycraft, C.J.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.J.; Yoder, B.K. Gli2 and Gli3 Localize to Cilia and Require the Intraflagellar Transport Protein Polaris for Processing and Function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.R.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Santos, N.; Reiter, J.F. A central region of Gli2 regulates its localization to the primary cilium and transcriptional activity. J. Cell. Sci. 2014, 127, 1500–1510. [Google Scholar] [CrossRef]
- Tuson, M.; He, M.; Anderson, K.V. Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development 2011, 138, 4921–4930. [Google Scholar] [CrossRef]
- Ruiz i Altaba, A.; Sánchez, P.; Dahmane, N. Gli and hedgehog in cancer: Tumours, embryos and stem cells. Nat. Rev. Cancer 2002, 2, 361–372. [Google Scholar] [CrossRef]
- Chen, J.-S.; Huang, X.; Wang, Q.; Huang, J.-Q.; Zhang, L.; Chen, X.-L.; Lei, J.; Cheng, Z.-X. Sonic hedgehog signaling pathway induces cell migration and invasion through focal adhesion kinase/AKT signaling-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9 in liver cancer. Carcinogenesis 2013, 34, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Yang, J.-Y. Targeting the Hedgehog Pathway in Pediatric Medulloblastoma. Cancers 2015, 7, 2110–2123. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, X.; Choy, E.; Mankin, H.; Hornicek, F.J.; Duan, Z. Targeting hedgehog-GLI-2 pathway in osteosarcoma. J. Orthop. Res. 2013, 31, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, J.; Feng, L. Hedgehog signaling pathway as a therapeutic target for ovarian cancer. Cancer Epidemiol. 2016, 40, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Bai, Y.; Dong, J.; Li, Q.; Jin, Y.; Chen, B.; Zhou, M. Hedgehog Signaling in Pancreatic Fibrosis and Cancer. Medicine 2016, 95, e2996. [Google Scholar] [CrossRef] [PubMed]
- Ramsbottom, S.A.; Pownall, M.E. Regulation of Hedgehog Signalling Inside and Outside the Cell. J. Dev. Biol. 2016, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Alman, B.A. The role of hedgehog signalling in skeletal health and disease. Nat. Rev. Rheumatol. 2015, 11, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Dellovade, T.; Romer, J.T.; Curran, T.; Rubin, L.L. The Hedgehog Pathway and Neurological Disorders. Annu. Rev. Neurosci. 2006, 29, 539–563. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.E.; Roessler, E.; Muenke, M. Human developmental disorders and the Sonic hedgehog pathway. Mol. Med. Today 1998, 4, 343–349. [Google Scholar] [CrossRef]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.-W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Sabol, M.; Trnski, D.; Musani, V.; Ozretić, P.; Levanat, S. Role of GLI Transcription Factors in Pathogenesis and Their Potential as New Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 2562. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.-C.; Slusarski, D.; Platt, K.A.; Holmgren, R.; Joyner, A.L. Expression of Three Mouse Homologs of the Drosophila Segment Polarity Gene cubitus interruptus, Gli, Gli-2, and Gli-3, in Ectoderm- and Mesoderm-Derived Tissues Suggests Multiple Roles during Postimplantation Development. Dev. Biol. 1994, 162, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar] [PubMed]
- Pan, Y.; Wang, B. A Novel Protein-processing Domain in Gli2 and Gli3 Differentially Blocks Complete Protein Degradation by the Proteasome. J. Biol. Chem. 2007, 282, 10846–10852. [Google Scholar] [CrossRef]
- Tempe, D.; Casas, M.; Karaz, S.; Blanchet-Tournier, M.-F.; Concordet, J.-P. Multisite Protein Kinase A and Glycogen Synthase Kinase 3 Phosphorylation Leads to Gli3 Ubiquitination by SCF TrCP. Mol. Cell. Biol. 2006, 26, 4316–4326. [Google Scholar] [CrossRef]
- Sheng, T.; Chi, S.; Zhang, X.; Xie, J. Regulation of Gli1 Localization by the cAMP/Protein Kinase A Signaling Axis through a Site Near the Nuclear Localization Signal. J. Biol. Chem. 2006, 281, 9–12. [Google Scholar] [CrossRef]
- Marks, S.A.; Kalderon, D. Regulation of mammalian Gli proteins by Costal 2 and PKA in Drosophila reveals Hedgehog pathway conservation. Development 2011, 138, 2533–2542. [Google Scholar] [CrossRef]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef]
- Mirza, A.N.; McKellar, S.A.; Urman, N.M.; Brown, A.S.; Hollmig, T.; Aasi, S.Z.; Oro, A.E. LAP2 Proteins Chaperone GLI1 Movement between the Lamina and Chromatin to Regulate Transcription. Cell 2018, 176, 198–212.e15. [Google Scholar] [CrossRef]
- Li, Y.-H.; Luo, J.; Mosley, Y.-Y.C.; Hedrick, V.E.; Paul, L.N.; Chang, J.; Zhang, G.; Wang, Y.-K.; Banko, M.R.; Brunet, A.; et al. AMP-Activated Protein Kinase Directly Phosphorylates and Destabilizes Hedgehog Pathway Transcription Factor GLI1 in Medulloblastoma. Cell Rep. 2015, 12, 599–609. [Google Scholar] [CrossRef]
- Antonucci, L.; Di Magno, L.; D’Amico, D.; Manni, S.; Serrao, S.M.; Di Pastena, F.; Bordone, R.; Yurtsever, Z.N.; Caimano, M.; Petroni, M.; et al. Mitogen-activated kinase kinase kinase 1 inhibits hedgehog signaling and medulloblastoma growth through GLI1 phosphorylation. Int. J. Oncol. 2019, 54, 505–514. [Google Scholar] [CrossRef]
- Shi, X.; Zhan, X.; Wu, J. A positive feedback loop between Gli1 and tyrosine kinase Hck amplifies shh signaling activities in medulloblastoma. Oncogenesis 2015, 4, e176. [Google Scholar] [CrossRef]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Y. Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 2006, 103, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Maye, P.; Kogerman, P.; Tejedor, F.J.; Toftgard, R.; Xie, W.; Wu, G.; Wu, D. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J. Biol. Chem. 2002, 277, 35156–35161. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget 2017, 8, 833–845. [Google Scholar] [CrossRef]
- Varjosalo, M.; Björklund, M.; Cheng, F.; Syvänen, H.; Kivioja, T.; Kilpinen, S.; Sun, Z.; Kallioniemi, O.; Stunnenberg, H.G.; He, W.-W.; et al. Application of active and kinase-deficient kinome collection for identification of kinases regulating hedgehog signaling. Cell 2008, 133, 537–548. [Google Scholar] [CrossRef]
- Maloverjan, A.; Piirsoo, M.; Michelson, P.; Kogerman, P.; Østerlund, T. Identification of a novel serine/threonine kinase ULK3 as a positive regulator of Hedgehog pathway. Exp. Cell Res. 2010, 316, 627–637. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Q.; Yen, C.-J.; Xia, W.; Izzo, J.G.; Lang, J.-Y.; Li, C.-W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef]
- Liu, X.; Pitarresi, J.R.; Cuitiño, M.C.; Kladney, R.D.; Woelke, S.A.; Sizemore, G.M.; Nayak, S.G.; Egriboz, O.; Schweickert, P.G.; Yu, L.; et al. Genetic ablation of Smoothened in pancreatic fibroblasts increases acinar-ductal metaplasia. Genes Dev. 2016, 30, 1943–1955. [Google Scholar]
- Shi, Y.; Chen, J.; Karner, C.M.; Long, F. Hedgehog signaling activates a positive feedback mechanism involving insulin-like growth factors to induce osteoblast differentiation. Proc. Natl. Acad. Sci. USA 2015, 112, 4678–4683. [Google Scholar] [CrossRef]
- Xing, Z.; Lin, A.; Li, C.; Liang, K.; Wang, S.; Liu, Y.; Park, P.K.; Qin, L.; Wei, Y.; Hawke, D.H.; et al. lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 2014, 159, 1110–1125. [Google Scholar] [CrossRef]
- Bhatia, N.; Thiyagarajan, S.; Elcheva, I.; Saleem, M.; Dlugosz, A.; Mukhtar, H.; Spiegelman, V.S. Gli2 is targeted for ubiquitination and degradation by beta-TrCP ubiquitin ligase. J. Biol. Chem. 2006, 281, 19320–19326. [Google Scholar] [CrossRef]
- Wang, C.; Pan, Y.; Wang, B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010, 137, 2001–2009. [Google Scholar] [CrossRef]
- Zhou, Z.; Yao, X.; Li, S.; Xiong, Y.; Dong, X.; Zhao, Y.; Jiang, J.; Zhang, Q. Deubiquitination of Ci/Gli by Usp7/HAUSP Regulates Hedgehog Signaling. Dev. Cell 2015, 34, 58–72. [Google Scholar] [CrossRef]
- Zhou, A.; Lin, K.; Zhang, S.; Ma, L.; Xue, J.; Morris, S.-A.; Aldape, K.D.; Huang, S. Gli1-induced deubiquitinase USP48 aids glioblastoma tumorigenesis by stabilizing Gli1. EMBO Rep. 2017, 18, 1318–1330. [Google Scholar] [CrossRef]
- Cox, B.; Briscoe, J.; Ulloa, F. SUMOylation by Pias1 Regulates the Activity of the Hedgehog Dependent Gli Transcription Factors. PLoS ONE 2010, 5, e11996. [Google Scholar] [CrossRef]
- Han, L.; Pan, Y.; Wang, B. Small Ubiquitin-like Modifier (SUMO) Modification Inhibits GLI2 Protein Transcriptional Activity in Vitro and in Vivo. J. Biol. Chem. 2012, 287, 20483–20489. [Google Scholar] [CrossRef]
