Cardiac Progenitor Cells from Stem Cells: Learning from Genetics and Biomaterials



Abstract
1. Cardiac Regeneration—A Problem to Solve or A Solution with Promise?
2. Cardiac Progenitor Cells (CPCs) In Vivo
2.1. c-KIT-Expressing CPCs
2.2. SCA1-Expressing CPCs
2.3. MESP1/2-Expressing CPCs
2.4. KDR/FLK1-Expressing CPCs
2.5. CPCs from the First and Second Heart Fields
2.6. Epicardium-Derived CPCs
2.7. Side Population-Derived CPCs
2.8. Cardiosphere-Derived CPCs
3. Generation of CPCs from Human iPSCs
4. Direct Reprogramming into CPCs
4.1. Partial Somatic Cell Reprogramming into CPCs
4.2. Direct Somatic Reprogramming into CPCs
4.3. Somatic Reprogramming into Cardiospheres
4.4. In Vivo Direct Reprogramming
5. In Vitro Culture of CPCs Derived Through Reprogramming Protocols
5.1. Isolation of CPCs
5.2. Expansion and Maintenance of iPSC-CPCs
5.3. Expansion and Maintenance of Transdifferentiated CPCs
6. Strategies to Improve CPC Reprogramming
6.1. Genetic Engineering with PIM1
6.2. CRISPR in Context with CPCs
6.3. Epigenetic Modulators
6.4. MicroRNAs
7. Tissue Engineering with CPCs and CPC-Derived Cardiomyocytes
7.1. Natural Scaffolds
7.2. Synthetic Scaffolds
8. In Vivo Applications of Human CPCs
9. Current Challenges and Limitations
10. Final Thoughts—Controversies Surrounding CPCs
11. Future Directions
Funding
Conflicts of Interest
References
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Executive summary: Heart disease and stroke statistics—2014 Update: A report from the American Heart Association. Circulation 2014, 129, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V. Global burden of cardiovascular disease. J. Am. Coll. Cardiol. 2014, 64, 520–522. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Witman, N.; Sahara, M. Cardiac progenitor cells in basic biology and regenerative medicine. Stem Cells Int. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Van Laake, L.W.; Davidson, S.M.; Engel, F.B.; Hausenloy, D.J.; Lecour, S.; Leor, J.; Perrino, C.; Schulz, R.; Ytrehus, K.; et al. Position paper of the European society of cardiology working group cellular biology of the heart: Cell-based therapies for myocardial repair and regeneration in ischemic heart disease and heart failure. Eur. Heart J. 2016, 37, 1789–1798. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; dos Remedios, C.; et al. Dynamics of cell generation and turnover in the human heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef]
- Graham, E.; Bergmann, O. Dating the heart: Exploring cardiomyocyte renewal in humans. Physiology 2017, 32, 33–41. [Google Scholar] [CrossRef]
- Urbanek, K.; Torella, D.; Sheikh, F.; De Angelis, A.; Nurzynska, D.; Silvestri, F.; Beltrami, C.A.; Bussani, R.; Beltrami, A.P.; Quaini, F.; et al. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 8692–8697. [Google Scholar] [CrossRef]
- Bloomekatz, J.; Galvez-Santisteban, M.; Chi, N.C. Myocardial plasticity: Cardiac development, regeneration and disease. Curr. Opin. Genet. Dev. 2016, 40, 120–130. [Google Scholar] [CrossRef]
- van Berlo, J.H.; Molkentin, J.D. An emerging consensus on cardiac regeneration. Nat. Med. 2014, 20, 1386–1393. [Google Scholar] [CrossRef]
- Liang, S.X.; Phillips, W.D. Migration of resident cardiac stem cells in myocardial infarction: Migration of cardiac stem cells. Anat. Rec. 2013, 296, 184–191. [Google Scholar] [CrossRef]
- Oh, H.; Bradfute, S.B.; Gallardo, T.D.; Nakamura, T.; Gaussin, V.; Mishina, Y.; Pocius, J.; Michael, L.H.; Behringer, R.R.; Garry, D.J.; et al. Cardiac progenitor cells from adult myocardium: Homing, differentiation, and fusion after infarction. Proc. Natl. Acad. Sci. USA 2003, 100, 12313–12318. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Lemcke, H.; David, R. Stem cell therapy in heart diseases—Cell types, mechanisms and improvement strategies. Cell. Physiol. Biochem. 2018, 48, 2607–2655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, N.; Huang, Y.; Spencer, C.I.; Fu, J.; Yu, C.; Liu, K.; Nie, B.; Xu, T.; Li, K.; et al. Expandable cardiovascular progenitor cells reprogrammed from fibroblasts. Cell Stem Cell 2016, 18, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Keller, G.; Gold, J.D.; Wu, J.C. Production of de novo cardiomyocytes: Human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell 2012, 10, 16–28. [Google Scholar] [CrossRef]
- Lam, J.T.; Moretti, A.; Laugwitz, K.-L. Multipotent progenitor cells in regenerative cardiovascular medicine. Pediatric Cardiol. 2009, 30, 690–698. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Ebrahimi, B. Cardiac progenitor reprogramming for heart regeneration. Cell Regen. 2018, 7, 1–6. [Google Scholar] [CrossRef]
- Birket, M.J.; Mummery, C.L. Pluripotent stem cell derived cardiovascular progenitors—A developmental perspective. Dev. Biol. 2015, 400, 169–179. [Google Scholar] [CrossRef]
- Kattman, S.J.; Huber, T.L.; Keller, G.M. Multipotent Flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev. Cell 2006, 11, 723–732. [Google Scholar] [CrossRef]
- Wu, S.M.; Fujiwara, Y.; Cibulsky, S.M.; Clapham, D.E.; Lien, C.; Schultheiss, T.M.; Orkin, S.H. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell 2006, 127, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.-L.; Liang, X.; Shi, Y.; Chu, P.-H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Henning, R.J. Stem cells in cardiac repair. Future Cardiol. 2011, 7, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Lalit, P.A.; Salick, M.R.; Nelson, D.O.; Squirrell, J.M.; Shafer, C.M.; Patel, N.G.; Saeed, I.; Schmuck, E.G.; Markandeya, Y.S.; Wong, R.; et al. Lineage reprogramming of fibroblasts into proliferative induced cardiac progenitor cells by defined factors. Cell Stem Cell 2016, 18, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Christoforou, N.; Miller, R.A.; Hill, C.M.; Jie, C.C.; McCallion, A.S.; Gearhart, J.D. Mouse ES cell–derived cardiac precursor cells are multipotent and facilitate identification of novel cardiac genes. J. Clin. Investig. 2008, 118, 894–903. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kawaguchi, N.; Ellison, G.M.; Matsuoka, R.; Shin’oka, T.; Kurosawa, H. Characterization of long-term cultured c-kit + cardiac stem cells derived from adult rat hearts. Stem Cells Dev. 2010, 19, 105–116. [Google Scholar] [CrossRef]
- Bearzi, C.; Rota, M.; Hosoda, T.; Tillmanns, J.; Nascimbene, A.; De Angelis, A.; Yasuzawa-Amano, S.; Trofimova, I.; Siggins, R.W.; LeCapitaine, N.; et al. Human cardiac stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 14068–14073. [Google Scholar] [CrossRef]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef]
- Yang, L.; Soonpaa, M.H.; Adler, E.D.; Roepke, T.K.; Kattman, S.J.; Kennedy, M.; Henckaerts, E.; Bonham, K.; Abbott, G.W.; Linden, R.M.; et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 2008, 453, 524–528. [Google Scholar] [CrossRef]
- Moretti, A.; Caron, L.; Nakano, A.; Lam, J.T.; Bernshausen, A.; Chen, Y.; Qyang, Y.; Bu, L.; Sasaki, M.; Martin-Puig, S.; et al. Multipotent embryonic Isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell 2006, 127, 1151–1165. [Google Scholar] [CrossRef]
- Tallini, Y.N.; Greene, K.S.; Craven, M.; Spealman, A.; Breitbach, M.; Smith, J.; Fisher, P.J.; Steffey, M.; Hesse, M.; Doran, R.M.; et al. c-Kit expression identifies cardiovascular precursors in the neonatal heart. Proc. Natl. Acad. Sci. USA 2009, 106, 1808–1813. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.J.; Schulman, S.P.; Hare, J.M. Stem cell therapy for cardiac repair: Ready for the next step. Circulation 2006, 114, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, D.; Schellings, M.; Pinto, Y.; Heymans, S. Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: A temporal and spatial window. Cardiovasc. Res. 2006, 69, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Deddens, J.C.; Sadeghi, A.H.; Hjortnaes, J.; van Laake, L.W.; Buijsrogge, M.; Doevendans, P.A.; Khademhosseini, A.; Sluijter, J.P.G. Modeling the human scarred heart in vitro: Toward new tissue engineered models. Adv. Healthc. Mater. 2017, 6, 1600571. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J. Mol. Cell. Cardiol. 2010, 48, 504–511. [Google Scholar] [CrossRef]
- Le, T.Y.L.; Thavapalachandran, S.; Kizana, E.; Chong, J.J. New developments in cardiac regeneration. Heart Lung Circ. 2017, 26, 316–322. [Google Scholar] [CrossRef]
- Van Vliet, P.; Roccio, M.; Smits, A.M.; van Oorschot, A. a. M.; Metz, C.H.G.; van Veen, T. a. B.; Sluijter, J.P.G.; Doevendans, P.A.; Goumans, M.-J. Progenitor cells isolated from the human heart: A potential cell source for regenerative therapy. Neth Heart J. 2008, 16, 163–169. [Google Scholar] [CrossRef]
- Sandstedt, J.; Jonsson, M.; Kajic, K.; Sandstedt, M.; Lindahl, A.; Dellgren, G.; Jeppsson, A.; Asp, J. Left atrium of the human adult heart contains a population of side population cells. Basic Res. Cardiol. 2012, 107, 255. [Google Scholar] [CrossRef]
- Messina, E.; De Angelis, L.; Frati, G.; Morrone, S.; Chimenti, S.; Fiordaliso, F.; Salio, M.; Battaglia, M.; Latronico, M.V.G.; Coletta, M.; et al. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ. Res. 2004, 95, 911–921. [Google Scholar] [CrossRef]
- Laugwitz, K.-L.; Moretti, A.; Lam, J.; Gruber, P.; Chen, Y.; Woodard, S.; Lin, L.-Z.; Cai, C.-L.; Lu, M.M.; Reth, M.; et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 2005, 433, 647–653. [Google Scholar] [CrossRef]
- Li, X.-H.; Li, Q.; Jiang, L.; Deng, C.; Liu, Z.; Fu, Y.; Zhang, M.; Tan, H.; Feng, Y.; Shan, Z.; et al. Generation of functional human cardiac progenitor cells by high-efficiency protein transduction: Protein-generated cardiac progenitor cells. Stem Cells Transl. Med. 2015, 4, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Pessina, A.; Gribaldo, L. The key role of adult stem cells: Therapeutic perspectives. Curr. Med. Res. Opin. 2006, 22, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Cedar, S. The function of stem cells and their future roles in healthcare. Br. J. Nurs. 2006, 15, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Cyranoski, D. ‘Reprogrammed’ stem cells approved to mend human hearts for the first time. Nature 2018, 557, 619–620. [Google Scholar] [CrossRef]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous induced stem-cell–derived retinal cells for macular degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Ronen, D.; Benvenisty, N. Genomic stability in reprogramming. Curr. Opin. Genet. Dev. 2012, 22, 444–449. [Google Scholar] [CrossRef]
- Margariti, A.; Kelaini, S.; Cochrane, A. Direct reprogramming of adult cells: Avoiding the pluripotent state. Stem Cells Cloning: Adv. Appl. 2014, 7, 19. [Google Scholar] [CrossRef]
- Xu, J.; Lian, W.; Li, L.; Huang, Z. Generation of induced cardiac progenitor cells via somatic reprogramming. Oncotarget 2017, 8, 29442. [Google Scholar] [CrossRef]
- Sassoli, C. Cardiac progenitor cells as target of cell and growth factor-based therapies for myocardial regeneration. J. Stem Cell Res. Ther. 2013, 9, 004. [Google Scholar] [CrossRef]
- Le, T.; Chong, J. Cardiac progenitor cells for heart repair. Cell Death Discov. 2016, 2, 16052. [Google Scholar] [CrossRef] [PubMed]
- Ellison, G.M.; Vicinanza, C.; Smith, A.J.; Aquila, I.; Leone, A.; Waring, C.D.; Henning, B.J.; Stirparo, G.G.; Papait, R.; Scarfò, M.; et al. Adult c-kitpos cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 2013, 154, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Edling, C.E.; Hallberg, B. c-Kit—A hematopoietic cell essential receptor tyrosine kinase. Int. J. Biochem. Cell Biol. 2007, 39, 1995–1998. [Google Scholar] [CrossRef] [PubMed]
- Vajravelu, B.N.; Hong, K.U.; Al-Maqtari, T.; Cao, P.; Keith, M.C.L.; Wysoczynski, M.; Zhao, J.; Moore IV, J.B.; Bolli, R. c-Kit promotes growth and migration of human cardiac progenitor cells via the PI3K-AKT and MEK-ERK pathways. PLoS ONE 2015, 10, e0140798. [Google Scholar] [CrossRef]
