Control of the Antitumor Immune Response by Cancer Metabolism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
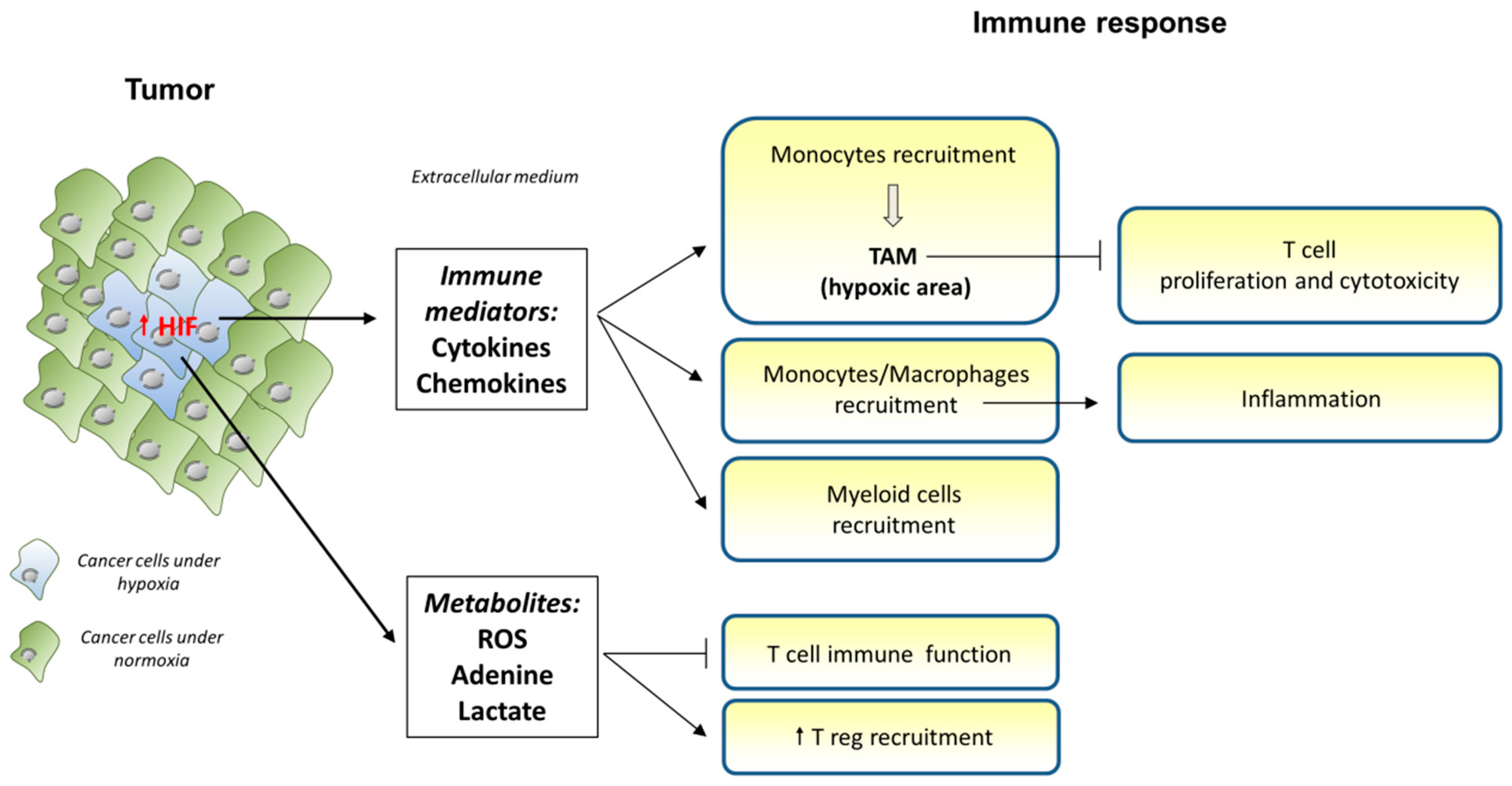
1. Introduction
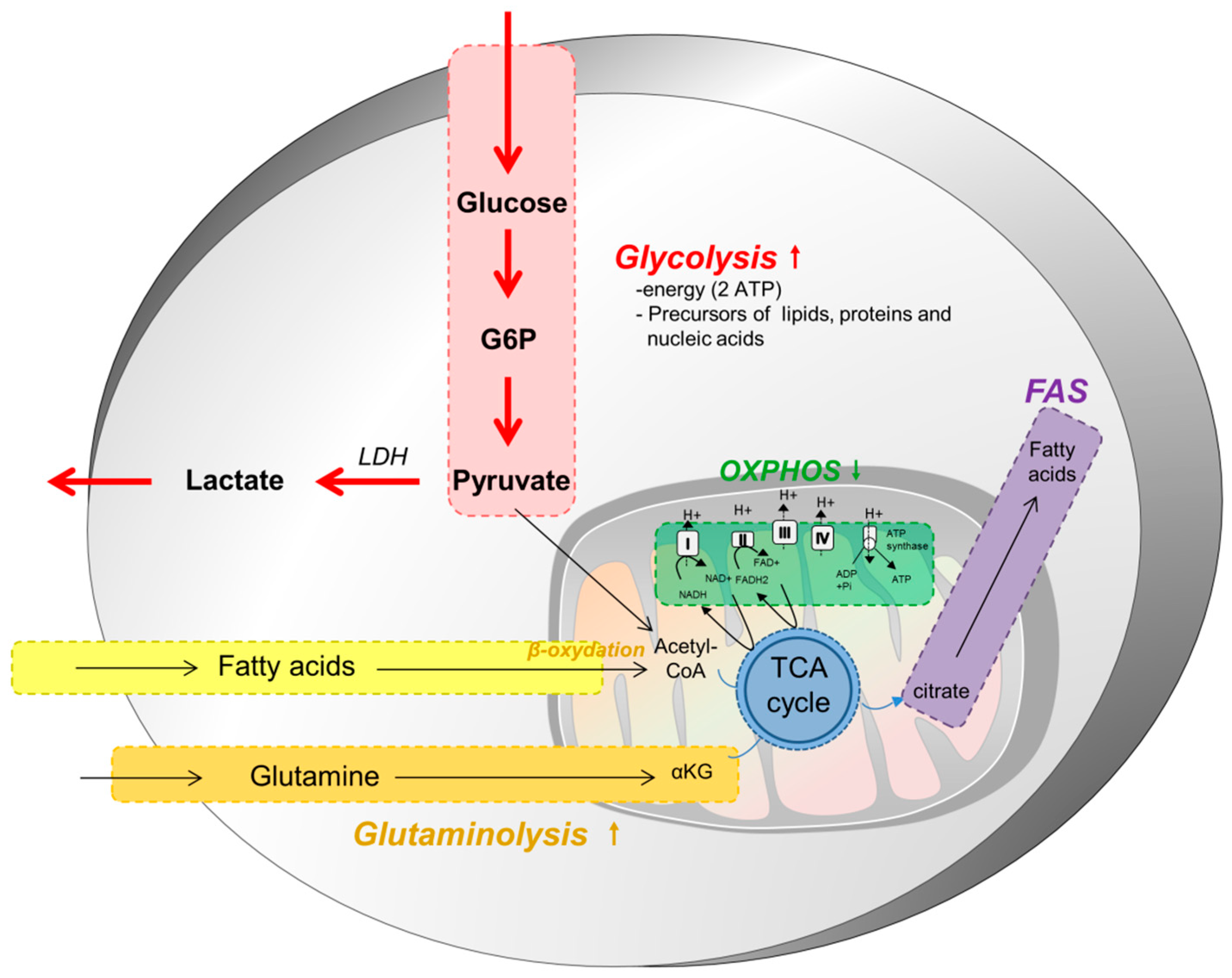
2. Metabolism of Tumor Cells
3. Effect of Cancer Metabolism on Infiltrating Immune Cells
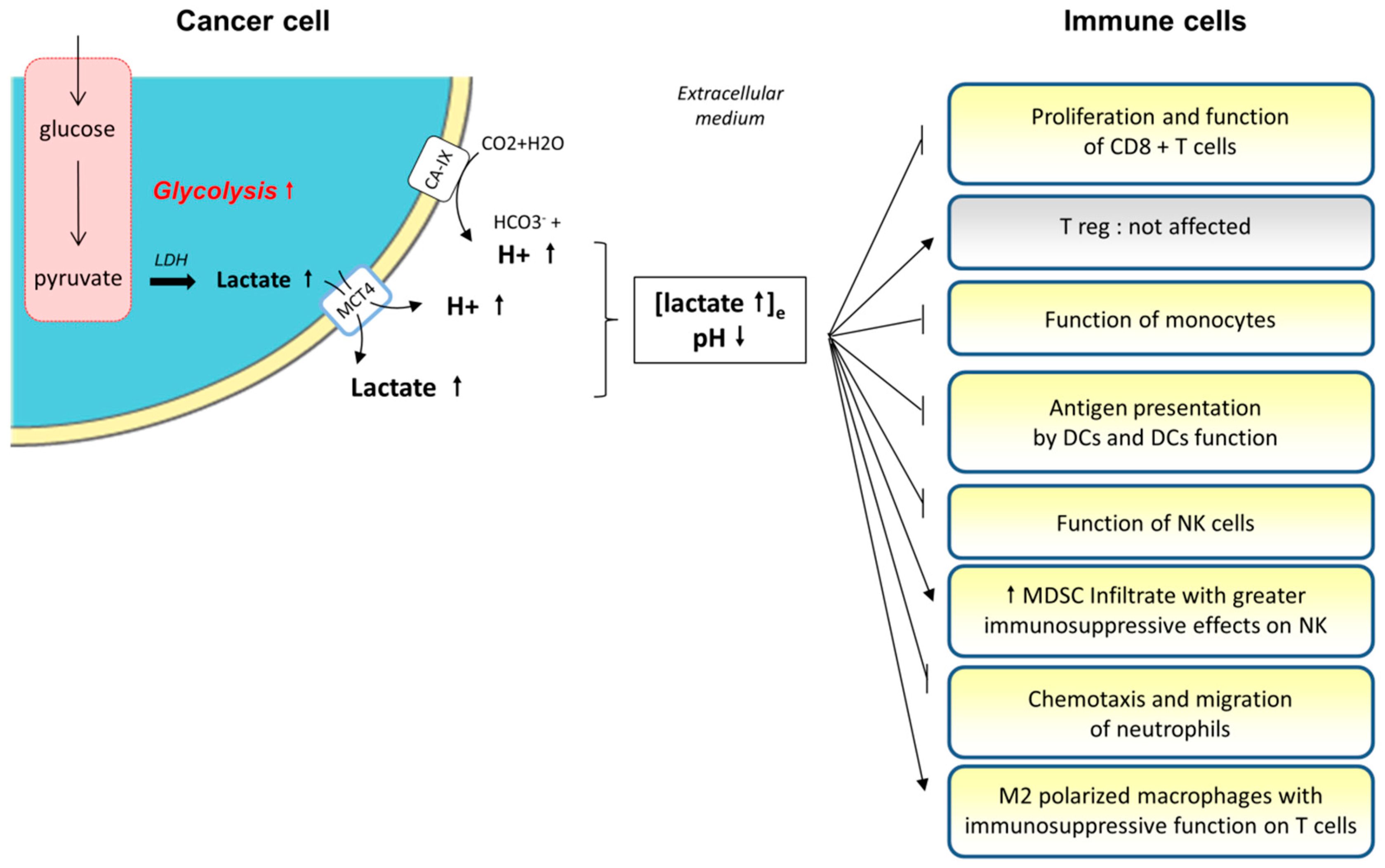
3.1. Medium Acidification and Lactate Accumulation
3.1.1. The Production of Lactate by Tumor Cells
3.1.2. Role of Lactate and Extracellular Medium Acidification on Immune Cells
3.1.3. Impact in Clinical Routines
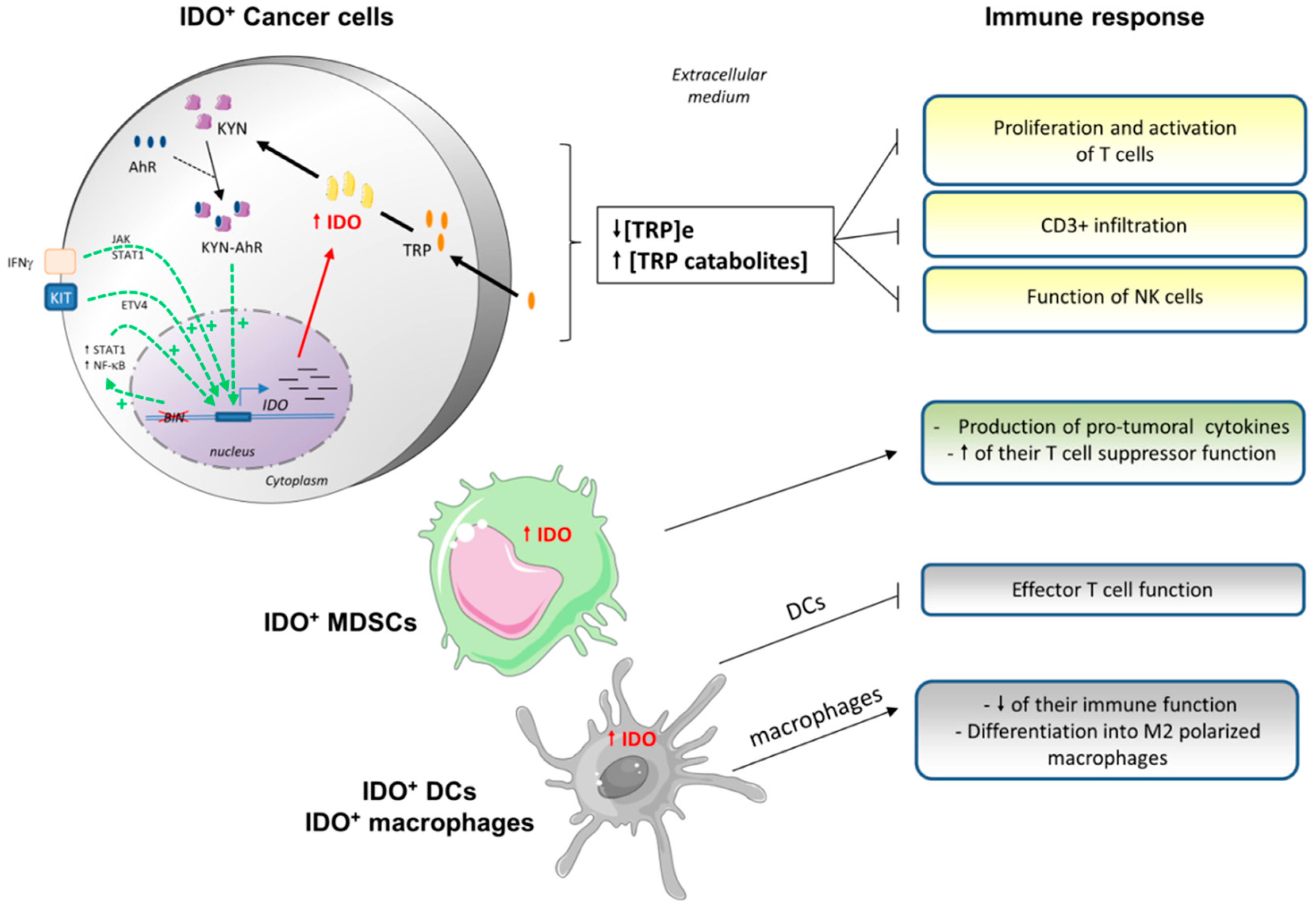
3.2. Indoleamine 2,3-Dioxygenase (IDO) and Tryptophan Dioxygenase (TDO)
3.2.1. IDO Characteristics
3.2.2. IDO Expression in Tumor Cells
3.2.3. Clinical Targeting of IDO
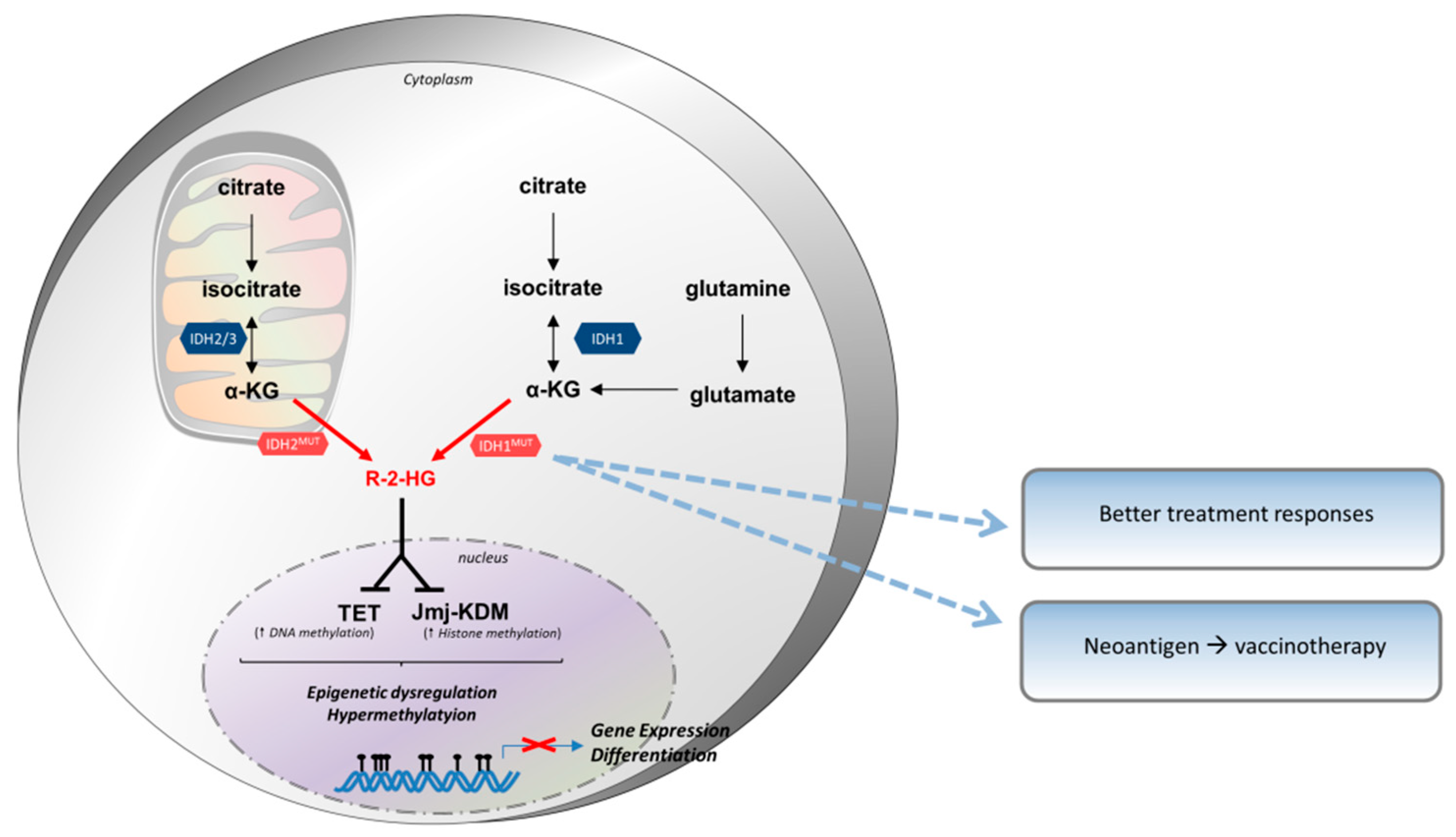
3.3. Isocitrate Dehydrogenase (IDH)
3.3.1. Biochemistry of IDH
3.3.2. IDH Expression in Tumor Cells
3.3.3. Impact in Clinical Routine
3.4. Hypoxic Conditions
3.4.1. Characteristics of Hypoxia Conditions
3.4.2. Hypoxia and Tumor Cells
3.4.3. Effect of Hypoxia on Immune Cell Types
3.4.4. Targeting Hypoxia
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Domblides, C.; Lartigue, L.; Faustin, B. Metabolic Stress in the Immune Function of T Cells, Macrophages and Dendritic Cells. Cells 2018, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, A.N.; Rathmell, J.C. Activated lymphocytes as a metabolic model for carcinogenesis. Cancer Metab. 2013, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef]
- Zambrano, A.; Jara, E.; Murgas, P.; Jara, C.; Castro, M.A.; Angulo, C.; Concha, I.I. Cytokine stimulation promotes increased glucose uptake via translocation at the plasma membrane of GLUT1 in HEK293 cells. J. Cell. Biochem. 2010, 110, 1471–1480. [Google Scholar] [CrossRef]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Clem, B. RB in glutamine metabolism. Oncoscience 2014, 1, 304–305. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Swamy, M.; Pathak, S.; Grzes, K.M.; Damerow, S.; Sinclair, L.V.; van Aalten, D.M.F.; Cantrell, D.A. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat. Immunol. 2016, 17, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Haleem, A.M.; Lewis, N.E.; Jamshidi, N.; Mineta, K.; Gao, X.; Gojobori, T. The Emerging Facets of Non-Cancerous Warburg Effect. Front. Endocrinol. 2017, 8, 279. [Google Scholar] [CrossRef]
- Ramapriyan, R.; Caetano, M.S.; Barsoumian, H.B.; Mafra, A.C.P.; Zambalde, E.P.; Menon, H.; Tsouko, E.; Welsh, J.W.; Cortez, M.A. Altered cancer metabolism in mechanisms of immunotherapy resistance. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.C.; Maddocks, O.D.K. One-carbon metabolism in cancer. Br. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017, 25, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Roy, A.; Dwarakanath, B.S. Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front. Oncol. 2017, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- De Milito, A.; Canese, R.; Marino, M.L.; Borghi, M.; Iero, M.; Villa, A.; Venturi, G.; Lozupone, F.; Iessi, E.; Logozzi, M.; et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int. J. Cancer 2010, 127, 207–219. [Google Scholar] [CrossRef]
- Dimmer, K.S.; Friedrich, B.; Lang, F.; Deitmer, J.W.; Bröer, S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 2000, 350 Pt 1, 219–227. [Google Scholar] [CrossRef]
- Pérez-Escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 2481–2497. [Google Scholar] [CrossRef]
- Chambard, J.C.; Pouyssegur, J. Intracellular pH controls growth factor-induced ribosomal protein S6 phosphorylation and protein synthesis in the G0—G1 transition of fibroblasts. Exp. Cell Res. 1986, 164, 282–294. [Google Scholar] [CrossRef]