- Liu, H.; Yan, S.; Ding, J.; Yu, T.-T.; Cheng, S.Y. DeSUMOylation of Gli1 by SENP1 Attenuates Sonic Hedgehog Signaling. Mol. Cell. Biol. 2017, 37, e00579-16. [Google Scholar] [CrossRef]
- Das, S.; Bailey, S.K.; Metge, B.J.; Hanna, A.; Hinshaw, D.C.; Mota, M.; Forero-Torres, A.; Chatham, J.C.; Samant, R.S.; Shevde, L.A. O-GlcNAcylation of GLI transcription factors in hyperglycemic conditions augments Hedgehog activity. Lab. Invest. 2019, 99, 260–270. [Google Scholar] [CrossRef]
- Fu, L.; Wu, H.; Cheng, S.Y.; Gao, D.; Zhang, L.; Zhao, Y. Set7 mediated Gli3 methylation plays a positive role in the activation of Sonic Hedgehog pathway in mammals. Elife 2016, 5, e15690. [Google Scholar] [CrossRef]
- Allen, M.D.; Zhang, J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem. Biophys. Res. Commun. 2006, 348, 716–721. [Google Scholar] [CrossRef]
- Niewiadomski, P.; Zhujiang, A.; Youssef, M.; Waschek, J.A. Interaction of PACAP with Sonic hedgehog reveals complex regulation of the hedgehog pathway by PKA. Cell. Signal. 2013, 25, 2222–2230. [Google Scholar] [CrossRef]
- Shi, Q.; Li, S.; Li, S.; Jiang, A.; Chen, Y.; Jiang, J. Hedgehog-induced phosphorylation by CK1 sustains the activity of Ci/Gli activator. Proc. Natl. Acad. Sci. USA 2014, 111, E5651–E5660. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgård, R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar] [CrossRef]
- Maloverjan, A.; Piirsoo, M.; Kasak, L.; Peil, L.; Osterlund, T.; Kogerman, P. Dual Function of UNC-51-like Kinase 3 (Ulk3) in the Sonic Hedgehog Signaling Pathway. J. Biol. Chem. 2010, 285, 30079–30090. [Google Scholar] [CrossRef]
- Mirza, A.N.; Fry, M.A.; Urman, N.M.; Atwood, S.X.; Roffey, J.; Ott, G.R.; Chen, B.; Lee, A.; Brown, A.S.; Aasi, S.Z.; et al. Combined inhibition of atypical PKC and histone deacetylase 1 is cooperative in basal cell carcinoma treatment. JCI Insight 2017, 2, 97071. [Google Scholar] [CrossRef]
- Krauss, S.; Foerster, J.; Schneider, R.; Schweiger, S. Protein Phosphatase 2A and Rapamycin Regulate the Nuclear Localization and Activity of the Transcription Factor GLI3. Cancer Res. 2008, 68, 4658–4665. [Google Scholar] [CrossRef]
- Pandolfi, S.; Montagnani, V.; Penachioni, J.Y.; Vinci, M.C.; Olivito, B.; Borgognoni, L.; Stecca, B. WIP1 phosphatase modulates the Hedgehog signaling by enhancing GLI1 function. Oncogene 2013, 32, 4737–4747. [Google Scholar] [CrossRef]
- Rape, M.; Jentsch, S. Productive RUPture: Activation of transcription factors by proteasomal processing. Biochim. Biophys. Acta 2004, 209–213. [Google Scholar] [CrossRef]
- Huntzicker, E.G.; Estay, I.S.; Zhen, H.; Lokteva, L.A.; Jackson, P.K.; Oro, A.E. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006, 20, 276–281. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, Y.; Lu, Q.; Chen, J.; Zhang, J.; Liu, T.; Lv, N.; Luo, S. SPOP suppresses tumorigenesis by regulating Hedgehog/Gli2 signaling pathway in gastric cancer. J. Exp. Clin. Cancer Res. 2014, 33, 75. [Google Scholar] [CrossRef]
- Zhang, Q.; Shi, Q.; Chen, Y.; Yue, T.; Li, S.; Wang, B.; Jiang, J. Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2009, 106, 21191–21196. [Google Scholar] [CrossRef]
- Cai, H.; Liu, A. Spop promotes skeletal development and homeostasis by positively regulating Ihh signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 14751–14756. [Google Scholar] [CrossRef]
- Yin, W.-C.; Satkunendran, T.; Mo, R.; Morrissy, S.; Zhang, X.; Huang, E.S.; Uusküla-Reimand, L.; Hou, H.; Son, J.E.; Liu, W.; et al. Dual Regulatory Functions of SUFU and Targetome of GLI2 in SHH Subgroup Medulloblastoma. Dev. Cell 2019, 48, 167–183. [Google Scholar] [CrossRef]
- Di Marcotullio, L.; Ferretti, E.; Greco, A.; De Smaele, E.; Po, A.; Sico, M.A.; Alimandi, M.; Giannini, G.; Maroder, M.; Screpanti, I.; et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat. Cell Biol. 2006, 8, 1415–1423. [Google Scholar] [CrossRef]
- Schneider, P.; Miguel Bayo-Fina, J.; Singh, R.; Kumar Dhanyamraju, P.; Holz, P.; Baier, A.; Fendrich, V.; Ramaswamy, A.; Baumeister, S.; Martinez, E.D.; et al. Identification of a novel actin-dependent signal transducing module allows for the targeted degradation of GLI1. Nat. Commun. 2015, 6, 8023. [Google Scholar] [CrossRef]
- Mazzà, D.; Infante, P.; Colicchia, V.; Greco, A.; Alfonsi, R.; Siler, M.; Antonucci, L.; Po, A.; De Smaele, E.; Ferretti, E.; et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013, 20, 1688–1697. [Google Scholar] [CrossRef]
- Malatesta, M.; Steinhauer, C.; Mohammad, F.; Pandey, D.P.; Squatrito, M.; Helin, K. Histone Acetyltransferase PCAF Is Required for Hedgehog–Gli-Dependent Transcription and Cancer Cell Proliferation. Cancer Res. 2013, 73, 6323–6333. [Google Scholar] [CrossRef]
- Heride, C.; Rigden, D.J.; Bertsoulaki, E.; Cucchi, D.; De Smaele, E.; Clague, M.J.; Urbé, S. The centrosomal deubiquitylase USP21 regulates Gli1 transcriptional activity and stability. J. Cell Sci. 2016, 129, 4001–4013. [Google Scholar] [CrossRef]
- Li, X.-Y.; Mao, X.-F.; Tang, X.-Q.; Han, Q.; Jiang, L.-X.; Qiu, Y.-M.; Dai, J.; Wang, Y.-X. Regulation of Gli2 stability by deubiquitinase OTUB2. Biochem. Biophys. Res. Commun. 2018, 505, 113–118. [Google Scholar] [CrossRef]
- Coni, S.; Mancuso, A.B.; Di Magno, L.; Sdruscia, G.; Manni, S.; Serrao, S.M.; Rotili, D.; Spiombi, E.; Bufalieri, F.; Petroni, M.; et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci. Rep. 2017, 7, 44079. [Google Scholar] [CrossRef]
- Caspary, T.; Larkins, C.E.; Anderson, K.V. The Graded Response to Sonic Hedgehog Depends on Cilia Architecture. Dev. Cell 2007, 12, 767–778. [Google Scholar] [CrossRef]
- Huangfu, D.; Anderson, K.V. Cilia and Hedgehog Responsiveness in the Mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 11325–11330. [Google Scholar] [CrossRef]
- Wen, X.; Lai, C.K.; Evangelista, M.; Hongo, J.-A.; de Sauvage, F.J.; Scales, S.J. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell. Biol. 2010, 30, 1910–1922. [Google Scholar] [CrossRef]
- Lin, C.; Yao, E.; Wang, K.; Nozawa, Y.; Shimizu, H.; Johnson, J.R.; Chen, J.-N.; Krogan, N.J.; Chuang, P.-T. Regulation of Sufu activity by p66β and Mycbp provides new insight into vertebrate Hedgehog signaling. Genes Dev. 2014, 28, 2547–2563. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, L.; Law, K.; Zhang, Z.; Liu, X.; Hua, H.; Li, S.; Huang, H.; Yue, S.; Hui, C.-C.; et al. Suppressor of Fused Chaperones Gli Proteins To Generate Transcriptional Responses to Sonic Hedgehog Signaling. Mol. Cell. Biol. 2017, 37, e00421-16. [Google Scholar] [CrossRef]
- Jia, J.; Kolterud, Å.; Zeng, H.; Hoover, A.; Teglund, S.; Toftgård, R.; Liu, A. Suppressor of Fused inhibits mammalian Hedgehog signaling in the absence of cilia. Dev. Biol. 2009, 330, 452–460. [Google Scholar] [CrossRef]
- Mick, D.U.; Rodrigues, R.B.; Leib, R.D.; Adams, C.M.; Chien, A.S.; Gygi, S.P.; Nachury, M.V. Proteomics of Primary Cilia by Proximity Labeling. Dev. Cell 2015, 35, 497–512. [Google Scholar] [CrossRef]
- Barzi, M.; Berenguer, J.; Menendez, A.; Alvarez-Rodriguez, R.; Pons, S. Sonic-hedgehog-mediated proliferation requires the localization of PKA to the cilium base. J. Cell Sci. 2010, 123, 62–69. [Google Scholar] [CrossRef]
- Li, J.; Wang, C.; Wu, C.; Cao, T.; Xu, G.; Meng, Q.; Wang, B. PKA-mediated Gli2 and Gli3 phosphorylation is inhibited by Hedgehog signaling in cilia and reduced in Talpid3 mutant. Dev. Biol. 2017, 429, 147–157. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Wen, X.; Ratti, N.; Loktev, A.; Rangell, L.; Scales, S.J.; Jackson, P.K. The Ciliary G-Protein-Coupled Receptor Gpr161 Negatively Regulates the Sonic Hedgehog Pathway via cAMP Signaling. Cell 2013, 152, 210–223. [Google Scholar] [CrossRef]
- Ocbina, P.J.R.; Anderson, K.V. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: Analysis in mouse embryonic fibroblasts. Dev. Dyn. 2008, 237, 2030–2038. [Google Scholar] [CrossRef]
- Chen, Y.; Yue, S.; Xie, L.; Pu, X.H.; Jin, T.; Cheng, S.Y. Dual Phosphorylation of Suppressor of Fused (Sufu) by PKA and GSK3 Regulates Its Stability and Localization in the Primary Cilium. J. Biol. Chem. 2011, 286, 13502–13511. [Google Scholar] [CrossRef]
- Breslow, D.K.; Koslover, E.F.; Seydel, F.; Spakowitz, A.J.; Nachury, M.V. An in vitro assay for entry into cilia reveals unique properties of the soluble diffusion barrier. J. Cell Biol. 2013, 203, 129–147. [Google Scholar] [CrossRef]