- Kuang, D.; Zhao, X.; Xiao, G.; Ni, J.; Feng, Y.; Wu, R.; Wang, G. Stem cell factor/c-kit signaling mediated cardiac stem cell migration via activation of p38 MAPK. Basic Res. Cardiol. 2008, 103, 265–273. [Google Scholar] [CrossRef]
- Ayach, B.B.; Yoshimitsu, M.; Dawood, F.; Sun, M.; Arab, S.; Chen, M.; Higuchi, K.; Siatskas, C.; Lee, P.; Lim, H.; et al. Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc. Natl. Acad. Sci. USA 2006, 103, 2304–2309. [Google Scholar] [CrossRef]
- van Berlo, J.H.; Kanisicak, O.; Maillet, M.; Vagnozzi, R.J.; Karch, J.; Lin, S.-C.J.; Middleton, R.C.; Marbán, E.; Molkentin, J.D. c-Kit+ cells minimally contribute cardiomyocytes to the heart. Nature 2014, 509, 337–341. [Google Scholar] [CrossRef]
- Jesty, S.A.; Steffey, M.A.; Lee, F.K.; Breitbach, M.; Hesse, M.; Reining, S.; Lee, J.C.; Doran, R.M.; Nikitin, A.Y.; Fleischmann, B.K.; et al. c-Kit+ precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc. Natl. Acad. Sci. USA 2012, 109, 13380–13385. [Google Scholar] [CrossRef]
- Zaruba, M.-M.; Soonpaa, M.; Reuter, S.; Field, L.J. Cardiomyogenic potential of c-Kit +–Expressing cells derived from neonatal and adult mouse hearts. Circulation 2010, 121, 1992–2000. [Google Scholar] [CrossRef]
- Sultana, N.; Zhang, L.; Yan, J.; Chen, J.; Cai, W.; Razzaque, S.; Jeong, D.; Sheng, W.; Bu, L.; Xu, M.; et al. Resident c-kit+ cells in the heart are not cardiac stem cells. Nat. Commun. 2015, 6, 8701. [Google Scholar] [CrossRef] [PubMed]
- Vicinanza, C.; Aquila, I.; Scalise, M.; Cristiano, F.; Marino, F.; Cianflone, E.; Mancuso, T.; Marotta, P.; Sacco, W.; Lewis, F.C.; et al. Adult cardiac stem cells are multipotent and robustly myogenic: C-Kit expression is necessary but not sufficient for their identification. Cell Death Differ. 2017, 24, 2101–2116. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Stanford, W.L. Concise review: Stem cell antigen-1: Expression, function, and enigma. Stem Cells 2007, 25, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Nagai, T.; Nishigaki, N.; Oyama, T.; Nishi, J.; Wada, H.; Sano, M.; Toko, H.; Akazawa, H.; Sato, T.; et al. Adult cardiac sca-1-positive cells differentiate into beating cardiomyocytes. J. Biol. Chem. 2004, 279, 11384–11391. [Google Scholar] [CrossRef]
- Tateishi, K.; Ashihara, E.; Takehara, N.; Nomura, T.; Honsho, S.; Nakagami, T.; Morikawa, S.; Takahashi, T.; Ueyama, T.; Matsubara, H.; et al. Clonally amplified cardiac stem cells are regulated by Sca-1 signaling for efficient cardiovascular regeneration. J. Cell Sci. 2007, 120, 1791–1800. [Google Scholar] [CrossRef]
- Wang, X.; Hu, Q.; Nakamura, Y.; Lee, J.; Zhang, G.; From, A.H.L.; Zhang, J. The role of the Sca-1 +/CD31 − cardiac progenitor cell population in postinfarction left ventricular remodeling. Stem Cells 2006, 24, 1779–1788. [Google Scholar] [CrossRef]
- Van Vliet, P.; Smits, A.M.; De Boer, T.P.; Korfage, T.H.; Metz, C.H.G.; Roccio, M.; Van Der Heyden, M.A.G.; Van Veen, T.A.B.; Sluijter, J.P.G.; Doevendans, P.A.; et al. Foetal and adult cardiomyocyte progenitor cells have different developmental potential. J. Cell. Mol. Med. 2010, 14, 861–870. [Google Scholar] [CrossRef][Green Version]
- Huang, C.; Gu, H.; Yu, Q.; Manukyan, M.C.; Poynter, J.A.; Wang, M. Sca-1+ cardiac stem cells mediate acute cardioprotection via paracrine factor SDF-1 following myocardial ischemia/reperfusion. PLoS ONE 2011, 6, e29246. [Google Scholar] [CrossRef]
- Takamiya, M.; Haider, K.H.; Ashraf, M. Identification and characterization of a novel multipotent sub-population of Sca-1+ cardiac progenitor cells for myocardial regeneration. PLoS ONE 2011, 6, e25265. [Google Scholar] [CrossRef]
- Uchida, S.; De Gaspari, P.; Kostin, S.; Jenniches, K.; Kilic, A.; Izumiya, Y.; Shiojima, I.; grosse Kreymborg, K.; Renz, H.; Walsh, K.; et al. Sca1-derived cells are a source of myocardial renewal in the murine adult heart. Stem Cell Rep. 2013, 1, 397–410. [Google Scholar] [CrossRef]
- Ge, Z.; Lal, S.; Le, T.Y.L.; dos Remedios, C.; Chong, J.J.H. Cardiac stem cells: Translation to human studies. Biophys. Rev. 2015, 7, 127–139. [Google Scholar] [CrossRef] [PubMed]
- David, R.; Jarsch, V.B.; Schwarz, F.; Nathan, P.; Gegg, M.; Lickert, H.; Franz, W.-M. Induction of MesP1 by Brachyury(T) generates the common multipotent cardiovascular stem cell. Cardiovasc. Res. 2011, 92, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schwartz, R.J. Transient Mesp1 expression: A driver of cardiac cell fate determination. Transcription 2013, 4, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Takagi, A.; Inoue, T.; Saga, Y. MesP1 and MesP2 are essential for the development of cardiac mesoderm. Development 2000, 127, 3215–3226. [Google Scholar]
- Habib, M.; Caspi, O.; Gepstein, L. Human embryonic stem cells for cardiomyogenesis. J. Mol. Cell. Cardiol. 2008, 45, 462–474. [Google Scholar] [CrossRef]
- Wu, S.M.; Chien, K.R.; Mummery, C. Origins and fates of cardiovascular progenitor cells. Cell 2008, 132, 537–543. [Google Scholar] [CrossRef]
- Saga, Y.; Miyagawa-Tomita, S.; Takagi, A.; Kitajima, S.; Miyazaki, J.; Inoue, T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development 1999, 126, 3437–3447. [Google Scholar]
- Chan, S.S.-K.; Shi, X.; Toyama, A.; Arpke, R.W.; Dandapat, A.; Iacovino, M.; Kang, J.; Le, G.; Hagen, H.R.; Garry, D.J.; et al. Mesp1 patterns mesoderm into cardiac, hematopoietic, or skeletal myogenic progenitors in a context-dependent manner. Cell Stem Cell 2013, 12, 587–601. [Google Scholar] [CrossRef]
- Bondue, A.; Tännler, S.; Chiapparo, G.; Chabab, S.; Ramialison, M.; Paulissen, C.; Beck, B.; Harvey, R.; Blanpain, C. Defining the earliest step of cardiovascular progenitor specification during embryonic stem cell differentiation. J. Cell Biol. 2011, 192, 751–765. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, L.; Diaz, A.D.; Benham, A.; Xu, X.; Wijaya, C.S.; Fa’ak, F.; Luo, W.; Soibam, B.; Azares, A.; et al. Mesp1 marked cardiac progenitor cells repair infarcted mouse hearts. Sci. Rep. 2016, 6, 31457. [Google Scholar] [CrossRef]
- Ema, M. Deletion of the selection cassette, but not cis-acting elements, in targeted Flk1-lacZ allele reveals Flk1 expression in multipotent mesodermal progenitors. Blood 2006, 107, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Kouskoff, V.; Lacaud, G.; Schwantz, S.; Fehling, H.J.; Keller, G. Sequential development of hematopoietic and cardiac mesoderm during embryonic stem cell differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 13170–13175. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Jiang, X.; Martin-Puig, S.; Caron, L.; Zhu, S.; Shao, Y.; Roberts, D.J.; Huang, P.L.; Domian, I.J.; Chien, K.R. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 2009, 460, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, G.; Lin, L.; Lowe, J.; Zhang, Q.; Bu, L.; Chen, Y.; Chen, J.; Sun, Y.; Evans, S.M. HCN4 dynamically marks the first heart field and conduction system precursors. Circ. Res. 2013, 113, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Später, D.; Abramczuk, M.K.; Buac, K.; Zangi, L.; Stachel, M.W.; Clarke, J.; Sahara, M.; Ludwig, A.; Chien, K.R. A HCN4+ cardiomyogenic progenitor derived from the first heart field and human pluripotent stem cells. Nat. Cell Biol. 2013, 15, 1098–1106. [Google Scholar] [CrossRef]
- Garcia-Frigola, C.; Shi, Y.; Evans, S.M. Expression of the hyperpolarization-activated cyclic nucleotide-gated cation channel HCN4 during mouse heart development. Gene Expr. Patterns 2003, 3, 777–783. [Google Scholar] [CrossRef]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; von Gise, A.; Ikeda, S.; Chien, K.R.; et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef]
- van Tuyn, J.; Atsma, D.E.; Winter, E.M.; van der Velde-van Dijke, I.; Pijnappels, D.A.; Bax, N.A.M.; Knaän-Shanzer, S.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; van der Laarse, A.; et al. Epicardial cells of human adults can undergo an epithelial-to-mesenchymal transition and obtain characteristics of smooth muscle cells in vitro. Stem Cells 2007, 25, 271–278. [Google Scholar] [CrossRef]
- Cai, C.-L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef]
- Smart, N.; Bollini, S.; Dubé, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Bollini, S.; Vieira, J.M.N.; Howard, S.; Dubè, K.N.; Balmer, G.M.; Smart, N.; Riley, P.R. Re-activated adult epicardial progenitor cells are a heterogeneous population molecularly distinct from their embryonic counterparts. Stem Cells Dev. 2014, 23, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Dubé, K.N.; Riley, P.R. Epicardial progenitor cells in cardiac regeneration and neovascularisation. Vasc. Pharmacol. 2013, 58, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.M.; Meeson, A.P.; Robertson, S.M.; Hawke, T.J.; Richardson, J.A.; Bates, S.; Goetsch, S.C.; Gallardo, T.D.; Garry, D.J. Persistent expression of the ATP-binding cassette transporter, Abcg2, identifies cardiac SP cells in the developing and adult heart. Dev. Biol. 2004, 265, 262–275. [Google Scholar] [CrossRef]
- Pfister, O.; Oikonomopoulos, A.; Sereti, K.-I.; Sohn, R.L.; Cullen, D.; Fine, G.C.; Mouquet, F.; Westerman, K.; Liao, R. Role of the ATP-binding cassette transporter Abcg2 in the phenotype and function of cardiac side population cells. Circ. Res. 2008, 103, 825–835. [Google Scholar] [CrossRef]
- Liang, S.X.; Tan, T.Y.L.; Gaudry, L.; Chong, B. Differentiation and migration of Sca1+/CD31− cardiac side population cells in a murine myocardial ischemic model. Int. J. Cardiol. 2010, 138, 40–49. [Google Scholar] [CrossRef]
- Oyama, T.; Nagai, T.; Wada, H.; Naito, A.T.; Matsuura, K.; Iwanaga, K.; Takahashi, T.; Goto, M.; Mikami, Y.; Yasuda, N.; et al. Cardiac side population cells have a potential to migrate and differentiate into cardiomyocytes in vitro and in vivo. J. Cell Biol. 2007, 176, 329–341. [Google Scholar] [CrossRef]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.-M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef]
- Alfakir, M.; Dawe, N.; Eyre, R.; Tyson-Capper, A.; Britton, K.; Robson, S.C.; Meeson, A.P. The temporal and spatial expression patterns of ABCG2 in the developing human heart. Int. J. Cardiol. 2012, 156, 133–138. [Google Scholar] [CrossRef][Green Version]
- Hierlihy, A.M.; Seale, P.; Lobe, C.G.; Rudnicki, M.A.; Megeney, L.A. The post-natal heart contains a myocardial stem cell population. FEBS Lett. 2002, 530, 239–243. [Google Scholar] [CrossRef]
- Pfister, O.; Mouquet, F.; Jain, M.; Summer, R.; Helmes, M.; Fine, A.; Colucci, W.S.; Liao, R. CD31− but not CD31+ cardiac side population cells exhibit functional cardiomyogenic differentiation. Circ. Res. 2005, 97, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Choi, S.-C.; Park, C.-Y.; Shim, W.-J.; Lim, D.-S. Cardiac side population cells exhibit endothelial differentiation potential. Exp. Mol. Med. 2007, 39, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; Harada, M.; McSweeney, S.; Leja, T.; Belian, E.; Stuckey, D.J.; Abreu Paiva, M.S.; Habib, J.; Macaulay, I.; de Smith, A.J.; et al. PDGFRα demarcates the cardiogenic clonogenic Sca1+ stem/progenitor cell in adult murine myocardium. Nat. Commun 2015, 6, 6930. [Google Scholar] [CrossRef] [PubMed]
- Yamahara, K.; Fukushima, S.; Coppen, S.R.; Felkin, L.E.; Varela-Carver, A.; Barton, P.J.R.; Yacoub, M.H.; Suzuki, K. Heterogeneic nature of adult cardiac side population cells. Biochem. Biophys. Res. Commun. 2008, 371, 615–620. [Google Scholar] [CrossRef]
- Chimenti, I.; Smith, R.R.; Li, T.-S.; Gerstenblith, G.; Messina, E.; Giacomello, A.; Marbán, E. Relative roles of direct regeneration versus paracrine effects of human cardiosphere-derived cells transplanted into infarcted mice. Circ. Res. 2010, 106, 971–980. [Google Scholar] [CrossRef]