- Pouysségur, J.; Sardet, C.; Franchi, A.; L’Allemain, G.; Paris, S. A specific mutation abolishing Na+/H+ antiport activity in hamster fibroblasts precludes growth at neutral and acidic pH. Proc. Natl. Acad. Sci. USA 1984, 81, 4833–4837. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Ganesan, R.; Reynolds, G.; Gross, L.; Stevens, A.; Pastorek, J.; Murray, P.G.; Perunovic, B.; Anwar, M.S.; Billingham, L.; et al. Hypoxia-regulated carbonic anhydrase IX expression is associated with poor survival in patients with invasive breast cancer. Br. J. Cancer 2007, 96, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Mazzio, E.A.; Boukli, N.; Rivera, N.; Soliman, K.F.A. Pericellular pH homeostasis is a primary function of the Warburg effect: Inversion of metabolic systems to control lactate steady state in tumor cells. Cancer Sci. 2012, 103, 422–432. [Google Scholar] [CrossRef]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Baumann, F.; Leukel, P.; Doerfelt, A.; Beier, C.P.; Dettmer, K.; Oefner, P.J.; Kastenberger, M.; Kreutz, M.; Nickl-Jockschat, T.; Bogdahn, U.; et al. Lactate promotes glioma migration by TGF-β2–dependent regulation of matrix metalloproteinase-2. Neuro-Oncology 2009, 11, 368–380. [Google Scholar] [CrossRef]
- Yang, X.; Wang, D.; Dong, W.; Song, Z.; Dou, K. Suppression of Na+/H + exchanger 1 by RNA interference or amiloride inhibits human hepatoma cell line SMMC-7721 cell invasion. Med. Oncol. 2011, 28, 385–390. [Google Scholar] [CrossRef]
- Rudrabhatla, S.R.; Mahaffey, C.L.; Mummert, M.E. Tumor Microenvironment Modulates Hyaluronan Expression: The Lactate Effect. J. Investig. Dermatol. 2006, 126, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Walenta, S.; Mueller-Klieser, W.F. Lactate: Mirror and motor of tumor malignancy. Semin. Radiat. Oncol. 2004, 14, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar] [PubMed]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- DeClerck, K.; Elble, R.C. The role of hypoxia and acidosis in promoting metastasis and resistance to chemotherapy. Front. Biosci. 2010, 15, 213–225. [Google Scholar] [CrossRef]
- Sattler, U.G.A.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef]
- Groussard, C.; Morel, I.; Chevanne, M.; Monnier, M.; Cillard, J.; Delamarche, A. Free radical scavenging and antioxidant effects of lactate ion: An in vitro study. J. Appl. Physiol. 1985 2000, 89, 169–175. [Google Scholar] [CrossRef]
- Kellum, J.A. Metabolic acidosis in patients with sepsis: Epiphenomenon or part of the pathophysiology? Crit. Care Resusc. J. Australas. Acad. Crit. Care Med. 2004, 6, 197–203. [Google Scholar]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Wegiel, B.; Vuerich, M.; Daneshmandi, S.; Seth, P. Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-cancer Therapy. Front. Oncol. 2018, 8, 284. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Mendler, A.N.; Hu, B.; Prinz, P.U.; Kreutz, M.; Gottfried, E.; Noessner, E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int. J. Cancer 2012, 131, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef]
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J.; et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J. Immunol. 2010, 184, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Peter, K.; Rehli, M.; Singer, K.; Renner-Sattler, K.; Kreutz, M. Lactic acid delays the inflammatory response of human monocytes. Biochem. Biophys. Res. Commun. 2015, 457, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Shime, H.; Yabu, M.; Akazawa, T.; Kodama, K.; Matsumoto, M.; Seya, T.; Inoue, N. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J. Immunol. 2008, 180, 7175–7183. [Google Scholar] [CrossRef] [PubMed]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 promotes tumour incidence and growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Akazawa, T.; Aoki, M.; Kuze, B.; Mizuta, K.; Ito, Y.; Inoue, N. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. J. Int. Cancer 2013, 133, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Lardner, A. The effects of extracellular pH on immune function. J. Leukoc. Biol. 2001, 69, 522–530. [Google Scholar] [PubMed]
- Loeffler, D.A.; Juneau, P.L.; Masserant, S. Influence of tumour physico-chemical conditions on interleukin-2-stimulated lymphocyte proliferation. Br. J. Cancer 1992, 66, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Severin, T.; Müller, B.; Giese, G.; Uhl, B.; Wolf, B.; Hauschildt, S.; Kreutz, W. pH-dependent LAK cell cytotoxicity. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 1994, 15, 304–310. [Google Scholar] [CrossRef]
- Walenta, S.; Wetterling, M.; Lehrke, M.; Schwickert, G.; Sundfør, K.; Rofstad, E.K.; Mueller-Klieser, W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000, 60, 916–921. [Google Scholar] [PubMed]