- Kee, H.L.; Dishinger, J.F.; Blasius, T.L.; Liu, C.-J.; Margolis, B.; Verhey, K.J. A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat. Cell Biol. 2012, 14, 431–437. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Niewiadomski, P.; Lin, B.; Nakamura, H.; Phua, S.C.; Jiao, J.; Levchenko, A.; Inoue, T.; Rohatgi, R.; Inoue, T. Chemically inducible diffusion trap at cilia reveals molecular sieve–like barrier. Nat. Chem. Biol. 2013, 9, 437–443. [Google Scholar] [CrossRef]
- Takao, D.; Dishinger, J.F.; Kee, H.L.; Pinskey, J.M.; Allen, B.L.; Verhey, K.J. An Assay for Clogging the Ciliary Pore Complex Distinguishes Mechanisms of Cytosolic and Membrane Protein Entry. Curr. Biol. 2014, 24, 2288–2294. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Badgandi, H.B.; Hwang, S.; Somatilaka, B.; Shimada, I.S.; Pal, K. Trafficking to the primary cilium membrane. Mol. Biol. Cell 2017, 28, 233–239. [Google Scholar] [CrossRef]
- Nachury, M.V.; Seeley, E.S.; Jin, H. Trafficking to the ciliary membrane: How to get across the periciliary diffusion barrier? Annu. Rev. Cell Dev. Biol. 2010, 26, 59–87. [Google Scholar] [CrossRef]
- Han, Y.; Xiong, Y.; Shi, X.; Wu, J.; Zhao, Y.; Jiang, J. Regulation of Gli ciliary localization and Hedgehog signaling by the PY-NLS/karyopherin-β2 nuclear import system. PLoS Biol. 2017, 15, e2002063. [Google Scholar] [CrossRef]
- Zeng, H.; Jia, J.; Liu, A. Coordinated translocation of mammalian Gli proteins and suppressor of fused to the primary cilium. PLoS ONE 2010, 5, e15900. [Google Scholar] [CrossRef]
- Cheung, H.O.-L.; Zhang, X.; Ribeiro, A.; Mo, R.; Makino, S.; Puviindran, V.; Law, K.K.; Briscoe, J.; Hui, C. The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci. Signal. 2009, 2, ra29. [Google Scholar] [CrossRef]
- Hsu, S.-H.C.; Zhang, X.; Yu, C.; Li, Z.J.; Wunder, J.S.; Hui, C.-C.; Alman, B.A. Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory function of Sufu. Development 2011, 138, 3791–3801. [Google Scholar] [CrossRef]
- He, M.; Subramanian, R.; Bangs, F.; Omelchenko, T.; Liem, K.F.; Kapoor, T.M.; Anderson, K.V. The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat. Cell Biol. 2014, 16, 663–672. [Google Scholar] [CrossRef]
- Pusapati, G.V.; Rohatgi, R. Location, location, and location: Compartmentalization of Hedgehog signaling at primary cilia. EMBO J. 2014, 33, 1852–1854. [Google Scholar] [CrossRef]
- Gupta, P.R.; Pendse, N.; Greenwald, S.H.; Leon, M.; Liu, Q.; Pierce, E.A.; Bujakowska, K.M. Ift172 conditional knock-out mice exhibit rapid retinal degeneration and protein trafficking defects. Hum. Mol. Genet. 2018, 27, 2012–2024. [Google Scholar]
- Eguether, T.; San Agustin, J.T.; Keady, B.T.; Jonassen, J.A.; Liang, Y.; Francis, R.; Tobita, K.; Johnson, C.A.; Abdelhamed, Z.A.; Lo, C.W.; et al. IFT27 Links the BBSome to IFT for Maintenance of the Ciliary Signaling Compartment. Dev. Cell 2014, 31, 279–290. [Google Scholar] [CrossRef]
- Yang, N.; Li, L.; Eguether, T.; Sundberg, J.P.; Pazour, G.J.; Chen, J. Intraflagellar transport 27 is essential for hedgehog signaling but dispensable for ciliogenesis during hair follicle morphogenesis. Development 2015, 142, 2194–2202. [Google Scholar] [CrossRef]
- Eguether, T.; Cordelieres, F.P.; Pazour, G.J. Intraflagellar transport is deeply integrated in hedgehog signaling. Mol. Biol. Cell 2018, 29, 1178–1189. [Google Scholar] [CrossRef]
- He, K.; Ma, X.; Xu, T.; Li, Y.; Hodge, A.; Zhang, Q.; Torline, J.; Huang, Y.; Zhao, J.; Ling, K.; et al. Axoneme polyglutamylation regulated by Joubert syndrome protein ARL13B controls ciliary targeting of signaling molecules. Nat. Commun. 2018, 9, 3310. [Google Scholar] [CrossRef]
- Hong, S.-R.; Wang, C.-L.; Huang, Y.-S.; Chang, Y.-C.; Chang, Y.-C.; Pusapati, G.V.; Lin, C.-Y.; Hsu, N.; Cheng, H.-C.; Chiang, Y.-C.; et al. Spatiotemporal manipulation of ciliary glutamylation reveals its roles in intraciliary trafficking and Hedgehog signaling. Nat. Commun. 2018, 9, 9. [Google Scholar] [CrossRef]
- Sorokin, A.V.; Kim, E.R.; Ovchinnikov, L.P. Nucleocytoplasmic transport of proteins. Biochemistry (Mosc) 2007, 72, 1439–1457. [Google Scholar] [CrossRef]
- Dingwall, C.; Laskey, R.A. Nuclear targeting sequences—A consensus? Trends Biochem. Sci. 1991, 16, 478–481. [Google Scholar] [CrossRef]
- Zhou, M.; Liu, H.; Xu, X.; Zhou, H.; Li, X.; Peng, C.; Shen, S.; Xiong, W.; Ma, J.; Zeng, Z.; et al. Identification of nuclear localization signal that governs nuclear import of BRD7 and its essential roles in inhibiting cell cycle progression. J. Cell. Biochem. 2006, 98, 920–930. [Google Scholar] [CrossRef]
- Lange, A.; McLane, L.M.; Mills, R.E.; Devine, S.E.; Corbett, A.H. Expanding the Definition of the Classical Bipartite Nuclear Localization Signal. Traffic 2010, 11, 311–323. [Google Scholar] [CrossRef]
- Xu, D.; Farmer, A.; Chook, Y.M. Recognition of nuclear targeting signals by Karyopherin-β proteins. Curr. Opin. Struct. Biol. 2010, 20, 782–790. [Google Scholar] [CrossRef]
- Lee, B.J.; Cansizoglu, A.E.; Süel, K.E.; Louis, T.H.; Zhang, Z.; Chook, Y.M. Rules for Nuclear Localization Sequence Recognition by Karyopherinβ2. Cell 2006, 126, 543–558. [Google Scholar] [CrossRef]
- Antoine, M.; Reimers, K.; Wirz, W.; Gressner, A.M.; Müller, R.; Kiefer, P. Identification of an unconventional nuclear localization signal in human ribosomal protein S2. Biochem. Biophys. Res. Commun. 2005, 335, 146–153. [Google Scholar] [CrossRef]
- Paraskeva, E.; Izaurralde, E.; Bischoff, F.R.; Huber, J.; Kutay, U.; Hartmann, E.; Lührmann, R.; Görlich, D. CRM1-mediated Recycling of Snurportin 1 to the Cytoplasm. J. Cell Biol. 1999, 145, 255–264. [Google Scholar] [CrossRef]
- La Cour, T.; Kiemer, L.; Mølgaard, A.; Gupta, R.; Skriver, K.; Brunak, S. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 2004, 17, 527–536. [Google Scholar] [CrossRef]
- Barnfield, P.C.; Zhang, X.; Thanabalasingham, V.; Yoshida, M.; Hui, C. Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple mechanisms. Differentiation 2005, 73, 397–405. [Google Scholar] [CrossRef]
- Hatayama, M.; Aruga, J. Chapter Four—Gli Protein Nuclear Localization Signal. In Vitamins & Hormones; Litwack, G., Ed.; Hedgehog Signaling; Academic Press: Cambridge, MA, USA, 2012; Volume 88, pp. 73–89. [Google Scholar]
- Torrado, B.; Graña, M.; Badano, J.L.; Irigoín, F. Ciliary Entry of the Hedgehog Transcriptional Activator Gli2 Is Mediated by the Nuclear Import Machinery but Differs from Nuclear Transport in Being Imp-α/β1-Independent. PLoS ONE 2016, 11, e0162033. [Google Scholar] [CrossRef]
- Shi, Q.; Han, Y.; Jiang, J. Suppressor of fused impedes Ci/Gli nuclear import by opposing Trn/Kap 2 in Hedgehog signaling. J. Cell Sci. 2014, 127, 1092–1103. [Google Scholar] [CrossRef]
- Szczepny, A.; Wagstaff, K.M.; Dias, M.; Gajewska, K.; Wang, C.; Davies, R.G.; Kaur, G.; Ly-Huynh, J.; Loveland, K.L.; Jans, D.A. Overlapping binding sites for importin β1 and suppressor of fused (SuFu) on glioma-associated oncogene homologue 1 (Gli1) regulate its nuclear localization. Biochem. J. 2014, 461, 469–476. [Google Scholar] [CrossRef]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Undén, A.B.; Sandstedt, B.; Toftg\a ard, R.; Zaphiropoulos, P.G. Mammalian suppressor-of-fused modulates nuclear–cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef]
- Fung, H.Y.J.; Fu, S.-C.; Brautigam, C.A.; Chook, Y.M. Structural determinants of nuclear export signal orientation in binding to exportin CRM1. Elife 2015, 4, 4. [Google Scholar] [CrossRef]
- Dunaeva, M.; Michelson, P.; Kogerman, P.; Toftgard, R. Characterization of the Physical Interaction of Gli Proteins with SUFU Proteins. J. Biol. Chem. 2003, 278, 5116–5122. [Google Scholar] [CrossRef]
- Asaoka, Y.; Kanai, F.; Ichimura, T.; Tateishi, K.; Tanaka, Y.; Ohta, M.; Seto, M.; Tada, M.; Ijichi, H.; Ikenoue, T.; et al. Identification of a suppressive mechanism for Hedgehog signaling through a novel interaction of Gli with 14-3-3. J. Biol. Chem. 2010, 285, 4185–4194. [Google Scholar] [CrossRef]
- Wong, S.Y.; Reiter, J.F. Chapter 9 The Primary Cilium. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 85, pp. 225–260. ISBN 978-0-12-374453-1. [Google Scholar]
- Hassounah, N.B.; Bunch, T.A.; McDermott, K.M. Molecular Pathways: The Role of Primary Cilia in Cancer Progression and Therapeutics with a Focus on Hedgehog Signaling. Clin. Cancer Res. 2012, 18, 2429–2435. [Google Scholar] [CrossRef]
- Yoon, J.W.; Lamm, M.; Iannaccone, S.; Higashiyama, N.; Leong, K.F.; Iannaccone, P.; Walterhouse, D. p53 modulates the activity of the GLI1 oncogene through interactions with the shared coactivator TAF9. DNA Repair 2015, 34, 9–17. [Google Scholar] [CrossRef]