- Li, T.-S.; Cheng, K.; Lee, S.-T.; Matsushita, S.; Davis, D.; Malliaras, K.; Zhang, Y.; Matsushita, N.; Smith, R.R.; Marbán, E. Cardiospheres recapitulate a niche-like microenvironment rich in stemness and cell-matrix interactions, rationalizing their enhanced functional potency for myocardial repair. Stem Cells 2010, 28, 2088–2098. [Google Scholar] [CrossRef]
- He, J.-Q.; Vu, D.M.; Hunt, G.; Chugh, A.; Bhatnagar, A.; Bolli, R. Human cardiac stem cells isolated from atrial appendages stably express c-kit. PLoS ONE 2011, 6, e27719. [Google Scholar] [CrossRef]
- Hesse, M.; Fleischmann, B.K.; Kotlikoff, M.I. Concise Review: The role of c-kit expressing cells in heart repair at the neonatal and adult stage: C-kit + cells in heart repair. Stem Cells 2014, 32, 1701–1712. [Google Scholar] [CrossRef]
- Freire, A.G.; Nascimento, D.S.; Forte, G.; Valente, M.; Resende, T.P.; Pagliari, S.; Abreu, C.; Carvalho, I.; Nardo, P.D.; Pinto-do-Ó, P. Stable phenotype and function of immortalized Lin− Sca-1+ cardiac progenitor cells in long-term culture: A step closer to standardization. Stem Cells Dev. 2014, 23, 1012–1026. [Google Scholar] [CrossRef]
- Yamashita, J.K.; Takano, M.; Hiraoka-Kanie, M.; Shimazu, C.; Peishi, Y.; Yanagi, K.; Nakano, A.; Inoue, E.; Kita, F.; Nishikawa, S.-I. Prospective identification of cardiac progenitors by a novel single cell-based cardiomyocyte induction. FASEB J. 2005, 19, 1534–1536. [Google Scholar] [CrossRef]
- Lescroart, F.; Chabab, S.; Lin, X.; Rulands, S.; Paulissen, C.; Rodolosse, A.; Auer, H.; Achouri, Y.; Dubois, C.; Bondue, A.; et al. Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat. Cell Biol. 2014, 16, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, T.I.; Appleby, N.; Tsay, E.; Martinez, J.J.; Bailey, L.; Hasaniya, N.; Kearns-Jonker, M. Human neonatal cardiovascular progenitors: Unlocking the secret to regenerative ability. PLoS ONE 2013, 8, e77464. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liang, X.; Najafi, N.; Cass, M.; Lin, L.; Cai, C.-L.; Chen, J.; Evans, S.M. Islet 1 is expressed in distinct cardiovascular lineages, including pacemaker and coronary vascular cells. Dev. Biol. 2007, 304, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.J.H.; Reinecke, H.; Iwata, M.; Torok-Storb, B.; Stempien-Otero, A.; Murry, C.E. Progenitor cells identified by PDGFR-alpha expression in the developing and diseased human heart. Stem Cells Dev. 2013, 22, 1932–1943. [Google Scholar] [CrossRef]
- Chong, J.J.H.; Chandrakanthan, V.; Xaymardan, M.; Asli, N.S.; Li, J.; Ahmed, I.; Heffernan, C.; Menon, M.K.; Scarlett, C.J.; Rashidianfar, A.; et al. Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell Stem Cell 2011, 9, 527–540. [Google Scholar] [CrossRef]
- Wessels, A.; Pérez-Pomares, J.M. The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells: Epicardially derived cells as cardiac stem cells. Anat. Rec. Part. A: Discov. Mol. Cell. Evol. Biol. 2004, 276A, 43–57. [Google Scholar] [CrossRef]
- Smits, A.; Riley, P. Epicardium-derived heart repair. J. Dev. Biol. 2014, 2, 84–100. [Google Scholar] [CrossRef]
- Emmert, M.Y.; Emmert, L.S.; Martens, A.; Ismail, I.; Schmidt-Richter, I.; Gawol, A.; Seifert, B.; Haverich, A.; Martin, U.; Gruh, I. Higher frequencies of BCRP+ cardiac resident cells in ischaemic human myocardium. Eur. Heart J. 2013, 34, 2830–2838. [Google Scholar] [CrossRef]
- Smith, R.R.; Barile, L.; Cho, H.C.; Leppo, M.K.; Hare, J.M.; Messina, E.; Giacomello, A.; Abraham, M.R.; Marbán, E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 2007, 115, 896–908. [Google Scholar] [CrossRef]
- Ye, J.; Boyle, A.; Shih, H.; Sievers, R.E.; Zhang, Y.; Prasad, M.; Su, H.; Zhou, Y.; Grossman, W.; Bernstein, H.S.; et al. Sca-1+ cardiosphere-derived cells are enriched for Isl1-expressing cardiac precursors and improve cardiac function after myocardial injury. PLoS ONE 2012, 7, e30329. [Google Scholar] [CrossRef]
- Klein, D. iPSCs-based generation of vascular cells: Reprogramming approaches and applications. Cell. Mol. Life Sci. 2018, 75, 1411–1433. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Thompson, S.; Millrod, M.A.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS ONE 2011, 6, e18293. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.H.; Mason, M.J.; Xie, W.; Volinia, S.; Singer, M.; Peterson, C.; Ambartsumyan, G.; Aimiuwu, O.; Richter, L.; Zhang, J.; et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell 2009, 5, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Mummery, C.L.; Zhang, J.; Ng, E.S.; Elliott, D.A.; Elefanty, A.G.; Kamp, T.J. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: A methods overview. Circ. Res. 2012, 111, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Olmer, R.; Haase, A.; Merkert, S.; Cui, W.; Paleček, J.; Ran, C.; Kirschning, A.; Scheper, T.; Glage, S.; Miller, K.; et al. Long term expansion of undifferentiated human iPS and ES cells in suspension culture using a defined medium. Stem Cell Res. 2010, 5, 51–64. [Google Scholar] [CrossRef]
- Kattman, S.J.; Witty, A.D.; Gagliardi, M.; Dubois, N.C.; Niapour, M.; Hotta, A.; Ellis, J.; Keller, G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 2011, 8, 228–240. [Google Scholar] [CrossRef]
- Drowley, L.; Koonce, C.; Peel, S.; Jonebring, A.; Plowright, A.T.; Kattman, S.J.; Andersson, H.; Anson, B.; Swanson, B.J.; Wang, Q.-D.; et al. Human induced pluripotent stem cell-derived cardiac progenitor cells in phenotypic screening: A transforming growth factor-β type 1 receptor kinase inhibitor induces efficient cardiac differentiation: iPSC-derived cardiac progenitors for phenotypic screening. Stem Cells Transl. Med. 2016, 5, 164–174. [Google Scholar]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef]
- Minami, I.; Yamada, K.; Otsuji, T.G.; Yamamoto, T.; Shen, Y.; Otsuka, S.; Kadota, S.; Morone, N.; Barve, M.; Asai, Y.; et al. A Small molecule that promotes cardiac differentiation of human pluripotent stem cells under defined, cytokine- and xeno-free conditions. Cell Rep. 2012, 2, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Kempf, H.; Olmer, R.; Kropp, C.; Rückert, M.; Jara-Avaca, M.; Robles-Diaz, D.; Franke, A.; Elliott, D.A.; Wojciechowski, D.; Fischer, M.; et al. Controlling expansion and cardiomyogenic differentiation of human pluripotent stem cells in scalable suspension culture. Stem Cell Rep. 2014, 3, 1132–1146. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Yan, P.; Otsuji, T.G.; Narazaki, G.; Uosaki, H.; Fukushima, H.; Kuwahara, K.; Harada, M.; Matsuda, H.; Matsuoka, S.; et al. Induction and enhancement of cardiac cell differentiation from mouse and human induced pluripotent stem cells with cyclosporin-A. PLoS ONE 2011, 6, e16734. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Nagasawa, A.; Uosaki, H.; Sugimoto, A.; Yamamizu, K.; Teranishi, M.; Matsuda, H.; Matsuoka, S.; Ikeda, T.; Komeda, M.; et al. Cyclosporin-A potently induces highly cardiogenic progenitors from embryonic stem cells. Biochem. Biophys. Res. Commun. 2009, 379, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Uosaki, H.; Andersen, P.; Shenje, L.T.; Fernandez, L.; Christiansen, S.L.; Kwon, C. Direct contact with endoderm-like cells efficiently induces cardiac progenitors from mouse and human pluripotent stem cells. PLoS ONE 2012, 7, e46413. [Google Scholar] [CrossRef] [PubMed]
- Xuan, W.; Wang, Y.; Tang, Y.; Ali, A.; Hu, H.; Maienschein-Cline, M.; Ashraf, M. Cardiac progenitors induced from human induced pluripotent stem cells with cardiogenic small molecule effectively regenerate infarcted hearts and attenuate fibrosis. Shock 2018, 50, 627–639. [Google Scholar] [CrossRef]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef]
- Gai, H. Generation and characterization of functional cardiomyocytes using induced pluripotent stem cells derived from human fibroblasts. Cell Biol. Int. 2009, 33, 1184–1193. [Google Scholar] [CrossRef]
- Uosaki, H.; Fukushima, H.; Takeuchi, A.; Matsuoka, S.; Nakatsuji, N.; Yamanaka, S.; Yamashita, J.K. Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by VCAM1 surface expression. PLoS ONE 2011, 6, e23657. [Google Scholar] [CrossRef]
- Zwi, L.; Caspi, O.; Arbel, G.; Huber, I.; Gepstein, A.; Park, I.-H.; Gepstein, L. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation 2009, 120, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Denning, C.; Borgdorff, V.; Crutchley, J.; Firth, K.S.A.; George, V.; Kalra, S.; Kondrashov, A.; Hoang, M.D.; Mosqueira, D.; Patel, A.; et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim. Et. Biophys. Acta Mol. Cell Res. 2016, 1863, 1728–1748. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Liu, Z.; Chen, Z.; Wang, J.; Chen, T.; Zhao, X.; Ma, Y.; Qin, L.; Kang, J.; Wei, B.; et al. Ascorbic acid enhances the cardiac differentiation of induced pluripotent stem cells through promoting the proliferation of cardiac progenitor cells. Cell Res. 2012, 22, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Liang, H.; Huang, J.; Wang, J.; Chen, Y.; Chen, Z.; Yang, H.-T. Highly efficient induction and long-term maintenance of multipotent cardiovascular progenitors from human pluripotent stem cells under defined conditions. Cell Res. 2013, 23, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Blin, G.; Nury, D.; Stefanovic, S.; Neri, T.; Guillevic, O.; Brinon, B.; Bellamy, V.; Rücker-Martin, C.; Barbry, P.; Bel, A.; et al. A purified population of multipotent cardiovascular progenitors derived from primate pluripotent stem cells engrafts in postmyocardial infarcted nonhuman primates. J. Clin. Investig. 2010, 120, 1125–1139. [Google Scholar] [CrossRef] [PubMed]
- Mauritz, C.; Martens, A.; Rojas, S.V.; Schnick, T.; Rathert, C.; Schecker, N.; Menke, S.; Glage, S.; Zweigerdt, R.; Haverich, A.; et al. Induced pluripotent stem cell (iPSC)-derived Flk-1 progenitor cells engraft, differentiate, and improve heart function in a mouse model of acute myocardial infarction. Eur. Heart J. 2011, 32, 2634–2641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Termglinchan, V.; Shao, N.-Y.; Itzhaki, I.; Liu, C.; Ma, N.; Tian, L.; Wang, V.Y.; Chang, A.C.Y.; Guo, H.; et al. A human iPSC double-reporter system enables purification of cardiac lineage subpopulations with distinct function and drug response profiles. Cell Stem Cell 2019, 24, 802–811. [Google Scholar] [CrossRef]
- Ren, Y.; Lee, M.Y.; Schliffke, S.; Paavola, J.; Amos, P.J.; Ge, X.; Ye, M.; Zhu, S.; Senyei, G.; Lum, L.; et al. Small molecule Wnt inhibitors enhance the efficiency of BMP-4-directed cardiac differentiation of human pluripotent stem cells. J. Mol. Cell. Cardiol. 2011, 51, 280–287. [Google Scholar] [CrossRef]
- Moretti, A.; Bellin, M.; Jung, C.B.; Thies, T.-M.; Takashima, Y.; Bernshausen, A.; Schiemann, M.; Fischer, S.; Moosmang, S.; Smith, A.G.; et al. Mouse and human induced pluripotent stem cells as a source for multipotent Isl1 + cardiovascular progenitors. FASEB J. 2010, 24, 700–711. [Google Scholar] [CrossRef]
- Lian, X.; Bao, X.; Zilberter, M.; Westman, M.; Fisahn, A.; Hsiao, C.; Hazeltine, L.B.; Dunn, K.K.; Kamp, T.J.; Palecek, S.P. Chemically defined, albumin-free human cardiomyocyte generation. Nat. Methods 2015, 12, 595–596. [Google Scholar] [CrossRef]
- Andersen, P.; Tampakakis, E.; Jimenez, D.V.; Kannan, S.; Miyamoto, M.; Shin, H.K.; Saberi, A.; Murphy, S.; Sulistio, E.; Chelko, S.P.; et al. Precardiac organoids form two heart fields via Bmp/Wnt signaling. Nat. Commun. 2018, 9, 3140. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Lian, X.; Qian, T.; Bhute, V.J.; Han, T.; Palecek, S.P. Directed differentiation and long-term maintenance of epicardial cells derived from human pluripotent stem cells under fully defined conditions. Nat. Protoc. 2017, 12, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Lian, X.; Hacker, T.A.; Schmuck, E.G.; Qian, T.; Bhute, V.J.; Han, T.; Shi, M.; Drowley, L.; Plowright, A.T.; et al. Long-term self-renewing human epicardial cells generated from pluripotent stem cells under defined xeno-free conditions. Nat. Biomed. Eng. 2017, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Witty, A.D.; Mihic, A.; Tam, R.Y.; Fisher, S.A.; Mikryukov, A.; Shoichet, M.S.; Li, R.-K.; Kattman, S.J.; Keller, G. Generation of the epicardial lineage from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1026–1035. [Google Scholar] [CrossRef]