- Hirschhaeuser, F.; Sattler, U.G.A.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef]
- Sola-Penna, M. Metabolic regulation by lactate. IUBMB Life 2008, 60, 605–608. [Google Scholar] [CrossRef]
- Wang, B.-Y.; Zhang, J.; Wang, J.-L.; Sun, S.; Wang, Z.-H.; Wang, L.-P.; Zhang, Q.-L.; Lv, F.-F.; Cao, E.-Y.; Shao, Z.-M.; et al. Intermittent high dose proton pump inhibitor enhances the antitumor effects of chemotherapy in metastatic breast cancer. J. Exp. Clin. Cancer Res. CR 2015, 34, 85. [Google Scholar] [CrossRef]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A.; et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef]
- Xie, H.; Valera, V.A.; Merino, M.J.; Amato, A.M.; Signoretti, S.; Linehan, W.M.; Sukhatme, V.P.; Seth, P. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol. Cancer Ther. 2009, 8, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, Z.; Chen, Z.; Chen, R.; Zhao, D.; Zhou, Y.; Qiao, L. Effects of the suppression of lactate dehydrogenase A on the growth and invasion of human gastric cancer cells. Oncol. Rep. 2015, 33, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Su, D.; Zhao, L.; Zhang, D.; Xu, J.; Wan, J.; Fan, S.; Chen, M. Different effects of LDH-A inhibition by oxamate in non-small cell lung cancer cells. Oncotarget 2014, 5, 11886–11896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Wang, X.; Gan, L.; Yu, G.; Chen, Y.; Liu, K.; Li, P.; Pan, J.; Wang, J.; et al. Inhibition of LDH-A by lentivirus-mediated small interfering RNA suppresses intestinal-type gastric cancer tumorigenicity through the downregulation of Oct4. Cancer Lett. 2012, 321, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Hayaishi, O. Tryptophan pyrrolase of rabbit intestine. D- and L-tryptophan-cleaving enzyme or enzymes. J. Biol. Chem. 1967, 242, 5260–5266. [Google Scholar] [PubMed]
- Théate, I.; van Baren, N.; Pilotte, L.; Moulin, P.; Larrieu, P.; Renauld, J.-C.; Hervé, C.; Gutierrez-Roelens, I.; Marbaix, E.; Sempoux, C.; et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol. Res. 2015, 3, 161–172. [Google Scholar] [CrossRef]
- Friberg, M.; Jennings, R.; Alsarraj, M.; Dessureault, S.; Cantor, A.; Extermann, M.; Mellor, A.L.; Munn, D.H.; Antonia, S.J. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int. J. Cancer 2002, 101, 151–155. [Google Scholar] [CrossRef]
- Takikawa, O.; Kuroiwa, T.; Yamazaki, F.; Kido, R. Mechanism of interferon-gamma action. Characterization of indoleamine 2,3-dioxygenase in cultured human cells induced by interferon-gamma and evaluation of the enzyme-mediated tryptophan degradation in its anticellular activity. J. Biol. Chem. 1988, 263, 2041–2048. [Google Scholar]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef]
- Soliman, H.; Mediavilla-Varela, M.; Antonia, S. Indoleamine 2,3-Dioxygenase. Cancer J. 2010, 16, 354–359. [Google Scholar] [CrossRef]
- Heng, B.; Lim, C.K.; Lovejoy, D.B.; Bessede, A.; Gluch, L.; Guillemin, G.J. Understanding the role of the kynurenine pathway in human breast cancer immunobiology. Oncotarget 2016, 7, 6506–6520. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Imanishi, J.; Oku, T.; Kishida, T.; Hayaishi, O. Induction of pulmonary indoleamine 2,3-dioxygenase by interferon. Proc. Natl. Acad. Sci. USA 1981, 78, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of Allogeneic Fetal Rejection by Tryptophan Catabolism. Science 1998, 281, 1191–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Spranger, S.; Binder, D.C.; Gritsina, G.; Lauing, K.L.; Giles, F.J.; Wainwright, D.A. Molecular Pathways: Targeting IDO1 and Other Tryptophan Dioxygenases for Cancer Immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 5427–5433. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.W.S.; Terentis, A.C.; King, N.J.C.; Thomas, S.R. Role of indoleamine 2,3-dioxygenase in health and disease. Clin. Sci. 2015, 129, 601–672. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; DuHadaway, J.B.; Chang, M.Y.; Ramalingam, A.; Sutanto-Ward, E.; Boulden, J.; Soler, A.P.; Mandik-Nayak, L.; Gilmour, S.K.; Prendergast, G.C. Non-hematopoietic expression of IDO is integrally required for inflammatory tumor promotion. Cancer Immunol. Immunother. CII 2010, 59, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; Sharma, M.D.; Chandler, P.R.; DuHadaway, J.B.; Everhart, M.E.; Johnson, B.A.; Kahler, D.J.; Pihkala, J.; Soler, A.P.; Munn, D.H.; et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc. Natl. Acad. Sci. USA 2008, 105, 17073–17078. [Google Scholar] [CrossRef]