- Yoon, J.W.; Liu, C.Z.; Yang, J.T.; Swart, R.; Iannaccone, P.; Walterhouse, D. GLI Activates Transcription through a Herpes Simplex Viral Protein 16-Like Activation Domain. J. Biol. Chem. 1998, 273, 3496–3501. [Google Scholar] [CrossRef]
- Zhou, H.; Kim, S.; Ishii, S.; Boyer, T.G. Mediator modulates Gli3-dependent Sonic hedgehog signaling. Mol. Cell. Biol. 2006, 26, 8667–8682. [Google Scholar] [CrossRef]
- Zhou, H.; Spaeth, J.M.; Kim, N.H.; Xu, X.; Friez, M.J.; Schwartz, C.E.; Boyer, T.G. MED12 mutations link intellectual disability syndromes with dysregulated GLI3-dependent Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 19763–19768. [Google Scholar] [CrossRef]
- Mosimann, C.; Hausmann, G.; Basler, K. The role of Parafibromin/Hyrax as a nuclear Gli/Ci-interacting protein in Hedgehog target gene control. Mech. Dev. 2009, 126, 394–405. [Google Scholar] [CrossRef]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Maekawa, T.; Nakafuku, M.; Ishii, S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 1999, 274, 8143–8152. [Google Scholar] [CrossRef]
- Petrij, F.; Giles, R.H.; Dauwerse, H.G.; Saris, J.J.; Hennekam, R.C.; Masuno, M.; Tommerup, N.; van Ommen, G.J.; Goodman, R.H.; Peters, D.J. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 1995, 376, 348–351. [Google Scholar] [CrossRef]
- Vortkamp, A.; Gessler, M.; Grzeschik, K.H. GLI3 zinc-finger gene interrupted by translocations in Greig syndrome families. Nature 1991, 352, 539–540. [Google Scholar] [CrossRef]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.-H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.-H.; Ayad, N.G.; et al. The BET bromodomain inhibitor I-BET151 acts downstream of Smoothened to abrogate the growth of Hedgehog driven cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, Z.; Zhan, X.; Cao, M.; Satoh, T.; Akira, S.; Shpargel, K.; Magnuson, T.; Li, Q.; Wang, R.; et al. An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nat. Commun. 2014, 5, 5425. [Google Scholar] [CrossRef]
- Zhan, X.; Shi, X.; Zhang, Z.; Chen, Y.; Wu, J.I. Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc. Natl. Acad. Sci. USA 2011, 108, 12758–12763. [Google Scholar] [CrossRef]
- Shi, X.; Wang, Q.; Gu, J.; Xuan, Z.; Wu, J.I. SMARCA4/Brg1 coordinates genetic and epigenetic networks underlying Shh-type medulloblastoma development. Oncogene 2016, 35, 5746–5758. [Google Scholar] [CrossRef]
- Jeon, S.; Seong, R.H. Anteroposterior Limb Skeletal Patterning Requires the Bifunctional Action of SWI/SNF Chromatin Remodeling Complex in Hedgehog Pathway. PLoS Genet. 2016, 12, e1005915. [Google Scholar] [CrossRef]
- Callahan, C.A.; Ofstad, T.; Horng, L.; Wang, J.K.; Zhen, H.H.; Coulombe, P.A.; Oro, A.E. MIM/BEG4, a Sonic hedgehog-responsive gene that potentiates Gli-dependent transcription. Genes Dev. 2004, 18, 2724–2729. [Google Scholar] [CrossRef]
- Bershteyn, M.; Atwood, S.X.; Woo, W.-M.; Li, M.; Oro, A.E. MIM and Cortactin Antagonism Regulates Ciliogenesis and Hedgehog Signaling. Dev. Cell 2010, 19, 270–283. [Google Scholar] [CrossRef]
- Infante, P.; Faedda, R.; Bernardi, F.; Bufalieri, F.; Severini, L.L.; Alfonsi, R.; Mazzà, D.; Siler, M.; Coni, S.; Po, A.; et al. Itch/β-arrestin2-dependent non-proteolytic ubiquitylation of SuFu controls Hedgehog signalling and medulloblastoma tumorigenesis. Nat. Commun. 2018, 9, 976. [Google Scholar] [CrossRef]
- Kise, Y.; Morinaka, A.; Teglund, S.; Miki, H. Sufu recruits GSK3beta for efficient processing of Gli3. Biochem. Biophys. Res. Commun. 2009, 387, 569–574. [Google Scholar] [CrossRef]
- Tsanev, R.; Vanatalu, K.; Jarvet, J.; Tanner, R.; Laur, K.; Tiigimägi, P.; Kragelund, B.B.; Østerlund, T.; Kogerman, P. The transcriptional repressor domain of Gli3 is intrinsically disordered. PLoS ONE 2013, 8, e76972. [Google Scholar] [CrossRef]
- Tsanev, R.; Tiigimägi, P.; Michelson, P.; Metsis, M.; Østerlund, T.; Kogerman, P. Identification of the gene transcription repressor domain of Gli3. FEBS Lett. 2009, 583, 224–228. [Google Scholar] [CrossRef]
- Dai, P. Ski is involved in transcriptional regulation by the repressor and full-length forms of Gli3. Genes Dev. 2002, 16, 2843–2848. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Bishop, J.M. Suppressor of Fused represses Gli-mediated transcription by recruiting the SAP18-mSin3 corepressor complex. PNAS 2002, 99, 5442–5447. [Google Scholar] [CrossRef]
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.L.; Tolstorukov, M.; et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433. [Google Scholar] [CrossRef]
- Aberger, F.; Kern, D.; Greil, R.; Hartmann, T.N. Canonical and Noncanonical Hedgehog/GLI Signaling in Hematological Malignancies. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2012; Volume 88, pp. 25–54. ISBN 978-0-12-394622-5. [Google Scholar]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical Hedgehog Signaling. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2012; Volume 88, pp. 55–72. ISBN 978-0-12-394622-5. [Google Scholar]
- Gu, D.; Xie, J. Non-Canonical Hh Signaling in Cancer—Current Understanding and Future Directions. Cancers 2015, 7, 1684–1698. [Google Scholar] [CrossRef]
- Regan, J.L.; Schumacher, D.; Staudte, S.; Steffen, A.; Haybaeck, J.; Keilholz, U.; Schweiger, C.; Golob-Schwarzl, N.; Mumberg, D.; Henderson, D.; et al. Non-Canonical Hedgehog Signaling Is a Positive Regulator of the WNT Pathway and Is Required for the Survival of Colon Cancer Stem Cells. Cell Rep. 2017, 21, 2813–2828. [Google Scholar] [CrossRef]
- Bermudez, O.; Hennen, E.; Koch, I.; Lindner, M.; Eickelberg, O. Gli1 mediates lung cancer cell proliferation and Sonic Hedgehog-dependent mesenchymal cell activation. PLoS ONE 2013, 8, e63226. [Google Scholar] [CrossRef]
- Hatton, B.A.; Knoepfler, P.S.; Kenney, A.M.; Rowitch, D.H.; de Alborán, I.M.; Olson, J.M.; Eisenman, R.N. N-myc Is an Essential Downstream Effector of Shh Signaling during both Normal and Neoplastic Cerebellar Growth. Cancer Res. 2006, 66, 8655–8661. [Google Scholar] [CrossRef]
- Bora-Singhal, N.; Perumal, D.; Nguyen, J.; Chellappan, S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non-Small Cell Lung Cancer. Neoplasia 2015, 17, 538–551. [Google Scholar] [CrossRef]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.-N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef]
- Louro, I.D.; Bailey, E.C.; Li, X.; South, L.S.; McKie-Bell, P.R.; Yoder, B.K.; Huang, C.C.; Johnson, M.R.; Hill, A.E.; Johnson, R.L.; et al. Comparative gene expression profile analysis of GLI and c-MYC in an epithelial model of malignant transformation. Cancer Res. 2002, 62, 5867–5873. [Google Scholar]
- Carpenter, R.L.; Paw, I.; Zhu, H.; Sirkisoon, S.; Xing, F.; Watabe, K.; Debinski, W.; Lo, H.-W. The gain-of-function GLI1 transcription factor TGLI1 enhances expression of VEGF-C and TEM7 to promote glioblastoma angiogenesis. Oncotarget 2015, 6, 22653–22665. [Google Scholar] [CrossRef]
- Pola, R.; Ling, L.E.; Silver, M.; Corbley, M.J.; Kearney, M.; Blake Pepinsky, R.; Shapiro, R.; Taylor, F.R.; Baker, D.P.; Asahara, T.; et al. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat. Med. 2001, 7, 706–711. [Google Scholar] [CrossRef]
- Regl, G.; Kasper, M.; Schnidar, H.; Eichberger, T.; Neill, G.W.; Philpott, M.P.; Esterbauer, H.; Hauser-Kronberger, C.; Frischauf, A.-M.; Aberger, F. Activation of the BCL2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res. 2004, 64, 7724–7731. [Google Scholar] [CrossRef]
- Fan, Q.; He, M.; Sheng, T.; Zhang, X.; Sinha, M.; Luxon, B.; Zhao, X.; Xie, J. Requirement of TGFβ Signaling for SMO-mediated Carcinogenesis. J. Biol. Chem. 2010, 285, 36570–36576. [Google Scholar] [CrossRef]
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.G.; Munoz, A.; et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004, 145, 3961–3970. [Google Scholar] [CrossRef]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Castillo, C.F.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef]
- Didiasova, M.; Schaefer, L.; Wygrecka, M. Targeting GLI Transcription Factors in Cancer. Molecules 2018, 23, 1003. [Google Scholar] [CrossRef]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef]
- Kimura, H.; Stephen, D.; Joyner, A.; Curran, T. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene 2005, 24, 4026–4036. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Ma, J.; Hawkins, C. GLI2 is a potential therapeutic target in pediatric medulloblastoma. J. Neuropathol. Exp. Neurol. 2011, 70, 430–437. [Google Scholar] [CrossRef]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 Signaling Regulates Human Glioma Growth, Cancer Stem Cell Self-Renewal, and Tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal Growth Factor Receptor Signaling Synergizes with Hedgehog/GLI in Oncogenic Transformation via Activation of the MEK/ERK/JUN Pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef]
