Pro-Thrombotic Activity of Blood Platelets in Multiple Sclerosis



Abstract
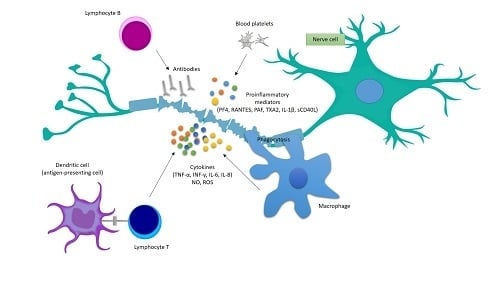
1. Introduction
2. Increased Risk of Cardiovascular Disorders
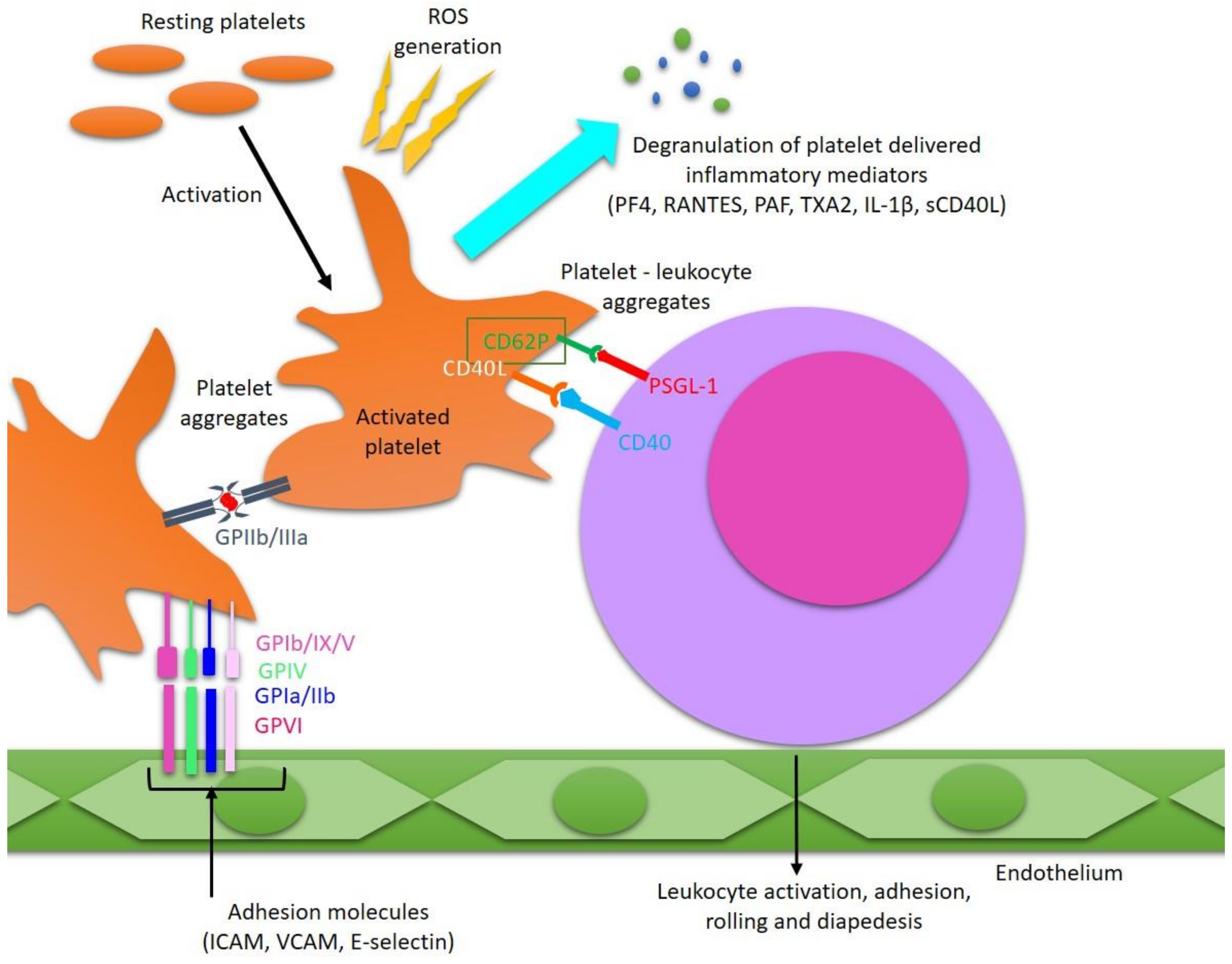
3. Pathological Activation of Platelets in MS
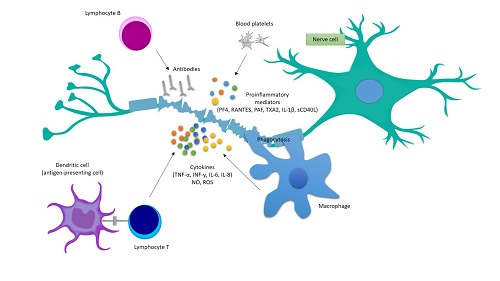
4. Platelets’ Involvement in the Development of Inflammatory Reactions in MS
Funding
Conflicts of Interest
References
- Lassmann, H.; van Horssen, J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011, 585, 3715–3723. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, D.; Simons, M. Chronic progressive multiple sclerosis-pathogenesis of neurodegeneration and therapeutic strategies. Curr. Neuropharmacol. 2010, 8, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Krieger, S.C.; Cook, K.; De Nino, S.; Fletcher, M. The topographical model of multiple sclerosis. Neurol.-Neuroimmunol. Neuroinflamm. 2016, e279. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Gaby, A. Multiple sclerosis. Glob. Adv. Health Med. 2013, 2, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Abboud, H.; Hill, E.; Siddiqui, J.; Serra, A.; Walter, B. Neuromodulation in multiple sclerosis. Mult. Scler. 2017, 23, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Motl, R.W. Lifestyle physical activity in persons wih multiple sclerosis: The new kind on the MS block. Mult. Scler. 2014, 20, 1025–1029. [Google Scholar] [CrossRef]
- Steinman, L. Multiple sclerosis: A two-stage disease. Nat. Immunol. 2001, 2, 762–764. [Google Scholar] [CrossRef]
- Engelhardt, B.; Ransohoff, R.M. The ins and outs of T-lymphocyte trafficking to the CNS: Anatomical sites and molecular mechanisms. Trends Immunol. 2005, 26, 485–495. [Google Scholar] [CrossRef]
- Wu, G.F.; Alvarez, E. The immunopathophysiology of multiple sclerosis. Neurol. Clin. 2011, 29, 257–278. [Google Scholar] [CrossRef]
- Klinger, M.H.; Jelkmann, W. Role of blood platelets in infection and inflammation. J. Interferon Cytokine Res. 2002, 22, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T. Platelets, inflammation and tissue regeneration. Thromb. Haemost. 2011, 105 (Suppl. S1), S13–S33. [Google Scholar] [CrossRef]
- Jenne, C.N.; Urrutia, R.; Kubes, P. Platelets: Bridging hemostasis, inflammation, and immunity. Int. J. Lab. Hematol. 2013, 35, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Steinhubl, S.R. Platelets as mediators of inflammation. Hematol. Oncol. Clin. N. Am. 2007, 21, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.P.; Simonsen, H.; Frederiksen, J.L.; Rostrup, E.; Larsson, H.B. Abnormal blood–brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. Neuroimage Clin. 2014, 4, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, C.F.; Christensen, S.; Farkas, D.K.; Miret, M.; Sørensen, H.T.; Pedersen, L. Risk of arterial cardiovascular diseases in patients with multiple sclerosis: A population-based cohort study. Neuroepidemiology 2010, 35, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.; Farkas, D.K.; Pedersen, L.; Miret, M.; Christiansen, C.F.; Sørensen, H.T. Multiple sclerosis and risk of venous thromboembolism: A population-based cohort study. Neuroepidemiology 2012, 38, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Jadidi, E.; Mohammadi, M.; Moradi, T. High risk of cardiovascular diseases after diagnosis of multiple sclerosis. Mult. Scler. 2013, 19, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Zöller, B.; Li, X.; Sundquist, J.; Sundquist, K. Risk of pulmonary embolism in patients with autoimmune disorders: A nationwide follow-up study from Sweden. Lancet 2012, 379, 244–249. [Google Scholar] [CrossRef]