- Smith, C.; Chang, M.Y.; Parker, K.H.; Beury, D.W.; DuHadaway, J.B.; Flick, H.E.; Boulden, J.; Sutanto-Ward, E.; Soler, A.P.; Laury-Kleintop, L.D.; et al. IDO Is a Nodal Pathogenic Driver of Lung Cancer and Metastasis Development. Cancer Discov. 2012, 2, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, T.; Kumar, S.; Markar, S.R.; Antonowicz, S.; Hanna, G.B. Tyrosine, phenylalanine, and tryptophan in gastroesophageal malignancy: A systematic review. Cancer Epidemiol. Biomark. Prev. 2015, 24, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Ferrara, G.B. T cell proliferation is blocked by indoleamine 2,3-dioxygenase. Transplant. Proc. 2001, 33, 428–430. [Google Scholar] [CrossRef]
- Hwu, P.; Du, M.X.; Lapointe, R.; Do, M.; Taylor, M.W.; Young, H.A. Indoleamine 2,3-Dioxygenase Production by Human Dendritic Cells Results in the Inhibition of T Cell Proliferation. J. Immunol. 2000, 164, 3596–3599. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Keskin, D.B.; Johnson, T.; Chandler, P.; Munn, D.H. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J. Immunol. 2002, 168, 3771–3776. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wei, F.; Yu, J.; Li, H.; Ren, X.; Hao, X. IDO inhibits T-cell function through suppressing Vav1 expression and activation. Cancer Biol. Ther. 2009, 8, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Metz, R.; Rust, S.; DuHadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR. Oncoimmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Brandacher, G.; Perathoner, A.; Ladurner, R.; Schneeberger, S.; Obrist, P.; Winkler, C.; Werner, E.R.; Werner-Felmayer, G.; Weiss, H.G.; Göbel, G.; et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: Effect on tumor-infiltrating T cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.K.; Park, H.J.; Macleod, M.; Chandler, P.; Munn, D.H.; Mellor, A.L. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology 2002, 107, 452–460. [Google Scholar] [CrossRef] [PubMed]
- McGaha, T.L.; Huang, L.; Lemos, H.; Metz, R.; Mautino, M.; Prendergast, G.C.; Mellor, A.L. Amino acid catabolism: A pivotal regulator of innate and adaptive immunity. Immunol. Rev. 2012, 249, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.; Chang, M.Y.; Mandik-Nayak, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase as a Modifier of Pathogenic Inflammation in Cancer and other Inflammation-Associated Diseases. Curr. Med. Chem. 2011, 18, 2257–2262. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived Catabolites Are Responsible for Inhibition of T and Natural Killer Cell Proliferation Induced by Indoleamine 2,3-Dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, G.-X.; Ciric, B.; Rostami, A. IDO: A double-edged sword for TH1/TH2 regulation. Immunol. Lett. 2008, 121, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Mo, J.-H.; Gong, X.; Rossetto, C.; Jang, A.; Beck, L.; Elliott, G.I.; Kufareva, I.; Abagyan, R.; Broide, D.H.; et al. 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 18619–18624. [Google Scholar] [CrossRef] [PubMed]
- Terness, P.; Bauer, T.M.; Röse, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of Allogeneic T Cell Proliferation by Indoleamine 2,3-Dioxygenase–expressing Dendritic Cells Mediation of Suppression by Tryptophan Metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef] [PubMed]
- Crellin, N.K.; Garcia, R.V.; Levings, M.K. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood 2007, 109, 2014–2022. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.D.; Shinde, R.; McGaha, T.L.; Huang, L.; Holmgaard, R.B.; Wolchok, J.D.; Mautino, M.R.; Celis, E.; Sharpe, A.H.; Francisco, L.M.; et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci. Adv. 2015, 1, e1500845. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Hwang, K.W.; Orabona, C.; Vacca, C.; Bianchi, R.; Belladonna, M.L.; Fioretti, M.C.; Alegre, M.-L.; Puccetti, P. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 2003, 4, 1206–1212. [Google Scholar] [CrossRef]