- Iyer, D.; Gambardella, L.; Bernard, W.G.; Serrano, F.; Mascetti, V.L.; Pedersen, R.A.; Talasila, A.; Sinha, S. Robust derivation of epicardium and its differentiated smooth muscle cell progeny from human pluripotent stem cells. Development 2015, 142, 1528–1541. [Google Scholar] [CrossRef]
- Zhao, J.; Cao, H.; Tian, L.; Huo, W.; Zhai, K.; Wang, P.; Ji, G.; Ma, Y. Efficient differentiation of TBX18+/WT1+ epicardial-like cells from human pluripotent stem cells using small molecular compounds. Stem Cells Dev. 2017, 26, 528–540. [Google Scholar] [CrossRef]
- Christoforou, N.; Liau, B.; Chakraborty, S.; Chellapan, M.; Bursac, N.; Leong, K.W. Induced pluripotent stem cell-derived cardiac progenitors differentiate to cardiomyocytes and form biosynthetic tissues. PLoS ONE 2013, 8, e65963. [Google Scholar] [CrossRef]
- Zhang, J.; Tao, R.; Campbell, K.F.; Carvalho, J.L.; Ruiz, E.C.; Kim, G.C.; Schmuck, E.G.; Raval, A.N.; da Rocha, A.M.; Herron, T.J.; et al. Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat. Commun. 2019, 10, 2238. [Google Scholar] [CrossRef]
- Efe, J.A.; Hilcove, S.; Kim, J.; Zhou, H.; Ouyang, K.; Wang, G.; Chen, J.; Ding, S. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat. Cell Biol. 2011, 13, 215–222. [Google Scholar] [CrossRef]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.-D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.-D.; Stone, N.R.; Liu, L.; Spencer, C.I.; Qian, L.; Hayashi, Y.; Delgado-Olguin, P.; Ding, S.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Rep. 2013, 1, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Berry, E.C.; Fu, J.; Ieda, M.; Srivastava, D. Reprogramming of mouse fibroblasts into cardiomyocyte-like cells in vitro. Nat. Protoc. 2013, 8, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Wada, R.; Muraoka, N.; Inagawa, K.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Kaneda, R.; Suzuki, T.; Kamiya, K.; et al. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12667–12672. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cao, N.; Spencer, C.I.; Nie, B.; Ma, T.; Xu, T.; Zhang, Y.; Wang, X.; Srivastava, D.; Ding, S. Small molecules enable cardiac reprogramming of mouse fibroblasts with a single factor, Oct4. Cell Rep. 2014, 6, 951–960. [Google Scholar] [CrossRef]
- Mathison, M.; Gersch, R.P.; Nasser, A.; Lilo, S.; Korman, M.; Fourman, M.; Hackett, N.; Shroyer, K.; Yang, J.; Ma, Y.; et al. In vivo cardiac cellular reprogramming efficacy is enhanced by angiogenic preconditioning of the infarcted myocardium with vascular endothelial growth factor. J Am Heart Assoc 2012, 1, e005652. [Google Scholar] [CrossRef]
- Protze, S.; Khattak, S.; Poulet, C.; Lindemann, D.; Tanaka, E.M.; Ravens, U. A new approach to transcription factor screening for reprogramming of fibroblasts to cardiomyocyte-like cells. J. Mol. Cell. Cardiol. 2012, 53, 323–332. [Google Scholar] [CrossRef]
- Addis, R.C.; Ifkovits, J.L.; Pinto, F.; Kellam, L.D.; Esteso, P.; Rentschler, S.; Christoforou, N.; Epstein, J.A.; Gearhart, J.D. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J. Mol. Cell. Cardiol. 2013, 60, 97–106. [Google Scholar] [CrossRef]
- Christoforou, N.; Chellappan, M.; Adler, A.F.; Kirkton, R.D.; Wu, T.; Addis, R.C.; Bursac, N.; Leong, K.W. Transcription factors MYOCD, SRF, Mesp1 and SMARCD3 enhance the cardio-inducing effect of GATA4, TBX5, and MEF2C during direct cellular reprogramming. PLoS ONE 2013, 8, e63577. [Google Scholar] [CrossRef]
- Hirai, H.; Katoku-Kikyo, N.; Keirstead, S.A.; Kikyo, N. Accelerated direct reprogramming of fibroblasts into cardiomyocyte-like cells with the MyoD transactivation domain. Cardiovasc. Res. 2013, 100, 105–113. [Google Scholar] [CrossRef]
- Ifkovits, J.L.; Addis, R.C.; Epstein, J.A.; Gearhart, J.D. Inhibition of TGFβ signaling increases direct conversion of fibroblasts to induced cardiomyocytes. PLoS ONE 2014, 9, e89678. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Z.; Yin, C.; Asfour, H.; Chen, O.; Li, Y.; Bursac, N.; Liu, J.; Qian, L. Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ. Res. 2015, 116, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Huang, C.; Xu, X.; Gu, H.; Ye, Y.; Jiang, C.; Qiu, Z.; Xie, X. Direct reprogramming of mouse fibroblasts into cardiomyocytes with chemical cocktails. Cell Res. 2015, 25, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Islas, J.F.; Liu, Y.; Weng, K.-C.; Robertson, M.J.; Zhang, S.; Prejusa, A.; Harger, J.; Tikhomirova, D.; Chopra, M.; Iyer, D.; et al. Transcription factors ETS2 and MESP1 transdifferentiate human dermal fibroblasts into cardiac progenitors. Proc. Natl. Acad. Sci. USA 2012, 109, 13016–13021. [Google Scholar] [CrossRef]
- Xu, J.-Y.; Lee, Y.-K.; Ran, X.; Liao, S.-Y.; Yang, J.; Au, K.-W.; Lai, W.-H.; Esteban, M.A.; Tse, H.-F. Generation of induced cardiospheres via reprogramming of skin fibroblasts for myocardial regeneration: Induced cardiospheres for myocardial regeneration. Stem Cells 2016, 34, 2693–2706. [Google Scholar] [CrossRef]
- Lian, W.; Jia, Y.; Li, L.; Huang, Z.; Xu, J. Generation of induced cardiospheres via reprogramming of mouse skin fibroblasts. Curr. Protoc. Stem Cell Biol. 2018, 46, e59. [Google Scholar] [CrossRef]
- Song, K.; Nam, Y.-J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef]
- Sadahiro, T.; Yamanaka, S.; Ieda, M. Direct cardiac reprogramming: Progress and challenges in basic biology and clinical applications. Circ. Res. 2015, 116, 1378–1391. [Google Scholar] [CrossRef]
- Srivastava, D.; DeWitt, N. In vivo cellular reprogramming: The next generation. Cell 2016, 166, 1386–1396. [Google Scholar] [CrossRef]
- Nam, Y.-J.; Song, K.; Luo, X.; Daniel, E.; Lambeth, K.; West, K.; Hill, J.A.; DiMaio, J.M.; Baker, L.A.; Bassel-Duby, R.; et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc. Natl. Acad. Sci. USA 2013, 110, 5588–5593. [Google Scholar] [CrossRef]
- Zhang, R.; Han, P.; Yang, H.; Ouyang, K.; Lee, D.; Lin, Y.-F.; Ocorr, K.; Kang, G.; Chen, J.; Stainier, D.Y.R.; et al. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature 2013, 498, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, T.-S.; Lee, S.-T.; Wawrowsky, K.A.; Cheng, K.; Galang, G.; Malliaras, K.; Abraham, M.R.; Wang, C.; Marbán, E. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS ONE 2010, 5, e12559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhong, J.F.; Qiu, H.; Robb MacLellan, W.; Marbán, E.; Wang, C. Epigenomic reprogramming of adult cardiomyocyte-derived cardiac progenitor cells. Sci. Rep. 2015, 5, 17686. [Google Scholar] [CrossRef]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-Mediated In Vitro and In Vivo Direct Reprogramming of Cardiac Fibroblasts to Cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wang, L.; Yin, C.; Liu, J.; Qian, L. In vivo cardiac reprogramming using an optimal single polycistronic construct: Figure 1. Cardiovasc. Res. 2015, 108, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Birket, M.J.; Ribeiro, M.C.; Verkerk, A.O.; Ward, D.; Leitoguinho, A.R.; den Hartogh, S.C.; Orlova, V.V.; Devalla, H.D.; Schwach, V.; Bellin, M.; et al. Expansion and patterning of cardiovascular progenitors derived from human pluripotent stem cells. Nat. Biotechnol. 2015, 33, 970–979. [Google Scholar] [CrossRef]
- Nsair, A.; Schenke-Layland, K.; Van Handel, B.; Evseenko, D.; Kahn, M.; Zhao, P.; Mendelis, J.; Heydarkhan, S.; Awaji, O.; Vottler, M.; et al. Characterization and therapeutic potential of induced pluripotent stem cell-derived cardiovascular progenitor cells. PLoS ONE 2012, 7, e45603. [Google Scholar] [CrossRef]
- Nelson, T.J.; Faustino, R.S.; Chiriac, A.; Crespo-Diaz, R.; Behfar, A.; Terzic, A. CXCR4+/FLK-1+ biomarkers select a cardiopoietic lineage from embryonic stem cells. Stem Cells 2008, 26, 1464–1473. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, J.; Guo, C.; Chang, W.; Zhuang, J.; Zhu, P.; Li, X. Temporally distinct Six2 -positive second heart field progenitors regulate mammalian heart development and disease. Cell Rep. 2017, 18, 1019–1032. [Google Scholar] [CrossRef]
- Torán, J.L.; López, J.A.; Gomes-Alves, P.; Aguilar, S.; Torroja, C.; Trevisan-Herraz, M.; Moscoso, I.; Sebastião, M.J.; Serra, M.; Brito, C.; et al. Definition of a cell surface signature for human cardiac progenitor cells after comprehensive comparative transcriptomic and proteomic characterization. Sci. Rep. 2019, 9, 4647. [Google Scholar] [CrossRef]
- Ardehali, R.; Ali, S.R.; Inlay, M.A.; Abilez, O.J.; Chen, M.Q.; Blauwkamp, T.A.; Yazawa, M.; Gong, Y.; Nusse, R.; Drukker, M.; et al. Prospective isolation of human embryonic stem cell-derived cardiovascular progenitors that integrate into human fetal heart tissue. Proc. Natl. Acad. Sci. USA 2013, 110, 3405–3410. [Google Scholar] [CrossRef] [PubMed]
- Skelton, R.J.P.; Costa, M.; Anderson, D.J.; Bruveris, F.; Finnin, B.W.; Koutsis, K.; Arasaratnam, D.; White, A.J.; Rafii, A.; Ng, E.S.; et al. SIRPA, VCAM1 and CD34 identify discrete lineages during early human cardiovascular development. Stem Cell Res. 2014, 13, 172–179. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qyang, Y.; Martin-Puig, S.; Chiravuri, M.; Chen, S.; Xu, H.; Bu, L.; Jiang, X.; Lin, L.; Granger, A.; Moretti, A.; et al. The renewal and differentiation of Isl1+ cardiovascular progenitors are controlled by a Wnt/β-catenin pathway. Cell Stem Cell 2007, 1, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.D.; Wang, Z.; Lepore, J.J.; Lu, M.M.; Taketo, M.M.; Epstein, D.J.; Morrisey, E.E. Wnt/β-catenin signaling promotes expansion of Isl-1–positive cardiac progenitor cells through regulation of FGF signaling. J. Clin. Investig. 2007, 117, 1794–1804. [Google Scholar] [CrossRef]
- Kwon, C.; Qian, L.; Cheng, P.; Nigam, V.; Arnold, J.; Srivastava, D. A regulatory pathway involving Notch1/β-catenin/Isl1 determines cardiac progenitor cell fate. Nat. Cell Biol. 2009, 11, 951–957. [Google Scholar] [CrossRef]
- Rosenblatt-Velin, N.; Lepore, M.G.; Cartoni, C.; Beermann, F.; Pedrazzini, T. FGF-2 controls the differentiation of resident cardiac precursors into functional cardiomyocytes. J. Clin. Investig. 2005, 115, 1724–1733. [Google Scholar] [CrossRef]
- Bylund, J.B.; Trinh, L.T.; Awgulewitsch, C.P.; Paik, D.T.; Jetter, C.; Jha, R.; Zhang, J.; Nolan, K.; Xu, C.; Thompson, T.B.; et al. Coordinated proliferation and differentiation of human-induced pluripotent stem cell-derived cardiac progenitor cells depend on bone morphogenetic protein signaling regulation by GREMLIN 2. Stem Cells Dev. 2017, 26, 678–693. [Google Scholar] [CrossRef]
- Ao, A.; Hao, J.; Hopkins, C.R.; Hong, C.C. DMH1, a novel BMP small molecule inhibitor, increases cardiomyocyte progenitors and promotes cardiac differentiation in mouse embryonic stem cells. PLoS ONE 2012, 7, e41627. [Google Scholar] [CrossRef]
- Gomes-Alves, P.; Serra, M.; Brito, C.; Ricardo, C.P.; Cunha, R.; Sousa, M.F.; Sanchez, B.; Bernad, A.; Carrondo, M.J.T.; Rodriguez-Borlado, L.; et al. In vitro expansion of human cardiac progenitor cells: Exploring ’omics tools for characterization of cell-based allogeneic products. Transl. Res. 2016, 171, 96–110.e3. [Google Scholar] [CrossRef]
- Dyer, L.A.; Makadia, F.A.; Scott, A.; Pegram, K.; Hutson, M.R.; Kirby, M.L. BMP signaling modulates hedgehog-induced secondary heart field proliferation. Dev. Biol. 2010, 348, 167–176. [Google Scholar] [CrossRef]