- Abe, Y.; Oda-Sato, E.; Tobiume, K.; Kawauchi, K.; Taya, Y.; Okamoto, K.; Oren, M.; Tanaka, N. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc. Natl. Acad. Sci. USA 2008, 105, 4838–4843. [Google Scholar] [CrossRef]
- Inaguma, S.; Riku, M.; Hashimoto, M.; Murakami, H.; Saga, S.; Ikeda, H.; Kasai, K. GLI1 interferes with the DNA mismatch repair system in pancreatic cancer through BHLHE41-mediated suppression of MLH1. Cancer Res. 2013, 73, 7313–7323. [Google Scholar] [CrossRef]
- Inaguma, S.; Kasai, K.; Ikeda, H. GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene 2011, 30, 714–723. [Google Scholar] [CrossRef]
- Villegas, V.E.; Rahman, M.F.-U.; Fernandez-Barrena, M.G.; Diao, Y.; Liapi, E.; Sonkoly, E.; Ståhle, M.; Pivarcsi, A.; Annaratone, L.; Sapino, A.; et al. Identification of novel non-coding RNA-based negative feedback regulating the expression of the oncogenic transcription factor GLI1. Mol. Oncol. 2014, 8, 912–926. [Google Scholar] [CrossRef]
- Ferretti, E.; De Smaele, E.; Miele, E.; Laneve, P.; Po, A.; Pelloni, M.; Paganelli, A.; Di Marcotullio, L.; Caffarelli, E.; Screpanti, I.; et al. Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J. 2008, 27, 2616–2627. [Google Scholar] [CrossRef]
- Peng, B.; Li, D.; Qin, M.; Luo, D.; Zhang, X.; Zhao, H.; Hu, S. MicroRNA218 inhibits glioma migration and invasion via inhibiting glioma-associated oncogene homolog 1 expression at N terminus. Tumor Biol. 2014, 35, 3831–3837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, J.; Zhang, L.; Qu, Y.; Li, J.; Yu, B.; Yan, M.; Yu, Y.; Liu, B.; Zhu, Z. MiR-133b is frequently decreased in gastric cancer and its overexpression reduces the metastatic potential of gastric cancer cells. BMC Cancer 2014, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, C.; Wang, M.; Su, L.; Qu, Y.; Li, J.; Yu, B.; Yan, M.; Yu, Y.; Liu, B.; et al. Decrease of miR-202-3p expression, a novel tumor suppressor, in gastric cancer. PLoS ONE 2013, 8, e69756. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Fang, Z.-Y.; Cui, S.-Z.; Zhang, X.-L.; Wu, Y.-B.; Tang, H.-S.; Tu, Y.-N.; Ding, Y. Thermo-chemotherapy Induced miR-218 upregulation inhibits the invasion of gastric cancer via targeting Gli2 and E-cadherin. Tumor Biol. 2015, 36, 5807–5814. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Z.; Li, M.-J.; Guo, H.-Y.; Jing, N.-C. Long non-coding RNA metastasis-associated lung adenocarcinoma transcript 1 regulates the expression of Gli2 by miR-202 to strengthen gastric cancer progression. Biomed. Pharmacother. 2017, 85, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Raza, S.S.; Knab, L.M.; Chow, C.R.; Kwok, B.; Bentrem, D.J.; Popovic, R.; Ebine, K.; Licht, J.D.; Munshi, H.G. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci. Rep. 2015, 5, 9489. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef]
- Benvenuto, M.; Masuelli, L.; Smaele, E.D.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Stefano, E.D.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.; Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.-J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, G.; Li, Q.; Wang, Z.; Hu, W.; Li, P.; Li, S.; Wu, H.; Kong, X.; Gao, J.; et al. Hedgehog Signaling Non-Canonical Activated by Pro-Inflammatory Cytokines in Pancreatic Ductal Adenocarcinoma. J. Cancer 2016, 7, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 Is a Direct Transcriptional Target of EWS-FLI1 Oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Gallant, M.; Lamm, M.L.G.; Iannaccone, S.; Vieux, K.-F.; Proytcheva, M.; Hyjek, E.; Iannaccone, P.; Walterhouse, D. Noncanonical regulation of the Hedgehog mediator GLI1 by c-MYC in Burkitt lymphoma. Mol. Cancer Res. 2013, 11, 604–615. [Google Scholar] [CrossRef]
- Flora, A.; Klisch, T.J.; Schuster, G.; Zoghbi, H.Y. Deletion of Atoh1 disrupts Sonic Hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science 2009, 326, 1424–1427. [Google Scholar] [CrossRef]
- Trnski, D.; Sabol, M.; Gojević, A.; Martinić, M.; Ozretić, P.; Musani, V.; Ramić, S.; Levanat, S. GSK3β and Gli3 play a role in activation of Hedgehog-Gli pathway in human colon cancer—Targeting GSK3β downregulates the signaling pathway and reduces cell proliferation. Biochim. Biophys. Acta 2015, 1852, 2574–2584. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, J.; Tian, T.; Fu, X.; Wang, W.; Li, S.; Shi, T.; Suo, A.; Ruan, Z.; Guo, H.; et al. SET7/9 inhibits oncogenic activities through regulation of Gli-1 expression in breast cancer. Tumor Biol. 2016, 37, 9311–9322. [Google Scholar] [CrossRef]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q.; et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-W.; Zhu, H.; Cao, X.; Aldrich, A.; Ali-Osman, F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009, 69, 6790–6798. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Geradts, J.; Dewhirst, M.W.; Lo, H.-W. Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene 2012, 31, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, T.; Rahman, M.F.-U.; Tostar, U.; Sonkoly, E.; Ståhle, M.; Pivarcsi, A.; Palaniswamy, R.; Zaphiropoulos, P.G. RNA editing of the GLI1 transcription factor modulates the output of Hedgehog signaling. RNA Biol. 2013, 10, 321–333. [Google Scholar] [CrossRef]
- Lazzari, E.; Mondala, P.K.; Santos, N.D.; Miller, A.C.; Pineda, G.; Jiang, Q.; Leu, H.; Ali, S.A.; Ganesan, A.-P.; Wu, C.N.; et al. Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat. Commun. 2017, 8, 1922. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, D.; Jiang, J.; Hu, J.; Zhang, W.; Chen, Y.; Cui, X.; Qi, Y.; Zou, H.; Zhang, W.; et al. Analysis of Molecular Cytogenetic Alteration in Rhabdomyosarcoma by Array Comparative Genomic Hybridization. PLoS ONE 2014, 9, e94924. [Google Scholar] [CrossRef] [PubMed]
- Naylor, T.L.; Greshock, J.; Wang, Y.; Colligon, T.; Yu, Q.C.; Clemmer, V.; Zaks, T.Z.; Weber, B.L. High resolution genomic analysis of sporadic breast cancer using array-based comparative genomic hybridization. Breast Cancer Res. 2005, 7, R1186–R1198. [Google Scholar] [CrossRef]
- Nessling, M.; Richter, K.; Schwaenen, C.; Roerig, P.; Wrobel, G.; Wessendorf, S.; Fritz, B.; Bentz, M.; Sinn, H.-P.; Radlwimmer, B.; et al. Candidate genes in breast cancer revealed by microarray-based comparative genomic hybridization of archived tissue. Cancer Res. 2005, 65, 439–447. [Google Scholar]
- Roberts, W.M.; Douglass, E.C.; Peiper, S.C.; Houghton, P.J.; Look, A.T. Amplification of the gli gene in childhood sarcomas. Cancer Res. 1989, 49, 5407–5413. [Google Scholar]
- Snijders, A.M.; Schmidt, B.L.; Fridlyand, J.; Dekker, N.; Pinkel, D.; Jordan, R.C.K.; Albertson, D.G. Rare amplicons implicate frequent deregulation of cell fate specification pathways in oral squamous cell carcinoma. Oncogene 2005, 24, 4232–4242. [Google Scholar] [CrossRef] [PubMed]
- Bridge, J.A.; Sanders, K.; Huang, D.; Nelson, M.; Neff, J.R.; Muirhead, D.; Walker, C.; Seemayer, T.A.; Sumegi, J. Pericytoma with t(7;12) and ACTB-GLI1 fusion arising in bone. Hum. Pathol. 2012, 43, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Dahlén, A.; Mertens, F.; Mandahl, N.; Panagopoulos, I. Molecular genetic characterization of the genomic ACTB-GLI fusion in pericytoma with t(7;12). Biochem. Biophys. Res. Commun. 2004, 325, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Kuromi, T.; Matsushita, M.; Iwasaki, T.; Nonaka, D.; Kuwamoto, S.; Nagata, K.; Kato, M.; Akizuki, G.; Kitamura, Y.; Hayashi, K. Association of expression of the hedgehog signal with Merkel cell polyomavirus infection and prognosis of Merkel cell carcinoma. Hum. Pathol. 2017, 69, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Dagklis, A.; Pauwels, D.; Lahortiga, I.; Geerdens, E.; Bittoun, E.; Cauwelier, B.; Tousseyn, T.; Uyttebroeck, A.; Maertens, J.; Verhoef, G.; et al. Hedgehog pathway mutations in T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, e102–e105. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Song, S.Y.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Intratumoral Heterogeneity of Frameshift Mutations of GLI1 Encoding a Hedgehog Signaling Protein in Colorectal Cancers. Pathol. Oncol. Res. 2018, 24, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Loh, M.; Liem, N.; Vaithilingam, A.; Lim, P.L.; Sapari, N.S.; Elahi, E.; Mok, Z.Y.; Cheng, C.L.; Yan, B.; Pang, B.; et al. DNA methylation subgroups and the CpG island methylator phenotype in gastric cancer: A comprehensive profiling approach. BMC Gastroenterol. 2014, 14, 55. [Google Scholar] [CrossRef]