- Grohmann, U.; Orabona, C.; Fallarino, F.; Vacca, C.; Calcinaro, F.; Falorni, A.; Candeloro, P.; Belladonna, M.L.; Bianchi, R.; Fioretti, M.C.; et al. CTLA-4–Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 2002, 3, 1097–1101. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef]
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: Much ado about IDO. Trends Immunol. 2003, 24, 242–248. [Google Scholar] [CrossRef]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Ravishankar, B.; Liu, H.; Shinde, R.; Chaudhary, K.; Xiao, W.; Bradley, J.; Koritzinsky, M.; Madaio, M.P.; McGaha, T.L. The amino acid sensor GCN2 inhibits inflammatory responses to apoptotic cells promoting tolerance and suppressing systemic autoimmunity. Proc. Natl. Acad. Sci. USA 2015, 112, 10774–10779. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Puccetti, P. Indoleamine 2,3-dioxygenase: From catalyst to signaling function. Eur. J. Immunol. 2012, 42, 1932–1937. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.L.; Chung, N.P.-Y.; Chan, J.K.-Y.; Lin, C.-L.S. Indoleamine 2,3-dioxygenase (IDO) is essential for dendritic cell activation and chemotactic responsiveness to chemokines. Cell Res. 2005, 15, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Bianchi, R.; Orabona, C.; Vacca, C.; Belladonna, M.L.; Fioretti, M.C.; Serreze, D.V.; Grohmann, U.; Puccetti, P. CTLA-4–Ig Activates Forkhead Transcription Factors and Protects Dendritic Cells from Oxidative Stress in Nonobese Diabetic Mice. J. Exp. Med. 2004, 200, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.; Hölzl, M.; Ahmadi, S.; Dillinger, B.; Pilat, N.; Fuchs, D.; Wekerle, T.; Heitger, A. CTLA4-Ig immunosuppressive activity at the level of dendritic cell/T cell crosstalk. Int. Immunopharmacol. 2013, 15, 638–645. [Google Scholar] [CrossRef]
- Wang, X.-F.; Wang, H.-S.; Wang, H.; Zhang, F.; Wang, K.-F.; Guo, Q.; Zhang, G.; Cai, S.-H.; Du, J. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: Focus on macrophage polarization of THP-1 cells. Cell. Immunol. 2014, 289, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, L.; Bradley, J.; Liu, K.; Bardhan, K.; Ron, D.; Mellor, A.L.; Munn, D.H.; McGaha, T.L. GCN2-dependent metabolic stress is essential for endotoxemic cytokine induction and pathology. Mol. Cell. Biol. 2014, 34, 428–438. [Google Scholar] [CrossRef]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef]
- Kita, H.; Shiraishi, Y.; Watanabe, K.; Suda, K.; Ohtsuka, K.; Koshiishi, Y.; Goya, T. Does Postoperative Serum Interleukin-6 Influence Early Recurrence after Curative Pulmonary Resection of Lung Cancer? Ann. Thorac. Cardiovasc. Surg. 2011, 17, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015, 13, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Newton, R.; Friedman, S.; Scherle, P. Indoleamine 2,3-Dioxygenase, an Emerging Target for Anti-Cancer Therapy. Curr. Cancer Drug Targets 2009, 9, 938–952. [Google Scholar] [CrossRef] [PubMed]
- Cady, S.G.; Sono, M. 1-Methyl-DL-tryptophan, beta-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and beta-[3-benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch. Biochem. Biophys. 1991, 291, 326–333. [Google Scholar] [CrossRef]
- Suzuki, S.; Toné, S.; Takikawa, O.; Kubo, T.; Kohno, I.; Minatogawa, Y. Expression of indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase in early concepti. Biochem. J. 2001, 355, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.H.; Minton, S.E.; Han, H.S.; Ismail-Khan, R.; Neuger, A.; Khambati, F.; Noyes, D.; Lush, R.; Chiappori, A.A.; Roberts, J.D.; et al. A Phase I study of indoximod in patients with advanced malignancies. Oncotarget 2016, 7, 22928. [Google Scholar] [CrossRef]
- Hanihara, M.; Kawataki, T.; Oh-Oka, K.; Mitsuka, K.; Nakao, A.; Kinouchi, H. Synergistic antitumor effect with indoleamine 2,3-dioxygenase inhibition and temozolomide in a murine glioma model. J. Neurosurg. 2015, 1–8. [Google Scholar] [CrossRef]
- Salvadori, M.L.B.; da Cunha Bianchi, P.K.F.; Gebrim, L.H.; Silva, R.S.; Kfoury, J.R. Effect of the association of 1-methyl-DL-tryptophan with paclitaxel on the expression of indoleamine 2,3-dioxygenase in cultured cancer cells from patients with breast cancer. Med. Oncol. 2015, 32, 248. [Google Scholar] [CrossRef]