- Gude, N.; Muraski, J.; Rubio, M.; Kajstura, J.; Schaefer, E.; Anversa, P.; Sussman, M.A. Akt promotes increased cardiomyocyte cycling and expansion of the cardiac progenitor cell population. Circ. Res. 2006, 99, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-S.; Cheng, K.; Malliaras, K.; Matsushita, N.; Sun, B.; Marbán, L.; Zhang, Y.; Marbán, E. Expansion of human cardiac stem cells in physiological oxygen improves cell production efficiency and potency for myocardial repair. Cardiovasc. Res. 2011, 89, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, S.; Khan, M.; Toko, H.; Bailey, B.; Cottage, C.T.; Wallach, K.; Nag, D.; Lee, A.; Siddiqi, S.; Lan, F.; et al. Human cardiac progenitor cells engineered with Pim-I kinase enhance myocardial repair. J. Am. Coll. Cardiol. 2012, 60, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, A.; Yuan, L.; Bréant, C.; Alitalo, K.; Koskinen, P.J. Developmental expression of Pim kinases suggests functions also outside of the hematopoietic system. Oncogene 2000, 19, 1215–1224. [Google Scholar] [CrossRef]
- Mohsin, S.; Khan, M.; Nguyen, J.; Alkatib, M.; Siddiqi, S.; Hariharan, N.; Wallach, K.; Monsanto, M.; Gude, N.; Dembitsky, W.; et al. Rejuvenation of human cardiac progenitor cells with Pim-1 kinase. Circ. Res. 2013, 113, 1169–1179. [Google Scholar] [CrossRef]
- Samse, K.; Emathinger, J.; Hariharan, N.; Quijada, P.; Ilves, K.; Völkers, M.; Ormachea, L.; De La Torre, A.; Orogo, A.M.; Alvarez, R.; et al. Functional effect of Pim1 depends upon intracellular localization in human cardiac progenitor cells. J. Biol. Chem. 2015, 290, 13935–13947. [Google Scholar] [CrossRef]
- Fischer, K.M.; Cottage, C.T.; Wu, W.; Din, S.; Gude, N.A.; Avitabile, D.; Quijada, P.; Collins, B.L.; Fransioli, J.; Sussman, M.A. Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing Pim-1 kinase. Circulation 2009, 120, 2077–2087. [Google Scholar] [CrossRef]
- Liu, N.; Wang, B.J.; Broughton, K.M.; Alvarez, R.; Siddiqi, S.; Loaiza, R.; Nguyen, N.; Quijada, P.; Gude, N.; Sussman, M.A. PIM1-minicircle as a therapeutic treatment for myocardial infarction. PLoS ONE 2017, 12, e0173963. [Google Scholar] [CrossRef]
- Hofsteen, P.; Robitaille, A.M.; Chapman, D.P.; Moon, R.T.; Murry, C.E. Quantitative proteomics identify DAB2 as a cardiac developmental regulator that inhibits WNT/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 1002–1007. [Google Scholar] [CrossRef]
- Hofsteen, P.; Robitaille, A.M.; Strash, N.; Palpant, N.; Moon, R.T.; Pabon, L.; Murry, C.E. ALPK2 promotes cardiogenesis in zebrafish and human pluripotent stem cells. iScience 2018, 2, 88–100. [Google Scholar] [CrossRef]
- Dupays, L.; Towers, N.; Wood, S.; David, A.; Stuckey, D.J.; Mohun, T. Furin, a transcriptional target of NKX2-5, has an essential role in heart development and function. PLoS ONE 2019, 14, e0212992. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, L.; Vaseghi, H.R.; Liu, Z.; Lu, R.; Alimohamadi, S.; Yin, C.; Fu, J.-D.; Wang, G.G.; Liu, J.; et al. Bmi1 is a key epigenetic barrier to direct cardiac reprogramming. Cell Stem Cell 2016, 18, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Lei, I.; Liu, L.; Sham, M.H.; Wang, Z. SWI/SNF in cardiac progenitor cell differentiation: SWI/SNF in Cardiac Progenitors. J. Cell. Biochem. 2013, 114, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fulcoli, F.G.; Ferrentino, R.; Martucciello, S.; Illingworth, E.A.; Baldini, A. Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 2012, 8, e1002571. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.D.; Miller, M.F.; Wang, Z.; Moon, R.T.; Morrisey, E.E. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development 2012, 139, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, N.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Isomi, M.; Nakashima, H.; Akiyama, M.; Wada, R.; Inagawa, K.; et al. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J. 2014, 33, 1565–1581. [Google Scholar] [CrossRef]
- Sluijter, J.P.G.; van Mil, A.; van Vliet, P.; Metz, C.H.G.; Liu, J.; Doevendans, P.A.; Goumans, M.-J. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 859–868. [Google Scholar] [CrossRef]
- Xiao, J.; Liang, D.; Zhang, H.; Liu, Y.; Zhang, D.; Liu, Y.; Pan, L.; Chen, X.; Doevendans, P.A.; Sun, Y.; et al. MicroRNA-204 is required for differentiation of human-derived cardiomyocyte progenitor cells. J. Mol. Cell. Cardiol. 2012, 53, 751–759. [Google Scholar] [CrossRef]
- Sirish, P.; López, J.E.; Li, N.; Wong, A.; Timofeyev, V.; Young, J.N.; Majdi, M.; Li, R.A.; Chen, H.V.; Chiamvimonvat, N. MicroRNA profiling predicts a variance in the proliferative potential of cardiac progenitor cells derived from neonatal and adult murine hearts. J. Mol. Cell. Cardiol. 2012, 52, 264–272. [Google Scholar] [CrossRef]
- Shen, X.; Soibam, B.; Benham, A.; Xu, X.; Chopra, M.; Peng, X.; Yu, W.; Bao, W.; Liang, R.; Azares, A.; et al. miR-322/-503 cluster is expressed in the earliest cardiac progenitor cells and drives cardiomyocyte specification. Proc. Natl. Acad. Sci. USA 2016, 113, 9551–9556. [Google Scholar] [CrossRef]
- Garate, X.; La Greca, A.; Neiman, G.; Blüguermann, C.; Santín Velazque, N.L.; Moro, L.N.; Luzzani, C.; Scassa, M.E.; Sevlever, G.E.; Romorini, L.; et al. Identification of the miRNAome of early mesoderm progenitor cells and cardiomyocytes derived from human pluripotent stem cells. Sci. Rep. 2018, 8, 8072. [Google Scholar] [CrossRef] [PubMed]
- Evseenko, D.; Zhu, Y.; Schenke-Layland, K.; Kuo, J.; Latour, B.; Ge, S.; Scholes, J.; Dravid, G.; Li, X.; MacLellan, W.R.; et al. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13742–13747. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Yang, J.; Zhao, X.; Zhang, E.; Zeng, Q.; Yu, Y.; Yang, L.; Wu, B.; Yi, G.; Mao, X.; et al. Circulating myocardial microRNAs from infarcted hearts are carried in exosomes and mobilise bone marrow progenitor cells. Nat. Commun. 2019, 10, 959. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Meng, X.; Zhang, L. microRNAs and cardiac stem cells in heart development and disease. Drug Discov. Today 2019, 24, 233–240. [Google Scholar] [CrossRef]
- Castellan, R.F.P.; Meloni, M. Mechanisms and therapeutic targets of cardiac regeneration: Closing the age gap. Front. Cardiovasc. Med. 2018, 5, 7. [Google Scholar] [CrossRef]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.-L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Cui, J.; Sun, M.; Du, W.; Chen, T.; Ming, X.; Zhang, L.; Tian, J.; Li, J.; et al. MiR218 modulates wnt signaling in mouse cardiac stem cells by promoting proliferation and inhibiting differentiation through a positive feedback loop. Sci. Rep. 2016, 6, 20968. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Chen, F.; Cao, N.; Zhou, Z.-W.; Yang, H.-T. miR-142-3p contributes to early cardiac fate decision of embryonic stem cells. Stem Cells Int. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Ivey, K.N.; Muth, A.; Arnold, J.; King, F.W.; Yeh, R.-F.; Fish, J.E.; Hsiao, E.C.; Schwartz, R.J.; Conklin, B.R.; Bernstein, H.S.; et al. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell 2008, 2, 219–229. [Google Scholar] [CrossRef]
- Purvis, N.; Bahn, A.; Katare, R. The role of microRNAs in cardiac stem cells. Stem Cells Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Deng, S.; Zhao, Q.; Zhou, X.; Zhang, L.; Bao, L.; Zhen, L.; Zhang, Y.; Fan, H.; Liu, Z.; Yu, Z. Neonatal heart-enriched miR-708 promotes differentiation of cardiac progenitor cells in rats. Int. J. Mol. Sci. 2016, 17, 875. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, Q.; He, S.; Yang, M.; Maguire, E.M.; An, W.; Afzal, T.A.; Luong, L.A.; Zhang, L.; Xiao, Q. miR-22 is a novel mediator of vascular smooth muscle cell phenotypic modulation and neointima formation. Circulation 2018, 137, 1824–1841. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Wu, Y.; Wang, Y.; Yu, D.; Yang, M.; Yang, F.; Feng, C.; Chen, T. MicroRNA-29a promotes smooth muscle cell differentiation from stem cells by targeting YY1. Stem Cell Res. 2016, 17, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Crippa, S.; Cassano, M.; Messina, G.; Galli, D.; Galvez, B.G.; Curk, T.; Altomare, C.; Ronzoni, F.; Toelen, J.; Gijsbers, R.; et al. miR669a and miR669q prevent skeletal muscle differentiation in postnatal cardiac progenitors. J. Cell Biol. 2011, 193, 1197–1212. [Google Scholar] [CrossRef] [PubMed]
- Limana, F.; Esposito, G.; D’Arcangelo, D.; Di Carlo, A.; Romani, S.; Melillo, G.; Mangoni, A.; Bertolami, C.; Pompilio, G.; Germani, A.; et al. HMGB1 attenuates cardiac remodelling in the failing heart via enhanced cardiac regeneration and miR-206-mediated inhibition of TIMP-3. PLoS ONE 2011, 6, e19845. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sun, Q.; Zhang, Y.; Teng, F.; Sun, J. Up-regulation of miRNA-21 expression promotes migration and proliferation of Sca-1+ cardiac stem cells in mice. Med. Sci. Monit. 2016, 22, 1724–1732. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, S.; Huang, M.; Nguyen, P.K.; Gong, Y.; Li, Z.; Jia, F.; Lan, F.; Liu, J.; Nag, D.; Robbins, R.C.; et al. Novel microRNA prosurvival cocktail for improving engraftment and function of cardiac progenitor cell transplantation. Circulation 2011, 124, S27–S34. [Google Scholar] [CrossRef]
- Liu, J.; van Mil, A.; Vrijsen, K.; Zhao, J.; Gao, L.; Metz, C.H.G.; Goumans, M.-J.; Doevendans, P.A.; Sluijter, J.P.G. MicroRNA-155 prevents necrotic cell death in human cardiomyocyte progenitor cells via targeting RIP1. J. Cell. Mol. Med. 2011, 15, 1474–1482. [Google Scholar] [CrossRef]
- Urbich, C.; Kuehbacher, A.; Dimmeler, S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008, 79, 581–588. [Google Scholar] [CrossRef]
- Li, Y.; Yang, C.-M.; Xi, Y.; Wu, G.; Shelat, H.; Gao, S.; Cheng, J.; Geng, Y.-J. MicroRNA-1/133 targeted dysfunction of potassium channels KCNE1 and KCNQ1 in human cardiac progenitor cells with simulated hyperglycemia. Int. J. Cardiol. 2013, 167, 1076–1078. [Google Scholar] [CrossRef]
- Mauretti, A.; Spaans, S.; Bax, N.A.M.; Sahlgren, C.; Bouten, C.V.C. Cardiac progenitor cells and the interplay with their microenvironment. Stem Cells Int. 2017, 2017, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, R.; Rizzitelli, G.; Chimenti, I.; Barile, L.; Forte, E.; Ionta, V.; Angelini, F.; Sluijter, J.P.G.; Barbetta, A.; Messina, E.; et al. Cardiospheres and tissue engineering for myocardial regeneration: Potential for clinical application. J. Cell. Mol. Med. 2010, 14, 1071–1077. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vunjak-Novakovic, G.; Tandon, N.; Godier, A.; Maidhof, R.; Marsano, A.; Martens, T.P.; Radisic, M. Challenges in cardiac tissue engineering. Tissue Eng. Part. B: Rev. 2010, 16, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Hwang, N.S.; Varghese, S.; Elisseeff, J. Controlled differentiation of stem cells. Adv. Drug Deliv. Rev. 2008, 60, 199–214. [Google Scholar] [CrossRef]
- Mendelson, K.; Schoen, F.J. Heart valve tissue engineering: Concepts, approaches, progress, and challenges. Ann. Biomed. Eng. 2006, 34, 1799–1819. [Google Scholar] [CrossRef]
- Dawson, E.; Mapili, G.; Erickson, K.; Taqvi, S.; Roy, K. Biomaterials for stem cell differentiation. Adv. Drug Deliv. Rev. 2008, 60, 215–228. [Google Scholar] [CrossRef]
- Bellamy, V.; Vanneaux, V.; Bel, A.; Nemetalla, H.; Emmanuelle Boitard, S.; Farouz, Y.; Joanne, P.; Perier, M.-C.; Robidel, E.; Mandet, C.; et al. Long-term functional benefits of human embryonic stem cell-derived cardiac progenitors embedded into a fibrin scaffold. J. Heart Lung Transplant. 2015, 34, 1198–1207. [Google Scholar] [CrossRef]
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Cacciapuoti, I.; Parouchev, A.; Benhamouda, N.; Tachdjian, G.; Tosca, L.; et al. Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: First clinical case report: Figure 1. Eur. Heart J. 2015, 36, 2011–2017. [Google Scholar] [CrossRef]
- Vallée, J.-P.; Hauwel, M.; Lepetit-Coiffé, M.; Bei, W.; Montet-Abou, K.; Meda, P.; Gardier, S.; Zammaretti, P.; Kraehenbuehl, T.P.; Herrmann, F.; et al. Embryonic stem cell-based cardiopatches improve cardiac function in infarcted rats. Stem Cells Transl. Med. 2012, 1, 248–260. [Google Scholar] [CrossRef]