- Peeters, P.J.; Bazelier, M.T.; Uitdehaag, B.M.; Leufkens, H.G.; De Bruin, M.L.; de Vries, F. The risk of venous thromboembolism in patients with multiple sclerosis: The Clinical Practice Research Datalink. J. Thromb. Haemost. 2014, 12, 444–451. [Google Scholar] [CrossRef]
- Koch-Henriksen, N.; Brønnum-Hansen, H.; Stenager, E. Underlying cause of death in Danish patients with multiple sclerosis: Results from the Danish Multiple Sclerosis Registry. J. Neurol. Neurosurg. Psychiatry 1998, 65, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Brønnum-Hansen, H.; Koch-Henriksen, N.; Stenager, E. Trends in survival and cause of death in Danish patients with multiple sclerosis. Brain 2004, 127, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.M.A.; Shah, S.M.S.; Khan, S.; Rehman, S.U.; Khan, Z.A.; Ahmed, W. “Addressing the impact of stroke risk factors in a case control study in tertiary care hospitals”: A case control study in Tertiary Care Hospitals of Peshawar, Khyber Phukhtoonkhwa (KPK) Pakistan. BMC Res. Notes 2013, 6, 268. [Google Scholar] [CrossRef] [PubMed]
- Thormann, A.; Magyari, M.; Koch-Henriksen, N.; Laursen, B.; Sorensen, P.S. Vascular comorbidities in multiple sclerosis: A nationwide study from Denmark. J. Neurol. 2016, 263, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Jakimovski, D.; Gandhi, S.; Paunkoski, I.; Bergsland, N.; Hagemeier, J.; Ramasamy, D.P.; Hojnacki, D.; Kolb, C.; Benedict, R.H.B.; Weinstock-Guttman, B.; et al. Hypertension and heart disease are associated with development of brain atrophy in multiple sclerosis: A 5-year longitudinal study. Eur. J. Neurol. 2019, 26, 87–e8. [Google Scholar] [CrossRef] [PubMed]
- Saroufim, P.; Zweig, S.A.; Conway, D.S.; Briggs, F.B.S. Cardiovascular conditions in persons with multiple sclerosis, neuromyelitis optica and transverse myelitis. Mult. Scler. Relat. Disord. 2018, 25, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Marrie, R.A. Comorbidity in multiple sclerosis: Implications for patient care. Nat. Rev. Neurol. 2017, 13, 375–382. [Google Scholar] [CrossRef]
- Racosta, J.M.; Kimpinski, K.; Morrow, S.A.; Kremenchutzky, M. Autonomic dysfunction in multiple sclerosis. Auton. Neurosci. 2015, 1–6. [Google Scholar] [CrossRef]
- Cygankiewicz, I.; Zareba, W. Heart rate variability. Handb. Clin. Neurol. 2013, 117, 379–393. [Google Scholar] [CrossRef]
- Damla, O.; Altug, C.; Pinar, K.K.; Alper, K.; Dilek, I.G.; Kadriye, A. Heart rate variability analysis in patients with multiple sclerosis. Mult. Scler. Relat. Disord. 2018, 24, 64–68. [Google Scholar] [CrossRef]