- Nakamura, N.; Hara, T.; Shimizu, M.; Mabuchi, R.; Nagano, J.; Ohno, T.; Kochi, T.; Kubota, M.; Shirakami, Y.; Goto, N.; et al. Effects of indoleamine 2,3-dioxygenase inhibitor in non-Hodgkin lymphoma model mice. Int. J. Hematol. 2015, 102, 327–334. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef]
- Koblish, H.K.; Hansbury, M.J.; Bowman, K.J.; Yang, G.; Neilan, C.L.; Haley, P.J.; Burn, T.C.; Waeltz, P.; Sparks, R.B.; Yue, E.W.; et al. Hydroxyamidine Inhibitors of Indoleamine-2,3-dioxygenase Potently Suppress Systemic Tryptophan Catabolism and the Growth of IDO-Expressing Tumors. Mol. Cancer Ther. 2010, 9, 489–498. [Google Scholar] [CrossRef]
- Gangadhar, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Luke, J.J.; Balmanoukian, A.S.; Kaufman, D.R.; Zhao, Y.; Maleski, J.; et al. Preliminary results from a Phase I/II study of epacadostat (incb024360) in combination with pembrolizumab in patients with selected advanced cancers. J. Immunother. Cancer 2015, 3, O7. [Google Scholar] [CrossRef]
- Sørensen, R.B.; Berge-Hansen, L.; Junker, N.; Hansen, C.A.; Hadrup, S.R.; Schumacher, T.N.M.; Svane, I.M.; Becker, J.C.; thor Straten, P.; Andersen, M.H. The Immune System Strikes Back: Cellular Immune Responses against Indoleamine 2,3-dioxygenase. PLoS ONE 2009, 4, e6910. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, R.B.; Hadrup, S.R.; Svane, I.M.; Hjortsø, M.C.; thor Straten, P.; Andersen, M.H. Indoleamine 2,3-dioxygenase specific, cytotoxic T cells as immune regulators. Blood 2011, 117, 2200–2210. [Google Scholar] [CrossRef]
- Iversen, T.Z.; Engell-Noerregaard, L.; Ellebaek, E.; Andersen, R.; Larsen, S.K.; Bjoern, J.; Zeyher, C.; Gouttefangeas, C.; Thomsen, B.M.; Holm, B.; et al. Long-lasting Disease Stabilization in the Absence of Toxicity in Metastatic Lung Cancer Patients Vaccinated with an Epitope Derived from Indoleamine 2,3 Dioxygenase. Clin. Cancer Res. 2014, 20, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Sayama, S.; Yoshida, R.; Oku, T.; Imanishi, J.; Kishida, T.; Hayaishi, O. Inhibition of interferon-mediated induction of indoleamine 2,3-dioxygenase in mouse lung by inhibitors of prostaglandin biosynthesis. Proc. Natl. Acad. Sci. USA 1981, 78, 7327–7330. [Google Scholar] [CrossRef]
- Basu, G.D.; Tinder, T.L.; Bradley, J.M.; Tu, T.; Hattrup, C.L.; Pockaj, B.A.; Mukherjee, P. Cyclooxygenase-2 inhibitor enhances the efficacy of a breast cancer vaccine: Role of IDO. J. Immunol. 2006, 177, 2391–2402. [Google Scholar] [CrossRef]
- Lee, S.Y.; Choi, H.K.; Lee, K.J.; Jung, J.Y.; Hur, G.Y.; Jung, K.H.; Kim, J.H.; Shin, C.; Shim, J.J.; In, K.H.; et al. The immune tolerance of cancer is mediated by IDO that is inhibited by COX-2 inhibitors through regulatory T cells. J. Immunother. 2009, 32, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; DuHadaway, J.B.; Jaller, D.; Curtis, P.; Metz, R.; Prendergast, G.C. Immunotherapeutic suppression of indoleamine 2,3-dioxygenase and tumor growth with ethyl pyruvate. Cancer Res. 2010, 70, 1845–1853. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef]
- Gottschalk, S.; Anderson, N.; Hainz, C.; Eckhardt, S.G.; Serkova, N.J. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 6661–6668. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Koropatnick, J.; Li, M.; Zhang, X.; Ling, F.; Ren, X.; Hao, X.; Sun, H.; Vladau, C.; Franek, J.A.; et al. Reinstalling antitumor immunity by inhibiting tumor-derived immunosuppressive molecule IDO through RNA interference. J. Immunol. 2006, 177, 5639–5646. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.-T.; Yen, M.-C.; Lin, C.-C.; Weng, T.-Y.; Chen, Y.-L.; Lin, C.-M.; Lai, M.-D. Skin delivery of short hairpin RNA of indoleamine 2,3 dioxygenase induces antitumor immunity against orthotopic and metastatic liver cancer. Cancer Sci. 2011, 102, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Dalziel, K. Isocitrate dehydrogenase and related oxidative decarboxylases. FEBS Lett. 1980, 117 (Suppl. 1), K45–K55. [Google Scholar] [CrossRef]
- Filipp, F.V.; Scott, D.A.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 2012, 25, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, L.; Hong, C.S.; Yang, C.; Zhuang, Z.; Heiss, J.D. New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int. J. Med. Sci. 2015, 12, 201–213. [Google Scholar] [CrossRef]