- Lucchesi, C.; Khalifa, E.; Laizet, Y.; Soubeyran, I.; Mathoulin-Pelissier, S.; Chomienne, C.; Italiano, A. Targetable Alterations in Adult Patients With Soft-Tissue Sarcomas: Insights for Personalized Therapy. JAMA Oncol. 2018, 4, 1398–1404. [Google Scholar] [CrossRef]
- Bai, C.B.; Auerbach, W.; Lee, J.S.; Stephen, D.; Joyner, A.L. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 2002, 129, 4753–4761. [Google Scholar] [PubMed]
- Drakopoulou, E.; Outram, S.V.; Rowbotham, N.J.; Ross, S.E.; Furmanski, A.L.; Saldana, J.I.; Hager-Theodorides, A.L.; Crompton, T. Non-redundant role for the transcription factor Gli1 at multiple stages of thymocyte development. Cell Cycle 2010, 9, 4144–4152. [Google Scholar] [CrossRef] [PubMed]
- Park, H.L.; Bai, C.; Platt, K.A.; Matise, M.P.; Beeghly, A.; Hui, C.C.; Nakashima, M.; Joyner, A.L. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 2000, 127, 1593–1605. [Google Scholar] [PubMed]
- Ding, Q.; Motoyama, J.; Gasca, S.; Mo, R.; Sasaki, H.; Rossant, J.; Hui, C.C. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 1998, 125, 2533–2543. [Google Scholar] [PubMed]
- Matise, M.P.; Epstein, D.J.; Park, H.L.; Platt, K.A.; Joyner, A.L. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 1998, 125, 2759–2770. [Google Scholar] [PubMed]
- Mo, R.; Freer, A.M.; Zinyk, D.L.; Crackower, M.A.; Michaud, J.; Heng, H.H.; Chik, K.W.; Shi, X.M.; Tsui, L.C.; Cheng, S.H.; et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 1997, 124, 113–123. [Google Scholar]
- Motoyama, J.; Milenkovic, L.; Iwama, M.; Shikata, Y.; Scott, M.P.; Hui, C. Differential requirement for Gli2 and Gli3 in ventral neural cell fate specification. Dev. Biol. 2003, 259, 150–161. [Google Scholar] [CrossRef]
- Bai, C.B.; Joyner, A.L. Gli1 can rescue the in vivo function of Gli2. Development 2001, 128, 5161–5172. [Google Scholar] [PubMed]
- Bowers, M.; Eng, L.; Lao, Z.; Turnbull, R.K.; Bao, X.; Riedel, E.; Mackem, S.; Joyner, A.L. Limb anterior-posterior polarity integrates activator and repressor functions of GLI2 as well as GLI3. Dev. Biol. 2012, 370, 110–124. [Google Scholar] [CrossRef]
- Hui, C.C.; Joyner, A.L. A mouse model of greig cephalopolysyndactyly syndrome: The extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 1993, 3, 241–246. [Google Scholar] [CrossRef]
- Litingtung, Y.; Dahn, R.D.; Li, Y.; Fallon, J.F.; Chiang, C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature 2002, 418, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Theil, T.; Alvarez-Bolado, G.; Walter, A.; Rüther, U. Gli3 is required for Emx gene expression during dorsal telencephalon development. Development 1999, 126, 3561–3571. [Google Scholar]
- Böse, J.; Grotewold, L.; Rüther, U. Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum. Mol. Genet. 2002, 11, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Stamataki, D. A gradient of Gli activity mediates graded Sonic Hedgehog signaling in the neural tube. Genes Dev. 2005, 19, 626–641. [Google Scholar] [CrossRef]
- Litingtung, Y.; Chiang, C. Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat. Neurosci. 2000, 3, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Wijgerde, M.; McMahon, J.A.; Rule, M.; McMahon, A.P. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002, 16, 2849–2864. [Google Scholar] [CrossRef] [PubMed]
- Oosterveen, T.; Kurdija, S.; Alekseenko, Z.; Uhde, C.W.; Bergsland, M.; Sandberg, M.; Andersson, E.; Dias, J.M.; Muhr, J.; Ericson, J. Mechanistic differences in the transcriptional interpretation of local and long-range Shh morphogen signaling. Dev. Cell 2012, 23, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Stamataki, D.; te Welscher, P.; Andersson, E.; Böse, J.; Rüther, U.; Ericson, J.; Briscoe, J. Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 2002, 16, 2865–2878. [Google Scholar] [CrossRef] [PubMed]
- Rallu, M.; Machold, R.; Gaiano, N.; Corbin, J.G.; McMahon, A.P.; Fishell, G. Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development 2002, 129, 4963–4974. [Google Scholar] [PubMed]
- Tickle, C.; Towers, M. Sonic Hedgehog Signaling in Limb Development. Front. Cell Dev. Biol. 2017, 5, 14. [Google Scholar] [CrossRef]
- Wang, B.; Fallon, J.F.; Beachy, P.A. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, C.; Wang, B. Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog-regulated mouse development. Dev. Biol. 2009, 326, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Rüther, U.; Wang, B. The Shh-independent activator function of the full-length Gli3 protein and its role in vertebrate limb digit patterning. Dev. Biol. 2007, 305, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Falkenstein, K.N.; Vokes, S.A. Transcriptional regulation of graded Hedgehog signaling. Semin. Cell Dev. Biol. 2014, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; He, G.; Lee, W.-C.; McKenzie, J.A.; Silva, M.J.; Long, F. Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat. Commun. 2017, 8, 2043. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Andre, P.; Ye, L.; Yang, Y.-Z. The Hedgehog signalling pathway in bone formation. Int. J. Oral. Sci. 2015, 7, 73–79. [Google Scholar] [CrossRef]
- Joeng, K.S.; Long, F. The Gli2 transcriptional activator is a crucial effector for Ihh signaling in osteoblast development and cartilage vascularization. Development 2009, 136, 4177–4185. [Google Scholar] [CrossRef]
- Miao, D.; Liu, H.; Plut, P.; Niu, M.; Huo, R.; Goltzman, D.; Henderson, J.E. Impaired endochondral bone development and osteopenia in Gli2-deficient mice. Exp. Cell Res. 2004, 294, 210–222. [Google Scholar] [CrossRef]
- Hilton, M.J.; Tu, X.; Cook, J.; Hu, H.; Long, F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development 2005, 132, 4339–4351. [Google Scholar] [CrossRef]
- Koziel, L.; Wuelling, M.; Schneider, S.; Vortkamp, A. Gli3 acts as a repressor downstream of Ihh in regulating two distinct steps of chondrocyte differentiation. Development 2005, 132, 5249–5260. [Google Scholar] [CrossRef]
- Shimoyama, A.; Wada, M.; Ikeda, F.; Hata, K.; Matsubara, T.; Nifuji, A.; Noda, M.; Amano, K.; Yamaguchi, A.; Nishimura, R.; et al. Ihh/Gli2 signaling promotes osteoblast differentiation by regulating Runx2 expression and function. Mol. Biol. Cell 2007, 18, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Regard, J.B.; Malhotra, D.; Gvozdenovic-Jeremic, J.; Josey, M.; Chen, M.; Weinstein, L.S.; Lu, J.; Shore, E.M.; Kaplan, F.S.; Yang, Y. Activation of Hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat. Med. 2013, 19, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Hojo, H.; Ohba, S.; Yano, F.; Saito, T.; Ikeda, T.; Nakajima, K.; Komiyama, Y.; Nakagata, N.; Suzuki, K.; Takato, T.; et al. Gli1 protein participates in Hedgehog-mediated specification of osteoblast lineage during endochondral ossification. J. Biol. Chem. 2012, 287, 17860–17869. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.P.C.; Connor, E.C.; Veltmaat, J.M.; Lana-Elola, E.; Veistinen, L.; Tanimoto, Y.; Bellusci, S.; Rice, R. Gli3Xt−J/Xt−J mice exhibit lambdoid suture craniosynostosis which results from altered osteoprogenitor proliferation and differentiation. Hum. Mol. Genet. 2010, 19, 3457–3467. [Google Scholar] [CrossRef] [PubMed]
- Veistinen, L.; Takatalo, M.; Kesper, D.A.; Vortkamp, A.; Rice, D.P. Deletion of Gli3 in mice causes abnormal frontal bone morphology and premature synostosis of the interfrontal suture. Front. Physiol. 2012, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Blaess, S. Sonic hedgehog regulates Gli activator and repressor functions with spatial and temporal precision in the mid/hindbrain region. Development 2006, 133, 1799–1809. [Google Scholar] [CrossRef]
- Corrales, J.D. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development 2004, 131, 5581–5590. [Google Scholar] [CrossRef]
- Corrales, J.D.; Blaess, S.; Mahoney, E.M.; Joyner, A.L. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development 2006, 133, 1811–1821. [Google Scholar] [CrossRef]
- Kim, J.J.; Gill, P.S.; Rotin, L.; van Eede, M.; Henkelman, R.M.; Hui, C.-C.; Rosenblum, N.D. Suppressor of fused controls mid-hindbrain patterning and cerebellar morphogenesis via GLI3 repressor. J. Neurosci. 2011, 31, 1825–1836. [Google Scholar] [CrossRef]