- Kalanie, H.; Harandi, A.A.; Alidaei, S.; Heidari, D.; Shahbeigi, S.; Ghorbani, M. Venous Thrombosis in Multiple Sclerosis Patients after High-Dose Intravenous Methylprednisolone: The Preventive Effect of Enoxaparin. Thrombosis 2011, 2011, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, G.; Bavera, P.M.; Caputo, D.; Mendozzi, L.; Cavarretta, R.; Agus, G.B.; Milani, M.; Ippolito, E.; Cimminiello, C. Risk of deep venous thrombosis (DVT) in bedridden or wheelchair-bound multiple sclerosis patients: A prospective study. Thromb. Res. 2010, 125, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. Coagulation and inflammation. J. Endotoxin Res. 2003, 9, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. Crosstalk between inflammation and thrombosis. Maturitas 2004, 47, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. Interactions between the innate immune and blood coagulation systems. Trends Immunol. 2004, 25, 536–542. [Google Scholar] [CrossRef]
- Horstman, L.L.; Jy, W.; Ahn, Y.S.; Zivadinov, R.; Maghzi, A.H.; Etemadifar, M.; Steven Alexander, J.; Minagar, A. Role of platelets in neuroinflammation: A wide-angle perspective. J. Neuroinflamm. 2010, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. Platelets provide a bounty of potential targets for therapy in multiple sclerosis. Circ. Res. 2012, 110, 1157–1158. [Google Scholar] [CrossRef]
- Behari, M.; Shrivastava, M. Role of platelets in neurodegenerative diseases: A universal pathophysiology. Int. J. Neurosci. 2013, 123, 287–299. [Google Scholar] [CrossRef]
- Laroni, A.; Signori, A.; Maniscalco, G.T.; Lanzillo, R.; Russo, C.V.; Binello, E.; Lo Fermo, S.; Repice, A.; Annovazzi, P.; Bonavita, S.; et al. Assessing association of comorbidities with treatment choice and persistence in MS. Neurology 2017, 89, 2222–2229. [Google Scholar] [CrossRef]
- Savage, B.; Almus-Jacobs, F.; Ruggeri, Z.M. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell 1998, 94, 657–666. [Google Scholar] [CrossRef]
- Morel, A.; Bijak, M.; Miller, E.; Rywaniak, J.; Miller, S.; Saluk, J. Relationship between the Increased Haemostatic Properties of Blood Platelets and Oxidative Stress Level in Multiple Sclerosis Patients with the Secondary Progressive Stage. Oxid. Med. Cell. Longev. 2015, 240918. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.; Miller, E.; Bijak, M.; Saluk, J. The increased level of COX-dependent arachidonic acid metabolism in blood platelets from secondary progressive multiple sclerosis patients. Mol. Cell. Biochem. 2016, 420, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.; Rywaniak, J.; Bijak, M.; Miller, E.; Niwald, M.; Saluk, J. Flow cytometric analysis reveals the high levels of platelet activation parameters in circulation of multiple sclerosis patients. Mol. Cell. Biochem. 2017, 430, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Chavakis, T. Platelets and neurovascular inflammation. Thromb. Haemost. 2013, 110, 888–893. [Google Scholar] [PubMed]
- Berghoff, S.A.; Düking, T.; Spieth, L.; Winchenbach, J.; Stumpf, S.K.; Gerndt, N.; Kusch, K.; Ruhwedel, T.; Möbius, W.; Saher, G. Blood–brain barrier hyperpermeability precedes demyelination in the cuprizone model. Acta Neuropathol. Commun. 2017, 5, 94. [Google Scholar] [CrossRef] [PubMed]