- Clark, O.; Yen, K.; Mellinghoff, I.K. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016. [Google Scholar] [CrossRef]
- Cohen, A.; Holmen, S.; Colman, H. IDH1 and IDH2 Mutations in Gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef]
- Ichimura, K. Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol. 2012, 29, 131–139. [Google Scholar] [CrossRef]
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Aghili, M.; Zahedi, F.; Rafiee, E. Hydroxyglutaric aciduria and malignant brain tumor: A case report and literature review. J. Neurooncol. 2009, 91, 233–236. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Jin, G.; Reitman, Z.J.; Duncan, C.G.; Spasojevic, I.; Gooden, D.M.; Rasheed, B.A.; Yang, R.; Lopez, G.Y.; He, Y.; McLendon, R.E.; et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013, 73, 496–501. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Latini, A.; Scussiato, K.; Rosa, R.B.; Llesuy, S.; Belló-Klein, A.; Dutra-Filho, C.S.; Wajner, M. D-2-hydroxyglutaric acid induces oxidative stress in cerebral cortex of young rats. Eur. J. Neurosci. 2003, 17, 2017–2022. [Google Scholar] [CrossRef]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Seo, S.I.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int. J. Cancer 2009, 125, 353–355. [Google Scholar] [CrossRef]
- Kats, L.M.; Reschke, M.; Taulli, R.; Pozdnyakova, O.; Burgess, K.; Bhargava, P.; Straley, K.; Karnik, R.; Meissner, A.; Small, D.; et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell 2014, 14, 329–341. [Google Scholar] [CrossRef]
- Wang, G.; Sai, K.; Gong, F.; Yang, Q.; Chen, F.; Lin, J. Mutation of isocitrate dehydrogenase 1 induces glioma cell proliferation via nuclear factor-κB activation in a hypoxia-inducible factor 1-α dependent manner. Mol. Med. Rep. 2014, 9, 1799–1805. [Google Scholar] [CrossRef]
- Richarson, A.D.; Scott, D.A.; Zagnitko, O.; Aza-Blanc, P.; Chang, C.-C.; Russler-Germain, D.A. Reproducibility Project: Cancer Biology. Registered report: IDH mutation impairs histone demethylation and results in a block to cell differentiation. eLife 2016, 5, e10860. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef]
- Dang, L.; Yen, K.; Attar, E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 2016, 27, 599–608. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Pellegatta, S.; Valletta, L.; Corbetta, C.; Patanè, M.; Zucca, I.; Riccardi Sirtori, F.; Bruzzone, M.G.; Fogliatto, G.; Isacchi, A.; Pollo, B.; et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol. Commun. 2015, 3, 4. [Google Scholar] [CrossRef]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef]
- Jiang, B.H.; Semenza, G.L.; Bauer, C.; Marti, H.H. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. 1996, 271, C1172–C1180. [Google Scholar] [CrossRef]
- Scholz, C.C.; Taylor, C.T. Targeting the HIF pathway in inflammation and immunity. Curr. Opin. Pharmacol. 2013, 13, 646–653. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Zhang, N.; Fu, Z.; Linke, S.; Chicher, J.; Gorman, J.J.; Visk, D.; Haddad, G.G.; Poellinger, L.; Peet, D.J.; Powell, F.; et al. The asparaginyl hydroxylase factor inhibiting HIF-1alpha is an essential regulator of metabolism. Cell Metab. 2010, 11, 364–378. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef]
- Jiang, B.H.; Jiang, G.; Zheng, J.Z.; Lu, Z.; Hunter, T.; Vogt, P.K. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 2001, 12, 363–369. [Google Scholar]
- Semenza, G. Signal transduction to hypoxia-inducible factor 1. Biochem. Pharmacol. 2002, 64, 993–998. [Google Scholar] [CrossRef]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Kaelin, W.G. Von Hippel-Lindau disease. Annu. Rev. Pathol. 2007, 2, 145–173. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Conway, E.M.; Collen, D.; Carmeliet, P. Molecular mechanisms of blood vessel growth. Cardiovasc. Res. 2001, 49, 507–521. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef]
- Roda, J.M.; Wang, Y.; Sumner, L.A.; Phillips, G.S.; Marsh, C.B.; Eubank, T.D. Stabilization of HIF-2α induces sVEGFR-1 production from tumor-associated macrophages and decreases tumor growth in a murine melanoma model. J. Immunol. 2012, 189, 3168–3177. [Google Scholar] [CrossRef]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef]