- Cain, J.E.; Islam, E.; Haxho, F.; Blake, J.; Rosenblum, N.D. GLI3 repressor controls functional development of the mouse ureter. J. Clin. Invest. 2011, 121, 1199–1206. [Google Scholar] [CrossRef]
- Cain, J.E.; Islam, E.; Haxho, F.; Chen, L.; Bridgewater, D.; Nieuwenhuis, E.; Hui, C.-C.; Rosenblum, N.D. GLI3 repressor controls nephron number via regulation of Wnt11 and Ret in ureteric tip cells. PLoS ONE 2009, 4, e7313. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Mo, R.; Bhella, S.; Wilson, C.W.; Chuang, P.-T.; Hui, C.-C.; Rosenblum, N.D. GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes disrupts renal morphogenesis. Development 2006, 133, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yeung, C.-K.; Ng, Y.-K.; Zhang, J.-R.; Hui, C.-C.; Kim, P.C.W. Sonic Hedgehog Mediator Gli2 Regulates Bladder Mesenchymal Patterning. J. Urol. 2008, 180, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Buttitta, L. Interplays of Gli2 and Gli3 and their requirement in mediating Shh-dependent sclerotome induction. Development 2003, 130, 6233–6243. [Google Scholar] [CrossRef] [PubMed]
- Grindley, J.C.; Bellusci, S.; Perkins, D.; Hogan, B.L.M. Evidence for the Involvement of theGliGene Family in Embryonic Mouse Lung Development. Dev. Biol. 1997, 188, 337–348. [Google Scholar] [CrossRef]
- Motoyama, J.; Liu, J.; Mo, R.; Ding, Q.; Post, M.; Hui, C.C. Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat. Genet. 1998, 20, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.B.; Gong, Y.; Ruan, H.F.; Pan, L.Y.; Wu, X.K.; Tang, C.; Wang, C.J.; Zhu, H.B.; Zhang, Z.M.; Tang, L.F.; et al. Sonic hedgehog through Gli2 and Gli3 is required for the proper development of placental labyrinth. Cell Death Dis. 2015, 6, e1653. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.J.; Cowin, P. Gli3-mediated repression of Hedgehog targets is required for normal mammary development. Development 2006, 133, 3661–3670. [Google Scholar] [CrossRef] [PubMed]
- Veltmaat, J.M.; Relaix, F.; Le, L.T.; Kratochwil, K.; Sala, F.G.; van Veelen, W.; Rice, R.; Spencer-Dene, B.; Mailleux, A.A.; Rice, D.P.; et al. Gli3-mediated somitic Fgf10 expression gradients are required for the induction and patterning of mammary epithelium along the embryonic axes. Development 2006, 133, 2325–2335. [Google Scholar] [CrossRef]
- Lewis, M.T.; Ross, S.; Strickland, P.A.; Sugnet, C.W.; Jimenez, E.; Hui, C.; Daniel, C.W. The Gli2 transcription factor is required for normal mouse mammary gland development. Dev. Biol. 2001, 238, 133–144. [Google Scholar] [CrossRef]
- Barsoum, I.; Yao, H.H.C. Redundant and Differential Roles of Transcription Factors Gli1 and Gli2 in the Development of Mouse Fetal Leydig Cells. Biol. Reprod. 2011, 84, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Mill, P.; Mo, R.; Fu, H.; Grachtchouk, M.; Kim, P.C.W.; Dlugosz, A.A.; Hui, C. Sonic hedgehog-dependent activation of Gli2 is essential for embryonic hair follicle development. Genes Dev. 2003, 17, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Hager-Theodorides, A.L.; Dessens, J.T.; Outram, S.V.; Crompton, T. The transcription factor Gli3 regulates differentiation of fetal CD4- CD8- double-negative thymocytes. Blood 2005, 106, 1296–1304. [Google Scholar] [CrossRef]
- Rowbotham, N.J.; Hager-Theodorides, A.L.; Furmanski, A.L.; Ross, S.E.; Outram, S.V.; Dessens, J.T.; Crompton, T. Sonic hedgehog negatively regulates pre-TCR-induced differentiation by a Gli2-dependent mechanism. Blood 2009, 113, 5144–5156. [Google Scholar] [CrossRef] [PubMed]
- Solanki, A.; Yanez, D.C.; Ross, S.; Lau, C.-I.; Papaioannou, E.; Li, J.; Saldaña, J.I.; Crompton, T. Gli3 in fetal thymic epithelial cells promotes thymocyte positive selection and differentiation by repression of Shh. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- McDermott, A.; Gustafsson, M.; Elsam, T.; Hui, C.-C.; Emerson, C.P.; Borycki, A.-G. Gli2 and Gli3 have redundant and context-dependent function in skeletal muscle formation. Development 2005, 132, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Brewster, R.; Mullor, J.L.; Ruiz i Altaba, A. Gli2 functions in FGF signaling during antero-posterior patterning. Development 2000, 127, 4395–4405. [Google Scholar]
- Pu, Y.; Huang, L.; Prins, G.S. Sonic hedgehog-patched Gli signaling in the developing rat prostate gland: Lobe-specific suppression by neonatal estrogens reduces ductal growth and branching. Dev. Biol. 2004, 273, 257–275. [Google Scholar] [CrossRef]
- Babu, D.; Fanelli, A.; Mellone, S.; Muniswamy, R.; Wasniewska, M.; Prodam, F.; Petri, A.; Bellone, S.; Salerno, M.C.; Giordano, M. Novel GLI2 mutations identified in patients with Combined Pituitary Hormone Deficiency (CPHD): Evidence for a pathogenic effect by functional characterization. Clin. Endocrinol. 2018. [Google Scholar] [CrossRef]
- França, M.M.; Jorge, A.A.L.; Carvalho, L.R.S.; Costalonga, E.F.; Otto, A.P.; Correa, F.A.; Mendonca, B.B.; Arnhold, I.J.P. Relatively high frequency of non-synonymous GLI2 variants in patients with congenital hypopituitarism without holoprosencephaly. Clin. Endocrinol. 2013, 78, 551–557. [Google Scholar] [CrossRef]
- Roessler, E.; Du, Y.-Z.; Mullor, J.L.; Casas, E.; Allen, W.P.; Gillessen-Kaesbach, G.; Roeder, E.R.; Ming, J.E.; Ruiz i Altaba, A.; Muenke, M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. PNAS 2003, 100, 13424–13429. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Sapp, J.C.; Turner, J.T.; Amor, D.; Aftimos, S.; Aleck, K.A.; Bocian, M.; Bodurtha, J.N.; Cox, G.F.; Curry, C.J.; et al. Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum. Mutat. 2010, 31, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Olivos-Glander, I.; Killoran, C.; Elson, E.; Turner, J.T.; Peters, K.F.; Abbott, M.H.; Aughton, D.J.; Aylsworth, A.S.; Bamshad, M.J.; et al. Molecular and Clinical Analyses of Greig Cephalopolysyndactyly and Pallister-Hall Syndromes: Robust Phenotype Prediction from the Type and Position of GLI3 Mutations. Am. J. Hum. Genet. 2005, 76, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Palencia-Campos, A.; Ullah, A.; Nevado, J.; Yildirim, R.; Unal, E.; Ciorraga, M.; Barruz, P.; Chico, L.; Piceci-Sparascio, F.; Guida, V.; et al. GLI1 inactivation is associated with developmental phenotypes overlapping with Ellis-van Creveld syndrome. Hum. Mol. Genet. 2017, 26, 4556–4571. [Google Scholar] [PubMed]
- Ihrie, R.A.; Shah, J.K.; Harwell, C.C.; Levine, J.H.; Guinto, C.D.; Lezameta, M.; Kriegstein, A.R.; Alvarez-Buylla, A. Persistent Sonic Hedgehog Signaling in Adult Brain Determines Neural Stem Cell Positional Identity. Neuron 2011, 71, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Petrova, R.; Garcia, A.D.R.; Joyner, A.L. Titration of GLI3 Repressor Activity by Sonic Hedgehog Signaling Is Critical for Maintaining Multiple Adult Neural Stem Cell and Astrocyte Functions. J. Neurosci. 2013, 33, 17490–17505. [Google Scholar] [CrossRef] [PubMed]
- Ringuette, R.; Atkins, M.; Lagali, P.S.; Bassett, E.A.; Campbell, C.; Mazerolle, C.; Mears, A.J.; Picketts, D.J.; Wallace, V.A. A Notch-Gli2 axis sustains Hedgehog responsiveness of neural progenitors and Müller glia. Dev. Biol. 2016, 411, 85–100. [Google Scholar] [CrossRef]
- Zhao, C.; Cai, S.; Shin, K.; Lim, A.; Kalisky, T.; Lu, W.-J.; Clarke, M.F.; Beachy, P.A. Stromal Gli2 activity coordinates a niche signaling program for mammary epithelial stem cells. Science 2017, 356, eaal3485. [Google Scholar] [CrossRef]
- Merchant, A.; Joseph, G.; Wang, Q.; Brennan, S.; Matsui, W. Gli1 regulates the proliferation and differentiation of HSCs and myeloid progenitors. Blood 2010, 115, 2391–2396. [Google Scholar] [CrossRef]
- Singh, B.N.; Koyano-Nakagawa, N.; Gong, W.; Moskowitz, I.P.; Weaver, C.V.; Braunlin, E.; Das, S.; van Berlo, J.H.; Garry, M.G.; Garry, D.J. A conserved HH-Gli1-Mycn network regulates heart regeneration from newt to human. Nat. Commun. 2018, 9, 4237. [Google Scholar] [CrossRef]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Type * | Enzyme | Residue Number | Effect on the Hh Pathway | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|
| mGli1 | hGli1 | mGli2 | hGli2 | mGli3 | hGli3 | ||||
| P | PKA | S789, S805, S817, S848, S923, S956 | S849, S865, S877, S907, S980, S1006 | inhibition, increased processing to GliR, reduced nuclear accumulation of Gli2/3 | [23] | ||||
| P | PKA | T374 | inhibition due to decreased nuclear localization | [47] | |||||
| P | PKA | S544, S560 | inhibition | [48] | |||||