- Sheremata, W.A.; Jy, W.; Horstman, L.L.; Ahn, Y.S.; Alexander, J.S.; Minagar, A. Evidence of platelet activation in multiple sclerosis. J. Neuroinflamm. 2008, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling during platelet adhesion and activation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef]
- Strukova, S.M. Role of platelets and serine proteinases in coupling of blood coagulation and inflammation. Biochem. (Mosc.) 2004, 69, 1067–1081. [Google Scholar] [CrossRef]
- Stakos, D.A.; Tziakas, D.N.; Stellos, K. Mechanisms of platelet activation in acute coronary syndromes. Curr. Vasc. Pharm. 2012, 10, 578–588. [Google Scholar] [CrossRef]
- Hisham, N.F.; Bayraktutan, U. Epidemiology, pathophysiology, and treatment of hypertension in ischaemic stroke patients. J. Stroke Cereb. Dis. 2013, 22, e4–e14. [Google Scholar] [CrossRef]
- Han, M.H.; Hwang, S.I.; Roy, D.B.; Lundgren, D.H.; Price, J.V.; Ousman, S.S.; Fernald, G.H.; Gerlitz, B.; Robinson, W.H.; Baranzini, S.E.; et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 2008, 451, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M.; Saluk, J.; Ponczek, M.B.; Nowak, P.; Wachowicz, B. The synthesis of proteins in unnucleated blood platelets. Post. Hig. Med. Dosw. 2013, 67, 672–679. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Gambaryan, S.; Watson, S.P.; Jurk, K.; Walter, U.; Sickmann, A.; Heemskerk, J.W.; Zahedi, R.P. What can proteomics tell us about platelets? Circ. Res. 2014, 114, 1204–1219. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Choi, E.Y.; Zhou, H.; Schleicher, R.; Chung, K.J.; Tang, Z.; Gobel, K.; Bdeir, K.; Chatzigeorgiou, A.; Wong, C.; et al. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ. Res. 2012, 110, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, A.S.; Lindemann, S.; Zimmerman, G.A. The evolving role of platelets in inflammation. J. Thromb. Haemost. 2003, 1, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Diacovo, T.G.; deFougerolles, A.R.; Bainton, D.F.; Springer, T.A. A functional integrin ligand on the surface of platelets: Intercellular adhesion molecule-2. J. Clin. Investig. 1994, 94, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Kuijper, P.H.; Gallardo Torres, H.I.; Houben, L.A.; Lammers, J.W.; Zwaginga, J.J.; Koenderman, L. P-selectin and MAC-1 mediate monocyte rolling and adhesion to ECM-bound platelets under flow conditions. J. Leukoc. Biol. 1998, 64, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh-Cognasse, H.; Cognasse, F.; Palle, S.; Chavarin, P.; Olivier, T.; Delézay, O.; Pozzetto, B.; Garraud, O. Direct contact of platelets and their released products exert different effects on human dendritic cell maturation. BMC Immunol. 2008, 9, 54. [Google Scholar] [CrossRef]
- Kuijper, P.H.; Gallardo Torres, H.I.; van der Linden, J.A.; Lammers, J.W.; Sixma, J.J.; Koenderman, L.; Zwaginga, J.J. Platelet-dependent primary hemostasis promotes selectin- and integrin-mediated neutrophil adhesion to damaged endothelium under flow conditions. Blood 1996, 87, 3271–3281. [Google Scholar]