- Lin, S.; Wan, S.; Sun, L.; Hu, J.; Fang, D.; Zhao, R.; Yuan, S.; Zhang, L. Chemokine C-C motif receptor 5 and C-C motif ligand 5 promote cancer cell migration under hypoxia. Cancer Sci. 2012, 103, 904–912. [Google Scholar] [CrossRef]
- Leek, R.D.; Hunt, N.C.; Landers, R.J.; Lewis, C.E.; Royds, J.A.; Harris, A.L. Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J. Pathol. 2000, 190, 430–436. [Google Scholar] [CrossRef]
- Grimshaw, M.J. Endothelins and hypoxia-inducible factor in cancer. Endocr. Relat. Cancer 2007, 14, 233–244. [Google Scholar] [CrossRef]
- Casanello, P.; Torres, A.; Sanhueza, F.; González, M.; Farías, M.; Gallardo, V.; Pastor-Anglada, M.; San Martín, R.; Sobrevia, L. Equilibrative nucleoside transporter 1 expression is downregulated by hypoxia in human umbilical vein endothelium. Circ. Res. 2005, 97, 16–24. [Google Scholar] [CrossRef]
- Lukashev, D.; Ohta, A.; Sitkovsky, M. Hypoxia-dependent anti-inflammatory pathways in protection of cancerous tissues. Cancer Metastasis Rev. 2007, 26, 273–279. [Google Scholar] [CrossRef]
- Palazón, A.; Aragonés, J.; Morales-Kastresana, A.; de Landázuri, M.O.; Melero, I. Molecular pathways: Hypoxia response in immune cells fighting or promoting cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1207–1213. [Google Scholar] [CrossRef]
- Kono, K.; Salazar-Onfray, F.; Petersson, M.; Hansson, J.; Masucci, G.; Wasserman, K.; Nakazawa, T.; Anderson, P.; Kiessling, R. Hydrogen peroxide secreted by tumor-derived macrophages down-modulates signal-transducing zeta molecules and inhibits tumor-specific T cell-and natural killer cell-mediated cytotoxicity. Eur. J. Immunol. 1996, 26, 1308–1313. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.-P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Hansen, W.; Hutzler, M.; Abel, S.; Alter, C.; Stockmann, C.; Kliche, S.; Albert, J.; Sparwasser, T.; Sakaguchi, S.; Westendorf, A.M.; et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J. Exp. Med. 2012, 209, 2001–2016. [Google Scholar] [CrossRef]
- Deng, B.; Zhu, J.-M.; Wang, Y.; Liu, T.-T.; Ding, Y.-B.; Xiao, W.-M.; Lu, G.-T.; Bo, P.; Shen, X.-Z. Intratumor hypoxia promotes immune tolerance by inducing regulatory T cells via TGF-β1 in gastric cancer. PLoS ONE 2013, 8, e63777. [Google Scholar] [CrossRef]
- Onnis, B.; Rapisarda, A.; Melillo, G. Development of HIF-1 inhibitors for cancer therapy. J. Cell. Mol. Med. 2009, 13, 2780–2786. [Google Scholar] [CrossRef]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef]
- Terzuoli, E.; Puppo, M.; Rapisarda, A.; Uranchimeg, B.; Cao, L.; Burger, A.M.; Ziche, M.; Melillo, G. Aminoflavone, a ligand of the aryl hydrocarbon receptor, inhibits HIF-1alpha expression in an AhR-independent fashion. Cancer Res. 2010, 70, 6837–6848. [Google Scholar] [CrossRef]
- Zhao, T.; Ren, H.; Jia, L.; Chen, J.; Xin, W.; Yan, F.; Li, J.; Wang, X.; Gao, S.; Qian, D.; et al. Inhibition of HIF-1α by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget 2015, 6, 2250–2262. [Google Scholar]
- Rapisarda, A.; Uranchimeg, B.; Sordet, O.; Pommier, Y.; Shoemaker, R.H.; Melillo, G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: Mechanism and therapeutic implications. Cancer Res. 2004, 64, 1475–1482. [Google Scholar] [CrossRef]
- Rapisarda, A.; Hollingshead, M.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Gehrs, B.; Raffeld, M.; Kinders, R.J.; Parchment, R.; et al. Increased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibition. Mol. Cancer Ther. 2009, 8, 1867–1877. [Google Scholar] [CrossRef]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domblides, C.; Lartigue, L.; Faustin, B. Control of the Antitumor Immune Response by Cancer Metabolism. Cells 2019, 8, 104. https://doi.org/10.3390/cells8020104
Domblides C, Lartigue L, Faustin B. Control of the Antitumor Immune Response by Cancer Metabolism. Cells. 2019; 8(2):104. https://doi.org/10.3390/cells8020104
Chicago/Turabian StyleDomblides, Charlotte, Lydia Lartigue, and Benjamin Faustin. 2019. "Control of the Antitumor Immune Response by Cancer Metabolism" Cells 8, no. 2: 104. https://doi.org/10.3390/cells8020104
APA StyleDomblides, C., Lartigue, L., & Faustin, B. (2019). Control of the Antitumor Immune Response by Cancer Metabolism. Cells, 8(2), 104. https://doi.org/10.3390/cells8020104