- Gaetani, R.; Doevendans, P.A.; Metz, C.H.G.; Alblas, J.; Messina, E.; Giacomello, A.; Sluijter, J.P.G. Cardiac tissue engineering using tissue printing technology and human cardiac progenitor cells. Biomaterials 2012, 33, 1782–1790. [Google Scholar] [CrossRef]
- Gaetani, R.; Feyen, D.A.M.; Verhage, V.; Slaats, R.; Messina, E.; Christman, K.L.; Giacomello, A.; Doevendans, P.A.F.M.; Sluijter, J.P.G. Epicardial application of cardiac progenitor cells in a 3D-printed gelatin/hyaluronic acid patch preserves cardiac function after myocardial infarction. Biomaterials 2015, 61, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.-Y.; Lin, B.; Kim, J.; Sullivan, M.; Tobita, K.; Salama, G.; Yang, L. Repopulation of decellularized mouse heart with human induced pluripotent stem cell-derived cardiovascular progenitor cells. Nat. Commun. 2013, 4, 2307. [Google Scholar] [CrossRef] [PubMed]
- Huby, A.-C.; Beigi, F.; Xiang, Q.; Gobin, A.; Taylor, D. Porcine decellularized heart tissue enhance the expression of contractile proteins in human cardiomyocytes and differentiated cardiac progenitor cells. Circ. Res. 2016, 119, A29. [Google Scholar]
- Padin-Iruegas, M.E.; Misao, Y.; Davis, M.E.; Segers, V.F.M.; Esposito, G.; Tokunou, T.; Urbanek, K.; Hosoda, T.; Rota, M.; Anversa, P.; et al. Cardiac progenitor cells and biotinylated insulin-like growth factor-1 nanofibers improve endogenous and exogenous myocardial regeneration after infarction. Circulation 2009, 120, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, M.; Liu, M.-L.; Nagai, T.; Iwanaga, K.; Matsuura, K.; Takahashi, T.; Kanda, M.; Kondo, N.; Wang, P.; Naito, A.T.; et al. Implantation of cardiac progenitor cells using self-assembling peptide improves cardiac function after myocardial infarction. J. Mol. Cell. Cardiol. 2010, 49, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, X.; Matsushita, S.; Guan, J. Differentiation of cardiosphere-derived cells into a mature cardiac lineage using biodegradable poly(N-isopropylacrylamide) hydrogels. Biomaterials 2011, 32, 3220–3232. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tian, S.; Zhao, C.; Chen, X.; Lei, I.; Wang, Z.; Ma, P.X. Porous nanofibrous poly(l-lactic acid) scaffolds supporting cardiovascular progenitor cells for cardiac tissue engineering. Acta Biomater. 2015, 26, 105–114. [Google Scholar] [CrossRef]
- Ciocci, M.; Mochi, F.; Carotenuto, F.; Di Giovanni, E.; Prosposito, P.; Francini, R.; De Matteis, F.; Reshetov, I.; Casalboni, M.; Melino, S.; et al. Scaffold-in-scaffold potential to induce growth and differentiation of cardiac progenitor cells. Stem Cells Dev. 2017, 26, 1438–1447. [Google Scholar] [CrossRef]
- Johnson, T.D.; DeQuach, J.A.; Gaetani, R.; Ungerleider, J.; Elhag, D.; Nigam, V.; Behfar, A.; Christman, K.L. Human versus porcine tissue sourcing for an injectable myocardial matrix hydrogel. Biomater. Sci. 2014, 2, 735–744. [Google Scholar] [CrossRef]
- van Marion, M.H.; Bax, N.A.M.; van Turnhout, M.C.; Mauretti, A.; van der Schaft, D.W.J.; Goumans, M.J.T.H.; Bouten, C.V.C. Behavior of CMPCs in unidirectional constrained and stress-free 3D hydrogels. J. Mol. Cell. Cardiol. 2015, 87, 79–91. [Google Scholar] [CrossRef][Green Version]
- Gaetani, R.; Yin, C.; Srikumar, N.; Braden, R.; Doevendans, P.A.; Sluijter, J.P.G.; Christman, K.L. Cardiac-derived extracellular matrix enhances cardiogenic properties of human cardiac progenitor cells. Cell Transplant. 2016, 25, 1653–1663. [Google Scholar] [CrossRef] [PubMed]
- French, K.M.; Boopathy, A.V.; DeQuach, J.A.; Chingozha, L.; Lu, H.; Christman, K.L.; Davis, M.E. A naturally derived cardiac extracellular matrix enhances cardiac progenitor cell behavior in vitro. Acta Biomater. 2012, 8, 4357–4364. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.L.J.; Narayanan, K.; Gao, S.; Wan, A.C.A. Lineage restricted progenitors for the repopulation of decellularized heart. Biomaterials 2011, 32, 7571–7580. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, S.; Pahlavan, S.; Ashtiani, M.K.; Ansari, H.; Abbasalizadeh, S.; Sayahpour, F.A.; Varzideh, F.; Kostin, S.; Aghdami, N.; Braun, T.; et al. Human embryonic stem cell-derived cardiovascular progenitor cells efficiently colonize in bFGF-tethered natural matrix to construct contracting humanized rat hearts. Biomaterials 2018, 154, 99–112. [Google Scholar] [CrossRef]
- Sánchez, P.L.; Fernández-Santos, M.E.; Costanza, S.; Climent, A.M.; Moscoso, I.; Gonzalez-Nicolas, M.A.; Sanz-Ruiz, R.; Rodríguez, H.; Kren, S.M.; Garrido, G.; et al. Acellular human heart matrix: A critical step toward whole heart grafts. Biomaterials 2015, 61, 279–289. [Google Scholar] [CrossRef]
- Bejleri, D.; Streeter, B.W.; Nachlas, A.L.Y.; Brown, M.E.; Gaetani, R.; Christman, K.L.; Davis, M.E. A bioprinted cardiac patch composed of cardiac-specific extracellular matrix and progenitor cells for heart repair. Adv. Healthc. Mater. 2018, 7, 1800672. [Google Scholar] [CrossRef]
- Silva, A.C.; Rodrigues, S.C.; Caldeira, J.; Nunes, A.M.; Sampaio-Pinto, V.; Resende, T.P.; Oliveira, M.J.; Barbosa, M.A.; Thorsteinsdóttir, S.; Nascimento, D.S.; et al. Three-dimensional scaffolds of fetal decellularized hearts exhibit enhanced potential to support cardiac cells in comparison to the adult. Biomaterials 2016, 104, 52–64. [Google Scholar] [CrossRef]
- Chamberland, C.; Martinez-Fernandez, A.; Beraldi, R.; Nelson, T.J. Embryonic decellularized cardiac scaffold supports embryonic stem cell differentiation to produce beating cardiac tissue. ISRN Stem Cells 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Rajabi-Zeleti, S.; Jalili-Firoozinezhad, S.; Azarnia, M.; Khayyatan, F.; Vahdat, S.; Nikeghbalian, S.; Khademhosseini, A.; Baharvand, H.; Aghdami, N. The behavior of cardiac progenitor cells on macroporous pericardium-derived scaffolds. Biomaterials 2014, 35, 970–982. [Google Scholar] [CrossRef]
- Chimenti, I.; Rizzitelli, G.; Gaetani, R.; Angelini, F.; Ionta, V.; Forte, E.; Frati, G.; Schussler, O.; Barbetta, A.; Messina, E.; et al. Human cardiosphere-seeded gelatin and collagen scaffolds as cardiogenic engineered bioconstructs. Biomaterials 2011, 32, 9271–9281. [Google Scholar] [CrossRef]
- Takehara, N.; Tsutsumi, Y.; Tateishi, K.; Ogata, T.; Tanaka, H.; Ueyama, T.; Takahashi, T.; Takamatsu, T.; Fukushima, M.; Komeda, M.; et al. Controlled delivery of basic fibroblast growth factor promotes human cardiosphere-derived cell engraftment to enhance cardiac repair for chronic myocardial infarction. J. Am. Coll. Cardiol. 2008, 52, 1858–1865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, X.; Sun, S.; Zhang, X. Implantation of engineered conduction tissue in the rat heart. Mol. Med. Rep. 2019, 19, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, C.; Nan, H.; Yu, Y.; Xiao, Y.; Poon, E.; Yang, G.; Wang, X.; Wang, C.; Li, L.; et al. Graphene sheet-induced global maturation of cardiomyocytes derived from human induced pluripotent stem cells. ACS Appl. Mater. Interfaces 2017, 9, 25929–25940. [Google Scholar] [CrossRef] [PubMed]
- Savchenko, A.; Cherkas, V.; Liu, C.; Braun, G.B.; Kleschevnikov, A.; Miller, Y.I.; Molokanova, E. Graphene biointerfaces for optical stimulation of cells. Sci. Adv. 2018, 4, eaat0351. [Google Scholar] [CrossRef]
- Feiner, R.; Engel, L.; Fleischer, S.; Malki, M.; Gal, I.; Shapira, A.; Shacham-Diamand, Y.; Dvir, T. Engineered hybrid cardiac patches with multifunctional electronics for online monitoring and regulation of tissue function. Nat. Mater. 2016, 15, 679–685. [Google Scholar] [CrossRef]
- Li, J.; Minami, I.; Shiozaki, M.; Yu, L.; Yajima, S.; Miyagawa, S.; Shiba, Y.; Morone, N.; Fukushima, S.; Yoshioka, M.; et al. Human pluripotent stem cell-derived cardiac tissue-like constructs for repairing the infarcted myocardium. Stem Cell Rep. 2017, 9, 1546–1559. [Google Scholar] [CrossRef]
- Nunes, S.S.; Miklas, J.W.; Liu, J.; Aschar-Sobbi, R.; Xiao, Y.; Zhang, B.; Jiang, J.; Massé, S.; Gagliardi, M.; Hsieh, A.; et al. Biowire: A platform for maturation of human pluripotent stem cell–derived cardiomyocytes. Nat. Methods 2013, 10, 781–787. [Google Scholar] [CrossRef]
- Asahi, Y.; Hamada, T.; Hattori, A.; Matsuura, K.; Odaka, M.; Nomura, F.; Kaneko, T.; Abe, Y.; Takasuna, K.; Sanbuissho, A.; et al. On-chip spatiotemporal electrophysiological analysis of human stem cell derived cardiomyocytes enables quantitative assessment of proarrhythmia in drug development. Sci. Rep. 2018, 8, 14536. [Google Scholar] [CrossRef]
- Qian, F.; Huang, C.; Lin, Y.-D.; Ivanovskaya, A.N.; O’Hara, T.J.; Booth, R.H.; Creek, C.J.; Enright, H.A.; Soscia, D.A.; Belle, A.M.; et al. Simultaneous electrical recording of cardiac electrophysiology and contraction on chip. Lab Chip 2017, 17, 1732–1739. [Google Scholar] [CrossRef]
- Banerjee, M.N.; Bolli, R.; Hare, J.M. Clinical studies of cell therapy in cardiovascular medicine: Recent developments and future directions. Circ. Res. 2018, 123, 266–287. [Google Scholar] [CrossRef]
- The Lancet Editors. Expression of concern: The SCIPIO trial. Lancet 2014, 383, 1279. [Google Scholar] [CrossRef]
- The Lancet Editors. Retraction—Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): Initial results of a randomised phase 1 trial. Lancet 2019, 393, 1084. [Google Scholar] [CrossRef]
- Makkar, R.R.; Smith, R.R.; Cheng, K.; Malliaras, K.; Thomson, L.E.; Berman, D.; Czer, L.S.; Marbán, L.; Mendizabal, A.; Johnston, P.V.; et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): A prospective, randomised phase 1 trial. Lancet 2012, 379, 895–904. [Google Scholar] [CrossRef]
- Yacoub, M.H.; Terrovitis, J. CADUCEUS, SCIPIO, ALCADIA: Cell therapy trials using cardiac-derived cells for patients with post myocardial infarction LV dysfunction, still evolving. Glob. Cardiol. Sci. Pract. 2013, 2013, 3. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takehara, N.; Ogata, T.; Nakata, M.; Kami, D.; Nakamura, T.; Matoba, S.; Gojo, S.; Sawada, T.; Yaku, H.; Matsubara, H. The alcadia (autologous human cardiac-derived stem cell to treat ischemic cardiomyopathy) trial. Circulation 2012, 126, 2776–2799. [Google Scholar]
- Malliaras, K.; Zhang, Y.; Seinfeld, J.; Galang, G.; Tseliou, E.; Cheng, K.; Sun, B.; Aminzadeh, M.; Marbán, E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol. Med. 2013, 5, 191–209. [Google Scholar] [CrossRef]
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Parouchev, A.; Cacciapuoti, I.; Al-Daccak, R.; Benhamouda, N.; Blons, H.; et al. Transplantation of human embryonic stem cell–derived cardiovascular progenitors for severe ischemic left ventricular dysfunction. J. Am. Coll. Cardiol. 2018, 71, 429–438. [Google Scholar] [CrossRef]
- Ishigami, S.; Ohtsuki, S.; Tarui, S.; Ousaka, D.; Eitoku, T.; Kondo, M.; Okuyama, M.; Kobayashi, J.; Baba, K.; Arai, S.; et al. Intracoronary autologous cardiac progenitor cell transfer in patients with hypoplastic left heart syndrome: The TICAP prospective phase 1 controlled trial. Circ. Res. 2015, 116, 653–664. [Google Scholar] [CrossRef]
- Ishigami, S.; Ohtsuki, S.; Eitoku, T.; Ousaka, D.; Kondo, M.; Kurita, Y.; Hirai, K.; Fukushima, Y.; Baba, K.; Goto, T.; et al. Intracoronary cardiac progenitor cells in single ventricle physiology: The PERSEUS (cardiac progenitor cell infusion to treat univentricular heart disease) randomized phase 2 trial. Circ. Res. 2017, 120, 1162–1173. [Google Scholar] [CrossRef]
- Tarui, S.; Ishigami, S.; Ousaka, D.; Kasahara, S.; Ohtsuki, S.; Sano, S.; Oh, H. Transcoronary infusion of cardiac progenitor cells in hypoplastic left heart syndrome: Three-year follow-up of the transcoronary infusion of cardiac progenitor cells in patients with single-ventricle physiology (TICAP) trial. J. Thorac. Cardiovasc. Surg. 2015, 150, 1198–1208. [Google Scholar] [CrossRef]
- Cardiac Stem/Progenitor Cell Infusion in Univentricular Physiology (APOLLON Trial). Available online: https://clinicaltrials.gov/ct2/show/NCT02781922 (accessed on 9 October 2019).