- Li, G.; Kim, Y.J.; Mantel, C.; Broxmeyer, H.E. P-selectin enhances generation of CD14+CD16+ dendritic-like cells and inhibits macrophage maturation from human peripheral blood monocytes. J. Immunol. 2003, 171, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.L.; Sheppard, J.A.; Feuerstein, I.A. Role of P-selectin and leukocyte activation in polymorphonuclear cell adhesion to surface adherent activated platelets under physiologic shear conditions (an injury vessel wall model). Blood 1994, 83, 2498–2507. [Google Scholar] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V. Dendritic cells in atherosclerosis: Current status of the problem and clinical relevance. Eur. Heart J. 2005, 26, 1700–1704. [Google Scholar] [CrossRef] [PubMed]
- Elzey, B.D.; Tian, J.; Jensen, R.J.; Swanson, A.K.; Lees, J.R.; Lentz, S.R.; Stein, C.S.; Nieswandt, B.; Wang, Y.; Davidson, B.L.; et al. Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity 2003, 19, 9–19. [Google Scholar] [CrossRef]
- Henn, V.; Slupsky, J.R.; Gräfe, M.; Anagnostopoulos, I.; Förster, R.; Müller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Saluk-Juszczak, J.; Krolewska, K. The role of CD40/CD40L pathway in biological activity of blood platelets: Part II. Przegląd Menopauzalny 2010, 9, 371–375. [Google Scholar]
- Saluk-Juszczak, J.; Krolewska, K. The role of CD40/CD40L pathway in the biological activity of blood platelets: Part I. Przegląd Menopauzalny 2010, 9, 305–308. [Google Scholar]
- Burman, J.; Fransson, M.; Tötterman, T.H.; Fagius, J.; Mangsbo, S.M.; Loskog, A.S. T-cell responses after haematopoietic stem cell transplantation for aggressive relapsing-remitting multiple sclerosis. Immunology 2013, 140, 211–219. [Google Scholar] [CrossRef]
- Rendu, F.; Brohard-Bohn, B. The platelet release reaction: granules’ constituents, secretion and functions. Platelets 2001, 12, 261–273. [Google Scholar] [CrossRef]
- Cananzi, A.R.; Ferro-Milone, F.; Grigoletto, F.; Toldo, M.; Meneghini, F.; Bortolon, F.; D’Andrea, G. Relevance of platelet factor four (PF4) plasma levels in multiple sclerosis. Acta Neurol. Scand. 1987, 76, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Webb, J.; Stuve, O.; Haskins, W.; Forsthuber, T. Body fluid biomarkers in multiple sclerosis: How far we have come and how they could affect the clinic now and in the future. Expert Rev. Clin. Immunol. 2015, 11, 69–91. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.H.; Sim, E.H.; Goh, R.Y.; Park, J.I.; Han, J.Y. Platelet Activation: The Mechanisms and Potential Biomarkers. Biomed. Res. Int. 2016, 9060143. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, I.I.; Predescu, S.A.; Neamu, R.F.; Gorovoy, M.S.; Knezevic, N.M.; Easington, C.; Malik, A.B.; Predescu, D.N. Tiam1 and Rac1 are required for platelet-activating factor-induced endothelial junctional disassembly and increase in vascular permeability. J. Biol. Chem. 2009, 284, 5381–5394. [Google Scholar] [CrossRef]
- Pisetsky, D.S. Microparticles as autoantigens: Making immune complexes big. Arthritis Rheum. 2012, 64, 958–961. [Google Scholar] [CrossRef]
- Ardoin, S.P.; Shanahan, J.C.; Pisetsky, D.S. The role of microparticles in inflammation and thrombosis. Scand. J. Immunol. 2007, 66, 159–165. [Google Scholar] [CrossRef]
- Cloutier, N.; Tan, S.; Boudreau, L.H.; Cramb, C.; Subbaiah, R.; Lahey, L.; Albert, A.; Shnayder, R.; Gobezie, R.; Nigrovic, P.A.; et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: The microparticle-associated immune complexes. EMBO Mol. Med. 2013, 5, 235–249. [Google Scholar] [CrossRef]