| P | aPKC | S246 **, S307 ** | S243, S304 | activation, increased DNA binding, promoting deacetylation | [49,50] | ||||
| P | AMPK | S105 **, T1079 ** | S102, S408, T1074 | inhibition, destabilization | [51] | ||||
| P | MEKK1 | + *** | inhibition | [52] | |||||
| P | Hck | Y800 | activation | [53] | |||||
| P | GSK3β | S861, S873, S903 | inhibition, increased GliR formation | [46] | |||||
| P | ? | S850, S894 | inhibition, increased GliR formation | [46] | |||||
| P | GSK3β | S801, S813, S844 | inhibition, destabilization | [54] | |||||
| P | CK1 | S852, S868, S880, S910 | inhibition, increased GliR formation | [55] | |||||
| P | CK1 | S792, S808, S820 S851 | inhibition, destabilization | [54] | |||||
| P | Dyrk1a | + *** | activation, increased nuclear accumulation | [56] | |||||
| P | Dyrk1b | + *** | non-canonical activation due to AKT phosphorylation | [57] | |||||
| P | Dyrk2 | S385, S1011 | inhibition, destabilization | [58] | |||||
| P | ULK3 | + *** | + *** | + *** | activation | [59] | |||
| P | S6K1 | S87 ** | S84 | - | - | non-canonical activation | [60] | ||
| P | AKT | - | S230 | S234 ** | - | non-canonical activation, stabilization | [61,62] | ||
| P | CIT | S145 ** | S149 | non-canonical activation, nuclear accumulation | [63] | ||||
| P | ? | S248 | activation (?) | [23] | |||||
| P | ? | - | S662 | - | inhibition, destabilization | [64] | |||
| Ub | Cul1/β-TrCP | K773, K779, K784, K800 | inhibition, GliR formation | [46] | |||||
| Ub | Cul3/Spop | + *** | + *** | inhibition, destabilization | [65] | ||||
| de-Ub | HAUSP | + *** | + *** | + *** | activation, stabilization | [66] | |||
| de-Ub | USP48 | + *** | activation, stabilization | [67] | |||||
| SUMO | Pias1 | K180, K815 | K376, K630, K716 | K87, K462, K696, K779 | activation, stabilization | [68] | |||
| SUMO | ? | K630, K716 | inhibition, binding to HDAC5 (?) | [69] | |||||
| de-SUMO | SENP1 | K180, K415, K815 | inhibition, destabilization, nuclear export | [70] | |||||
| Ac | p300, HATs | K518 | inhibition | [24] | |||||
| de-Ac | HDAC1 | K518 | activation | [24] | |||||
| G | ? | + *** | + *** | activation, nuclear accumulation | [71] | ||||
| Me | Set7 | K436, K595 | stabilization, increased DNA binding | [72] | |||||
| Member of Gli Family | Tumor Type | Experimental Model | Activation Cause | Effect | Reference |
|---|---|---|---|---|---|
| Canonical Activation via Hh/Ptch1/Smo | |||||
| Gli1 | melanoma | primary cell culture, short term culture xenograft | Smo-dependent | cancer stem cell self-renewal, tumor initiation | [183] |
| Gli1, Gli2 | medulloblastoma | genetic mouse model | Ptch1 mutation | malignant transformation | [184] |
| Gli2 | medulloblastoma | cell culture | Smo-dependent | proliferation, viability | [185] |
| Gli1 | colon cancer | primary cell culture, short term culture xenograft | Smo-dependent | cancer stem cell self-renewal, proliferation, xenograft growth, metastasis | [186] |
| Gli1, Gli2 | glioma | primary cell culture | Smo-dependent | cancer stem cell self-renewal, proliferation, viability | [187] |
| Gli1 | pancreatic and colon cancer | cell co-culture, patient-derived xenograft | Hh ligand secretion | stroma induction | [188] |
| Gli1 | prostate cancer | cell co-culture, cell line xenograft | Hh ligand secretion | stroma induction | [180] |
| ? | skin cancer (BCC-like) | genetic mouse model | mutant Smo | immune inhibition via TGFβ | [179] |
| Gli1, Gli2 | basal cell carcinoma | cell culture | Smo-dependent | synergy with EGFR/AP-1, malignant transformation, viability | [189] |
| Gli1, Gli2 | breast cancer | cell culture | Smo-dependent | p53 inhibition through activation of Mdm2 | [190] |
| Overexpression of Gli Proteins/Aberrant microRNA Silencing | |||||
| Gli1, Gli2 | pancreatic cancer | cell culture | Gli protein overexpression | migration, invasion, DNA damage | [191,192] |
| Gli1 | non-small cell lung cancer | cell culture | Gli1/Gli2 overexpression | cancer stem cell self-renewal | [173] |
| Gli1 | rhabdomyosarcoma | cell culture, CAM assay | loss of GLI1 antisense RNA | proliferation, tumor growth in CAM assay | [193] |
| Gli1 | medulloblastoma | cell culture | loss of miR-324-5p, also regulates Smo | proliferation, colony growth on soft agar | [194] |
| Gli1 | glioma | cell culture | loss of miR-218 | migration, invasion | [195] |
| Gli1 | gastric cancer | cell culture, cell line xenograft | loss of miR-202-3b and miR-133b | migration, invasion, xenograft growth and metastasis | [196,197] |
| Gli2 | gastric cancer | cell culture | loss of miR-218 | proliferation, migration, invasion | [198] |
| Gli2 | gastric cancer | cell culture | loss of miR-202 | proliferation, viability | [199] |
| Cross-Talk with Other Pathways | |||||
| Gli2 | pancreatic cancer | cell culture | Wnt | c-Myc expression, colony growth in collagen, drug resistance | [200] |
| Gli1 | pancreatic cancer | cell culture, genetic mouse model | TGFβ, KRAS | viability, malignant transformation | [201] |
| ? | Prostate cancer | cell line xenograft | Smo-independent | xenograft growth | [202] |
| ? | breast cancer | cell culture, cell line xenograft | Smo-independent | proliferation, viability, xenograft growth | [203] |
| Gli1 | pancreatic cancer | cell culture | KRAS | growth on soft agar | [204] |
| Gli1 | melanoma | cell line xenograft | NRAS, but also Smo-dependent | xenograft growth, recurrence, metastasis | [205] |
| Gli2 | pancreatic cancer | cell culture | TGFβ | proliferation | [206] |
| Gli1 | esophageal cancer | cell culture, cell line xenograft | TNFα/mTOR, but also Smo-dependent | viability | [60] |
| Gli1 | pancreatic cancer | cell culture | TNFα/IL1β | migration, invasion, EMT and drug resistance | [207] |
| Gli1 | breast cancer | cell culture, cell line xenograft | NFκB | viability, motility, EMT, clonogenicity, self-renewal | [208] |
| Gli1 | melanoma, breast cancer | cell culture, cell line xenograft | WIP1, but also Smo-dependent | proliferation, self-renewal, xenograft growth | [80] |
| Gli1 | glioblastoma | cell culture, cell line xenograft | p53, but also Smo-dependent | feedback loop on p53, proliferation, self-renewal, xenograft growth | [209] |
| Gli1 | Ewing sarcoma family of tumors | cell culture | EWS-FLI1 | proliferation, growth on soft agar | [210] |
| Gli1 | Burkitt lymphoma | cell culture | MYC | viability | [211] |
| Gli2 | medulloblastoma | genetic mouse model | ATOH1, but also Smo-dependent | proliferation | [212] |
| Gli3 | colon cancer | cell culture | GSK3B | proliferation, viability | [213] |
| Gli1 | breast cancer | cell culture | loss of SETD7, but also Smo-dependent | proliferation, migration, invasion | [214] |
| Gli3 | non-small cell lung cancer | cell culture, cell line xenograft | SETD7 | proliferation, xenograft growth and metastasis | [72] |
| Gli2 | breast cancer | cell culture, cell line xenograft | BCAR4 lncRNA | growth on Matrigel, xenograft metastasis | [63] |
| Gli2 | breast cancer | cell culture, cell line xenograft | FOXC1 | self-renewal, xenograft growth | [215] |
| RNA Splicing or Editing | |||||
| Gli1 | glioblastoma | cell culture | tGLI1 splice variant | cell migration, invasion | [216] |
| Gli1 | glioblastoma | cell culture | tGLI1 splice variant | angiogenesis | [176] |
| Gli1 | breast cancer | cell culture | tGLI1 splice variant | angiogenesis, migration, growth on soft agar | [217] |
| Gli1 | medulloblastoma | cell culture | RNA editing | proliferation | [218] |
| Gli1 | multiple myeloma | serial transplant xenograft | RNA editing | proliferation, transplant growth | [219] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niewiadomski, P.; Niedziółka, S.M.; Markiewicz, Ł.; Uśpieński, T.; Baran, B.; Chojnowska, K. Gli Proteins: Regulation in Development and Cancer. Cells 2019, 8, 147. https://doi.org/10.3390/cells8020147
Niewiadomski P, Niedziółka SM, Markiewicz Ł, Uśpieński T, Baran B, Chojnowska K. Gli Proteins: Regulation in Development and Cancer. Cells. 2019; 8(2):147. https://doi.org/10.3390/cells8020147
Chicago/Turabian StyleNiewiadomski, Paweł, Sylwia M. Niedziółka, Łukasz Markiewicz, Tomasz Uśpieński, Brygida Baran, and Katarzyna Chojnowska. 2019. "Gli Proteins: Regulation in Development and Cancer" Cells 8, no. 2: 147. https://doi.org/10.3390/cells8020147
APA StyleNiewiadomski, P., Niedziółka, S. M., Markiewicz, Ł., Uśpieński, T., Baran, B., & Chojnowska, K. (2019). Gli Proteins: Regulation in Development and Cancer. Cells, 8(2), 147. https://doi.org/10.3390/cells8020147