- Transcoronary Infusion of Cardiac Progenitor Cells in Pediatric Dilated Cardiomyopathy. Available online: https://clinicaltrials.gov/ct2/show/NCT03129568 (accessed on 9 October 2019).
- Malliaras, K.; Makkar, R.R.; Smith, R.R.; Cheng, K.; Wu, E.; Bonow, R.O.; Marbán, L.; Mendizabal, A.; Cingolani, E.; Johnston, P.V.; et al. Intracoronary cardiosphere-derived cells after myocardial infarction. J. Am. Coll. Cardiol. 2014, 63, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Allogeneic Heart Stem Cells to Achieve Myocardial Regeneration. Available online: https://clinicaltrials.gov/ct2/show/NCT01458405 (accessed on 9 October 2019).
- Sanz-Ruiz, R.; Casado Plasencia, A.; Borlado, L.R.; Fernández-Santos, M.E.; Al-Daccak, R.; Claus, P.; Palacios, I.; Sádaba, R.; Charron, D.; Bogaert, J.; et al. Rationale and design of a clinical trial to evaluate the safety and efficacy of intracoronary infusion of allogeneic human cardiac stem cells in patients with acute myocardial infarction and left ventricular dysfunction: The randomized multicenter double-blind controlled CAREMI trial (cardiac stem cells in patients with acute myocardial infarction). Circ. Res. 2017, 121, 71–80. [Google Scholar] [PubMed]
- Dilated CardiomYopathy iNtervention with Allogeneic MyocardIally-Regenerative Cells (DYNAMIC). Available online: https://clinicaltrials.gov/ct2/show/NCT02293603 (accessed on 9 October 2019).
- Bolli, R.; Hare, J.M.; March, K.L.; Pepine, C.J.; Willerson, J.T.; Perin, E.C.; Yang, P.C.; Henry, T.D.; Traverse, J.H.; Mitrani, R.D.; et al. Rationale and design of the CONCERT-HF trial (combination of mesenchymal and c-kit + cardiac stem cells as regenerative therapy for heart failure). Circ. Res. 2018, 122, 1703–1715. [Google Scholar] [CrossRef] [PubMed]
- Regression of Fibrosis & Reversal of Diastolic Dysfunction in HFPEF Patients Treated with Allogeneic CDCs. Available online: https://clinicaltrials.gov/ct2/show/NCT02941705 (accessed on 9 October 2019).
- Sahara, M.; Santoro, F.; Chien, K.R. Programming and reprogramming a human heart cell. EMBO J. 2015, 34, 710–738. [Google Scholar] [CrossRef]
- Amini, H.; Rezaie, J.; Vosoughi, A.; Rahbarghazi, R.; Nouri, M. Cardiac progenitor cells application in cardiovascular disease. J. Cardiovasc. Thorac. Res. 2017, 9, 127–132. [Google Scholar] [CrossRef]
- Sanchez-Freire, V.; Lee, A.S.; Hu, S.; Abilez, O.J.; Liang, P.; Lan, F.; Huber, B.C.; Ong, S.-G.; Hong, W.X.; Huang, M.; et al. Effect of human donor cell source on differentiation and function of cardiac induced pluripotent stem cells. J. Am. Coll. Cardiol. 2014, 64, 436–448. [Google Scholar] [CrossRef]
- Martens, T.P.; Godier, A.F.G.; Parks, J.J.; Wan, L.Q.; Koeckert, M.S.; Eng, G.M.; Hudson, B.I.; Sherman, W.; Vunjak-Novakovic, G. Percutaneous cell delivery into the heart using hydrogels polymerizing in situ. Cell Transplant. 2009, 18, 297–304. [Google Scholar] [CrossRef]
- Beeres, S.L.M.A.; Atsma, D.E.; van Ramshorst, J.; Schalij, M.J.; Bax, J.J. Cell therapy for ischaemic heart disease. Heart 2008, 94, 1214–1226. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, R.; Huang, X.; Zhang, H.; He, L.; Zhang, L.; Tian, X.; Nie, Y.; Hu, S.; Yan, Y.; et al. Genetic lineage tracing identifies in situ Kit-expressing cardiomyocytes. Cell Res. 2016, 26, 119–130. [Google Scholar] [CrossRef]
- He, L.; Li, Y.; Li, Y.; Pu, W.; Huang, X.; Tian, X.; Wang, Y.; Zhang, H.; Liu, Q.; Zhang, L.; et al. Enhancing the precision of genetic lineage tracing using dual recombinases. Nat. Med. 2017, 23, 1488–1498. [Google Scholar] [CrossRef]
- Li, Y.; He, L.; Huang, X.; Bhaloo, S.I.; Zhao, H.; Zhang, S.; Pu, W.; Tian, X.; Li, Y.; Liu, Q.; et al. Genetic lineage tracing of nonmyocyte population by dual recombinases. Circulation 2018, 138, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Marino, F.; Scalise, M.; Cianflone, E.; Mancuso, T.; Aquila, I.; Agosti, V.; Torella, M.; Paolino, D.; Mollace, V.; Nadal-Ginard, B.; et al. Role of c-Kit in myocardial regeneration and aging. Front. Endocrinol. 2019, 10, 371. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.-L.; Molkentin, J.D. The elusive progenitor cell in cardiac regeneration: Slip slidin’ away. Circ. Res. 2017, 120, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Eschenhagen, T.; Bolli, R.; Braun, T.; Field, L.J.; Fleischmann, B.K.; Frisén, J.; Giacca, M.; Hare, J.M.; Houser, S.; Lee, R.T.; et al. Cardiomyocyte regeneration: A consensus statement. Circulation 2017, 136, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.W.; Witten, C.M.; Califf, R.M. Clarifying stem-cell therapy’s benefits and risks. N. Engl. J. Med. 2017, 376, 1007–1009. [Google Scholar] [CrossRef] [PubMed]
- Maliken, B.D.; Molkentin, J.D. Undeniable evidence that the adult mammalian heart lacks an endogenous regenerative stem cell. Circulation 2018, 138, 806–808. [Google Scholar] [CrossRef]
- Writing Group Members; Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; et al. Heart disease and stroke statistics—2012 Update: A report from the American Heart Association. Circulation 2012, 125, e2–e220. [Google Scholar]
- Cesselli, D.; Beltrami, A.P.; D’Aurizio, F.; Marcon, P.; Bergamin, N.; Toffoletto, B.; Pandolfi, M.; Puppato, E.; Marino, L.; Signore, S.; et al. Effects of age and heart failure on human cardiac stem cell function. Am. J. Pathol. 2011, 179, 349–366. [Google Scholar] [CrossRef]
- Yao, Y.-G.; Ellison, F.M.; McCoy, J.P.; Chen, J.; Young, N.S. Age-dependent accumulation of mtDNA mutations in murine hematopoietic stem cells is modulated by the nuclear genetic background. Hum. Mol. Genet. 2007, 16, 286–294. [Google Scholar] [CrossRef][Green Version]
- Mohsin, S.; Siddiqi, S.; Collins, B.; Sussman, M.A. Empowering adult stem cells for myocardial regeneration. Circ. Res. 2011, 109, 1415–1428. [Google Scholar] [CrossRef]
- Frati, C.; Savi, M.; Graiani, G.; Lagrasta, C.; Cavalli, S.; Prezioso, L.; Rossetti, P.; Mangiaracina, C.; Ferraro, F.; Madeddu, D.; et al. Resident cardiac stem cells. Curr. Pharm. Des. 2011, 17, 3252–3257. [Google Scholar] [PubMed]
- Leonardini, A.; Avogaro, A. Abnormalities of the cardiac stem and progenitor cell compartment in experimental and human diabetes. Arch. Physiol. Biochem. 2013, 119, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Kurazumi, H.; Kubo, M.; Ohshima, M.; Yamamoto, Y.; Takemoto, Y.; Suzuki, R.; Ikenaga, S.; Mikamo, A.; Udo, K.; Hamano, K.; et al. The effects of mechanical stress on the growth, differentiation, and paracrine factor production of cardiac stem cells. PLoS ONE 2011, 6, e28890. [Google Scholar] [CrossRef] [PubMed]
- Torella, D.; Rota, M.; Nurzynska, D.; Musso, E.; Monsen, A.; Shiraishi, I.; Zias, E.; Walsh, K.; Rosenzweig, A.; Sussman, M.A.; et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ. Res. 2004, 94, 514–524. [Google Scholar] [CrossRef]
- Anversa, P.; Rota, M.; Urbanek, K.; Hosoda, T.; Sonnenblick, E.H.; Leri, A.; Kajstura, J.; Bolli, R. Myocardial aging: A stem cell problem. Basic Res. Cardiol. 2005, 100, 482–493. [Google Scholar] [CrossRef]
- Urbanek, K.; Quaini, F.; Tasca, G.; Torella, D.; Castaldo, C.; Nadal-Ginard, B.; Leri, A.; Kajstura, J.; Quaini, E.; Anversa, P. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2003, 100, 10440–10445. [Google Scholar] [CrossRef]



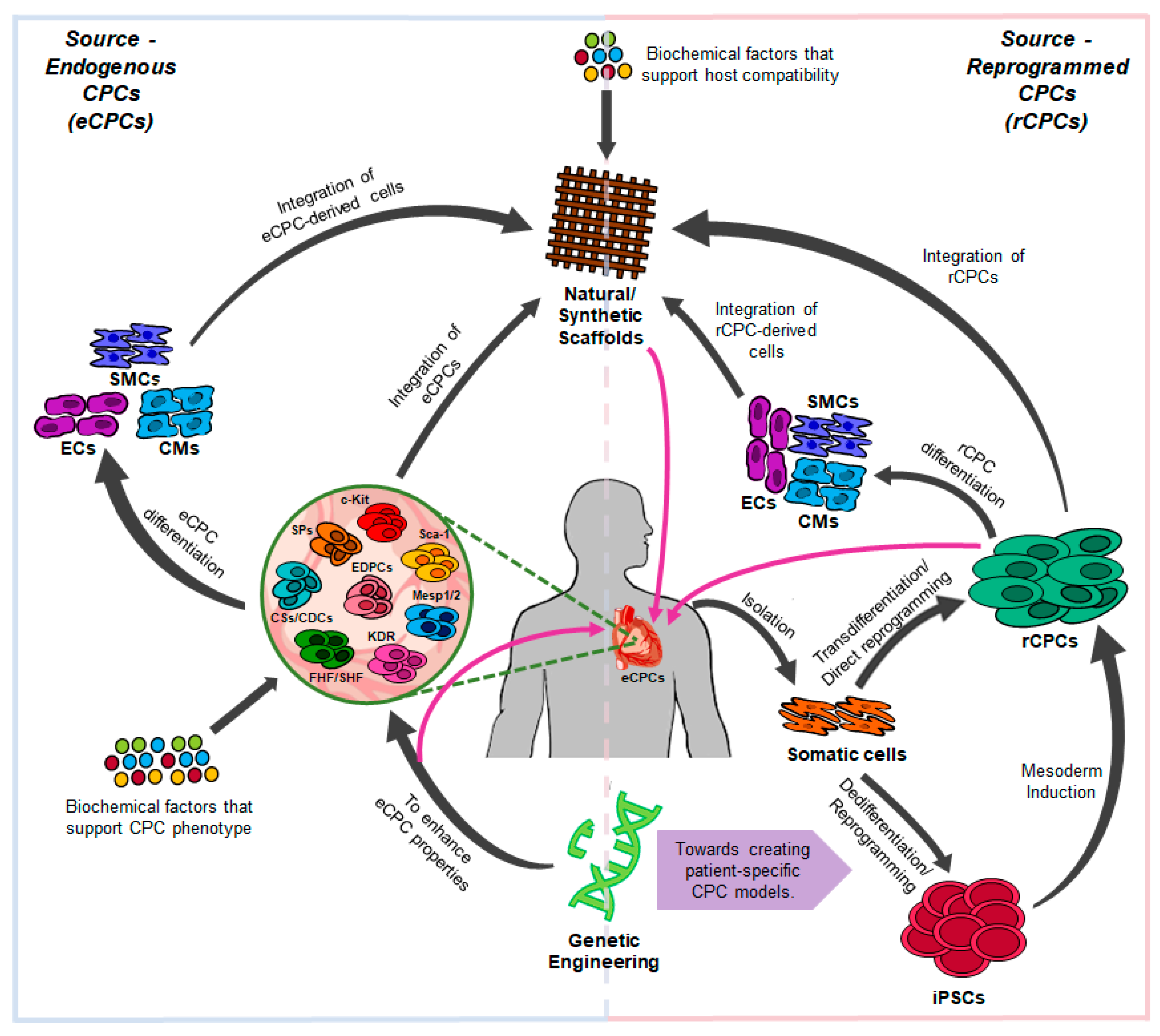{kind=link}
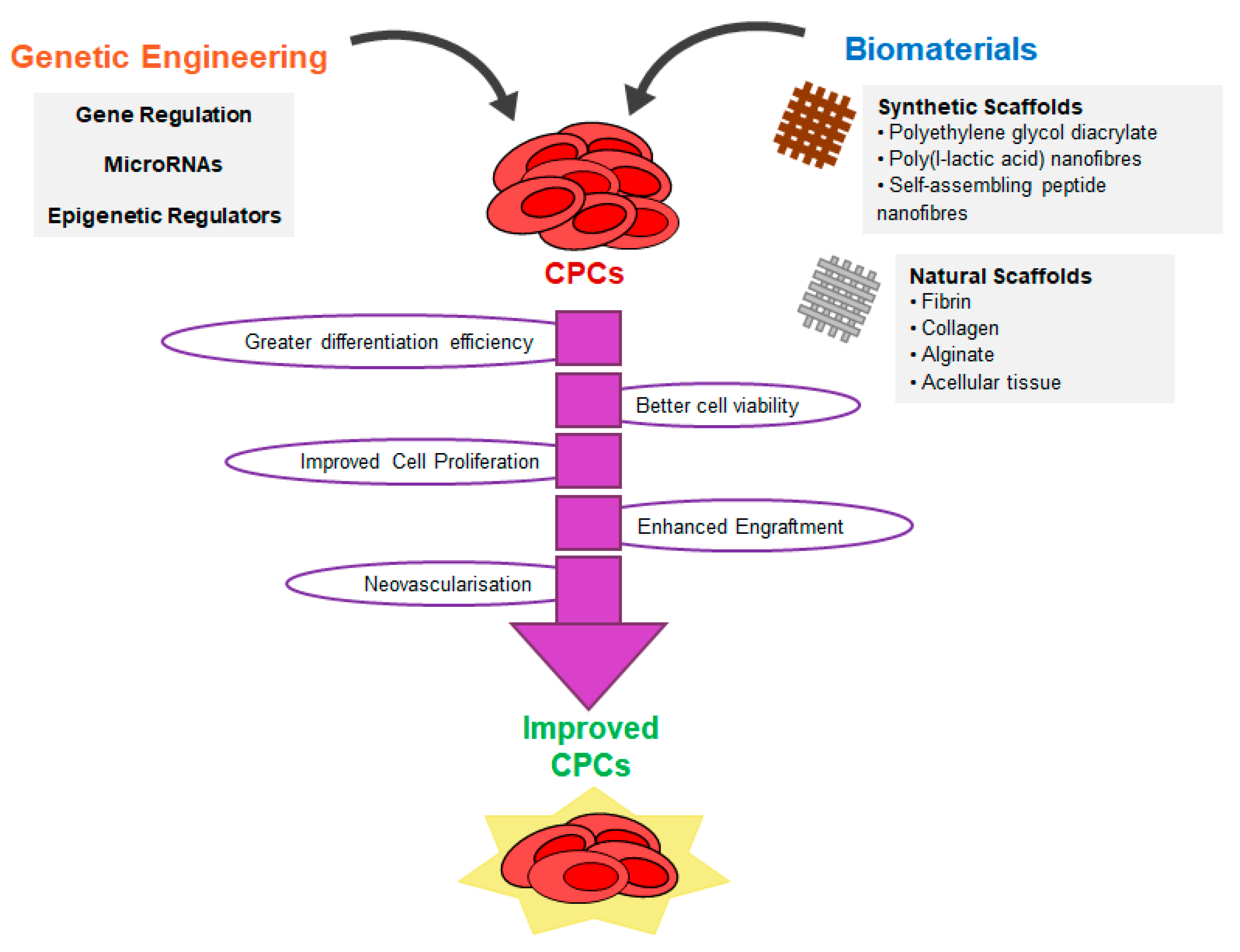{kind=link}
| CPC Type | Marker Expression | Differential Potential | Functionality of the Differentiated Cells | Applied to Disease In Vivo | Concerns | Ref. |
|---|---|---|---|---|---|---|
| c-KIT | Ki67+ NKX2.5+ GATA4/5+ MEF2C+ TBX5+ CD45− CD34− CD31+/− | -Differentiation trend towards CMs *, ** -Few fibroblasts * -ECs * | In vitro: -Atrial and ventricular CMs and cells of the conduction system * -CMs show a disorganized structure, no sarcomeres, and smaller size than their adult counterparts *, ** In vivo: -CMs couple with host cells and display spontaneous beating and striated structures *, ** | -Formation of structural and functional CMs and contribution to coronary vessels in MI rats ** -Reconstitution of a myocardial wall that encompassed up to 70% of LV in MI rats *** | -CPC population is heterogeneous with cells at distinct stage of differentiation and with different commitment to the cardiac lineages *, ** -Differentiated cells show an immature phenotype *, ** -No consensus regarding the regenerative capability of c-KIT CPCs and their lineage marker expression *, ** -Distinct differential potential between neonatal and adult c-KIT+ CPCs and between species *, ** -Benefits are mainly a result of paracrine factors *, ** | [21,27,28,31,53,58,59,107,108] |