- Villar-Vesga, J.; Grajales, C.; Burbano, C.; Vanegas-García, A.; Muñoz-Vahos, C.H.; Vásquez, G.; Rojas, M.; Castaño, D. Platelet-derived microparticles generated in vitro resemble circulating vesicles of patients with rheumatoid arthritis and activate monocytes. Cell. Immunol. 2018. [Google Scholar] [CrossRef]
- Xue, L.J.; Cui, B.B.; Li, X.; Huang, Q.R.; Liu, Y.; Lin, H. Association of Elevated Platelet Microparticles with Disease Activity in Rheumatoid Arthritis. Sichuan Da Xue Xue Bao Yi Xue Ban 2017, 48, 405–409. [Google Scholar]
- Mobarrez, F.; Svenungsson, E.; Pisetsky, D.S. Microparticles as Autoantigens in Systemic Lupus Erythematosus. Eur. J. Clin. Investig. 2018, e13010. [Google Scholar] [CrossRef] [PubMed]
- Seizer, P.; May, A.E. Platelets and matrix metalloproteinases. Thromb. Haemost. 2013, 110, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Bültmann, A.; Fischel, S.; Gillitzer, A.; Cullen, P.; Walch, A.; Jost, P.; Ungerer, M.; Tolley, N.D.; Lindemann, S.; et al. Extracellular matrix metalloproteinase inducer (CD147) is a novel receptor on platelets, activates platelets, and augments nuclear factor kappaB-dependent inflammation in monocytes. Circ. Res. 2008, 102, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Abou-Saleh, H.; Théorêt, J.F.; Yacoub, D.; Merhi, Y. Neutrophil P-selectin-glycoprotein-ligand-1 binding to platelet P-selectin enhances metalloproteinase 2 secretion and platelet-neutrophil aggregation. Thromb. Haemost. 2005, 94, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- May, A.E.; Kälsch, T.; Massberg, S.; Herouy, Y.; Schmidt, R.; Gawaz, M. Engagement of glycoprotein IIb/IIIa (alpha(IIb)beta3) on platelets upregulates CD40L and triggers CD40L-dependent matrix degradation by endothelial cells. Circulation 2002, 106, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Stokes, K.Y.; Granger, D.N. Platelets: A critical link between inflammation and microvascular dysfunction. J. Physiol. 2012, 590, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Hargett, L.A.; Bauer, N.N. On the Origin of Microparticles: From “Platelet Dust” to Mediators of Intercellular Communication. Pulm. Circ. 2013, 3, 329–340. [Google Scholar] [CrossRef]
- Dürk, T.; Duerschmied, D.; Müller, T.; Grimm, M.; Reuter, S.; Vieira, R.P.; Ayata, K.; Cicko, S.; Sorichter, S.; Walter, D.J.; et al. Production of Serotonin by Tryptophan Hydroxylase 1 and Release via Platelets Contribute to Allergic Airway Inflammation. Am. J. Respir. Crit. Care Med. 2013, 187, 476–485. [Google Scholar] [CrossRef]
- Hamidi, V.; Couto, E.; Ringerike, T.; Klemp, M. A Multiple Treatment Comparison of Eleven Disease-Modifying Drugs Used for Multiple Sclerosi. J. Clin. Med. Res. 2018, 10, 88–105. [Google Scholar] [CrossRef]
- Sáenz-Cuesta, M.; Irizar, H.; Castillo-Triviño, T.; Muñoz-Culla, M.; Osorio-Querejeta, I.; Prada, A.; Sepúlveda, L.; López-Mato, M.P.; López de Munain, A.; Comabella, M.; et al. Circulating microparticles reflect treatment effects and clinical status in multiple sclerosis. Biomark. Med. 2014, 8, 653–661. [Google Scholar] [CrossRef]
- Wright, H.P.; Thompson, R.H.; Zilkha, K.J. Platelet adhesiveness in multiple sclerosis. Lancet 1965, 2, 1109–1110. [Google Scholar] [CrossRef]
- Kuenz, B.; Lutterotti, A.; Khalil, M.; Ehling, R.; Gneiss, C.; Deisenhamme, R.F.; Reindl, M.; Berger, T. Plasma levels of soluble adhesion molecules sPECAM-1, sP-selectin and sE-selectin are associated with relapsing-remitting disease course of multiple sclerosis. J. Neuroimmunol. 2005, 167, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Callea, L.; Arese, M.; Orlandini, A.; Bargnani, C.; Priori, A.; Bussolino, F. Platelet activating factor is elevated in cerebral spinal fluid and plasma of patients with relapsing-remitting multiple sclerosis. J. Neuroimmunol. 1999, 94, 212–221. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Platelet Investigation in MS and Major Findings | Stage of MS | References |
|---|---|---|
| Increased plasma level of β-TG and PF4 (role of platelet degranulation in increasing BBB permeability) | RRMS | [71] |
| Higher percentage of circulating PMPs (proven pro-inflammatory and prothrombotic properties of PMPs) | RRMS | [46,90] |
| Elevated level of platelet aggregation (increased platelet hemostatic function) | RRMS | [46] |
| Raised surface exposure of P-selectin (marker of platelet activation, receptor crucial for cellular interactions) | RRMS | [90] |
| Increased platelet adhesiveness (changes of platelet hemostatic function) | RRMS | [91] |
| Augmented plasma level of sP-selectin (marker of permanent activation and consumption of platelets) | RRMS | [92] |
| Elevated level of PAF in cerebral spinal fluid and plasma (platelet activator and mediator) | RRMS | [93] |
| Increased expression of P-selectin | SPMS | [43] |
| Enhanced activation of GPIIb/IIIa (receptor responsible for platelet aggregation) | SPMS | [43] |
| Higher percentage of circulating PMPs | SPMS | [43] |
| Augmented formation of platelet aggregates | SPMS | [41,42,43] |
| Increased platelet adhesiveness | SPMS | [41] |
| Extensively ROS generation (blood platelets actively participate in oxidative stress existing in SPMS) | SPMS | [41] |
| Increased cyclooxygenase-dependent arachidonic acid metabolism (the main metabolism pathway in platelets) | SPMS | [42] |
| High platelet reactivity in response to action of physiological agonists (excessive excitability and sensitivity of platelets) | SPMS | [41,42,43] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saluk-Bijak, J.; Dziedzic, A.; Bijak, M. Pro-Thrombotic Activity of Blood Platelets in Multiple Sclerosis. Cells 2019, 8, 110. https://doi.org/10.3390/cells8020110
Saluk-Bijak J, Dziedzic A, Bijak M. Pro-Thrombotic Activity of Blood Platelets in Multiple Sclerosis. Cells. 2019; 8(2):110. https://doi.org/10.3390/cells8020110
Chicago/Turabian StyleSaluk-Bijak, Joanna, Angela Dziedzic, and Michal Bijak. 2019. "Pro-Thrombotic Activity of Blood Platelets in Multiple Sclerosis" Cells 8, no. 2: 110. https://doi.org/10.3390/cells8020110
APA StyleSaluk-Bijak, J., Dziedzic, A., & Bijak, M. (2019). Pro-Thrombotic Activity of Blood Platelets in Multiple Sclerosis. Cells, 8(2), 110. https://doi.org/10.3390/cells8020110