| SCA1 | ISL1+ c-KIT+/− PDGFRα+ CD105+ CD90+ CD44+ GATA4+ MEF2C+ NKX2.5+/− TEF-1+ CD31+/− CD34− ABCG2+ | -CMs, SMCs, and ECs *, ** -Foetal SCA1+ CPCs tend to differentiate into ECs, whereas adult CPCs have more efficiency towards CMs ** | In vitro: -CMs display spontaneous beating, myofilaments and expressed connexin 43 *, ** -Immature CMs and SMCs *, ** -ECs form tube-like structures *, ** -Foetal SCA1+ CPCs exhibit more spontaneous beating than adult SCA1+ CPCs ** In vivo: -ECs contribute to capillaries and CMs display defined striated structures * | -Knockdown of SCA1 led to larger LV volume, increased infarct rate and limited angiogenesis in MI mice * -SCA1+/CD31− cell population numbers increased in the LV following MI * -Transplantation of SCA1+/CD31− in MI mice attenuates adverse LV remodeling * | -No human homolog of SCA1 identified ** -SCA1 does not discriminate between proliferating and differentiating cells *, ** -SCA1+ CPCs represent a heterogeneous population with subpopulations displaying different lineage potential *, ** -Distinct potency between neonatal and adult SCA1+ CPCs ** -Differentiation into CMs requires co-culture with adult/neonatal CMs ** -Benefits are mainly a result of paracrine factors *, ** | [11,21,37,64,65,66,67,69,70,109] |
| KDR/FLK1low/− | T+ MESP1+ c-KIT− GATA4+ TBX5+/ NKX2.5+/− CD31+/− SL1+/− SMA+ PDGFRα+ | -Highest efficiency for SMCs, followed by CMs and then ECs *, ** -KDR+/CXCR4+ has better efficiency towards CMs * | In vitro: -CMs display spontaneous Beating *, ** -Predominantly atrial and ventricular CMs ** -Few pacemaker and conduction system cells * -Electrical coupling is observed ** -ECs display LDL-uptake capacity ** -ECs and SMCs form tube-like structures ** In vivo: -Human ESC-derived KDR+ CPCs differentiate into CMs and ECs ** | -Human ESC-derived KDR+ progenitors increased ejection fraction in infarcted hearts of NOD/SCID mice ** | -Hematopoietic tendency *, ** -FLK1/KDR marks two populations with distinct cardiac potential that develop at different temporal stages of mesoderm differentiation * | [20,29,79,82,110] |
| MESP1/2 | SSEA1+ OCT4+ T+ KDR+ ISL1+ TBX5/6/18/20+ GATA4/6+ NKX2.5+ MEF2C+ MYOCD+ PDGFRα/β+ CXCR4+ WNT8A+ FGF8+ HAND2+ | -More efficiency towards SMCs and ECs *, ** -Some CMs *, ** | In vitro: -Formation of ventricular CMs * -CMs express sarcomeric structures when co-cultured with human cardiac fibroblasts and CMs ** In vivo: -CMs display organized myofibrillar striations and express CX43, and SMCs and ECs form tube-like structures and contribute to neovasculogenesis * | -Murine ESC-derived MESP1 CPCs decreased LV-EDV, scar size, and improved LV ejection fraction, stroke volume and cardiac function in MI mice hearts * | -Not fully committed to the cardiac lineages *, ** -Not thoroughly investigated as CPCs *, ** -MESP1 marks a mixed population of CPCs with different multilineage differentiation potential *, ** -MESP1 CPC might be a subset of KDR+/PDGFRα+ cells *, ** -MESP1 is transiently expressed, making it difficult to track the expansion and differentiation of the CPCs *, ** | [72,76,79,80,111] |
| From First Heart Field (FHF) | NKX2.5+ HAND1+ TBX5+ HCN4+ | -More efficiency towards CMs *, ** -Some SMCs *, ** | In vitro: -Atrial, left ventricle and conduction myocytes *, ** -Presence of both mature and immature CMs * -Some spontaneous beating *, ** -Most CMs display a ventricular-like action potential * -Some atrial-like and nodal-like action potentials are formed * In vivo: -ESC-derived CPCs differentiate into SMCs and CMs, which display beating and form myofibrils * | -Not yet applied in vivo in a disease context | -Difficult to identify and characterized due to lack of markers *, ** -FHF have limited potency *, ** -Not thoroughly investigated as CPCs *, ** | [21,84,85,86] |
| From Second Heart Field (SHF) | ISL1+ c-KIT−/+ NKX2.5+/− TBX1+ GATA4+ KDR+/− FGF8/10+ FOXH1+ MEF2C+ WT1+ | -Majority to CMs, including pacemaker *, ** -Some cardiac fibroblasts, SMCs and ECs *, ** -ISL1+/KDR+ into ECs and SMCs * -NKX2.5+/ISL1+ into CMs *, ** -NKX2.5+/KDR+ into SMCs * | In vitro: -Remarkable contribution to the sino-atrial node * -Only a few towards atrial-ventricular node * -CMs exhibit synchronized calcium transients * In vivo: -Contribution to the coronary arterial system * -SMCs are in the most proximal outflow tract * -ESC-derived ISL1+ CPCs differentiate into pacemaker and ventricular CMs, SMCs and ECs * -Knockdown of ISL1 led to a reduction in cardiac tissue formation and affects CPC proliferation, survival and migration * | -Not yet applied in vivo in a disease context | -Majority of contribution to the conduction system is restricted to the sino-atrial node * -EC and SMC contribution is limited to the proximal area of the great vessels * -Embryo-derived SHF show a significant reduction in differentiation into CMs and tripotency was rare * | [22,30,40,83,84,112,113] |
| Epicardial-derived | WT1+ TBX18+ SLUG RALDH2 SCA1+ PDGFRα+ | -Vascular SMCs *, ** -CMs under certain in vitro conditions *, ** -Some cardiac fibroblasts (perivascular and interstitial) *, ** | In vitro: -SMCs and fibroblasts *,** -Atrial and ventricular CMs, with striated cytoarchitecture, spontaneous contraction, native calcium oscillations and electrical coupling * In vivo: -SMCs contribute to the coronary arteries * -Differentiation into fibroblasts, SMCs and coronary endothelial cells; CMs can be formed when subjected to the stimulation of exogenous factors * | -Epicardial-derived CPCs increased vessel formation and stimulate angiogenesis in murine MI models * -Epicardial-derived CPC conditioned medium reduced infarcted size and improved heart function in MI mice models *,** -Priming of the epicardium with Tβ4 prior to injury led to enhanced migration of epicardial-derived CPCs and generation of CMs in MI mice * | -Epicardial-derived CPCs descend from NKX2.5-and ISL1-expressing cells *, ** -No EC differentiation *, ** -Epicardial-derived CPCs are difficult to culture in Vitro *, ** -No consensus about the level of contribution of the epicardium in cardiac repair *,** | [88,89,90,91,114,115,116,117] |
| Side Population (SP) | ABCG2+ SCA1+ CD34+/− CD31+/− c-KIT− NKX2.5+/− GATA4+/− MEF2C+ CD45− VE-cadherin− | -Fibroblasts & SMCs *, ** -SCA1+/CD31− SPs into CMs * -SCA1+/CD31+ SPs + VEGF into ECs * -CD45− SPs into ECs * | In vitro: -CMs show spontaneous beating and striations on staining * -Electrical coupling is observed when SPs are co-cultured with adult CMs * In vivo: -Differentiation into CMs, forming striated sarcomere structures, SMCs, ECs, and fibroblasts *, *** | -Cardiac SP numbers are significantly increased, particularly in the left ventricle, following acute ischemia ** -Myocardial injury facilitated migration and homing of cardiac SPs *, *** | -Hematopoietic differentiation tendency * -Low percentage of CMs reach advanced maturity *, ** -Contradictory results between different studies on the maturity of the SP-derived CMs *, ** -SPs represent an extremely heterogeneous population * -Complete differentiation requires both cell-intrinsic and -extrinsic factors * | [38,94,96,97,100,101,102,104,118] |
| Cardiosphere (CS)-derived cells (CDCs) | KDR+ c-KIT+ SCA1+ CD34+/− CD45− CD133− NKX2.5+ GATA4+ ISL1+ CD105+/CD31+/ CD90+/c-KIT− supporting cells | CMs, SMCs & ECs *, ** | In vitro: -CMs display spontaneous beating, but lack sarcomeric structure * -Differentiation into ECs and SMCs with VEGF treatment *, ** In vivo: -Differentiation into SMCs and ECs, some potential towards CMs lineages *, ** -Formation of tubular-like structures * | -Transplantation of CDCs/CSs improved cell survival, engraftment and LV ejection fraction, stimulated angiogenesis, inhibited adverse LV remodeling and reduced infarct size in the infarcted mice ** | -Human CSs/CDCs require co-culture with adult CMs to stimulate contraction and advance maturity ** -Stemness decreases in monolayer cultures ** -CSs/CDCs represent a mixed cell population *, ** -Benefits result from paracrine factors *, ** -Low CDC engraftment and differentiation efficiency ** -Different markers used, which isolate cells with distinct differentiation potential *, ** | [39,106,119,120] |
| Protocol | CPC-Associated Markers Identified | CPCs as Target or Intermediate | Differentiation and Functionality Potential | Limitations | Ref. | ||
|---|---|---|---|---|---|---|---|
| Pluripotent Culture | Mesoderm Differentiation | Cardiac Specification | |||||
| Mouse iPSCs on feeder-layers and human iPSCs in hESC culture medium without bFGF | Differentiation medium with 20% FBS + gelatin-coated plates + AA between day 2 and 6 | NKX2.5+ TBX5+ & FLK1+ CXCR4+ | Intermediate | -Synchronous beating and better-organized striated myofilaments in CMs | -AA is not able to promote mesodermal differentiation and CM proliferation -No reports on CPC potential into SMCs and ECs | [143] | |
| Human iPSCs in monolayer culture (mTeSR1 + Matrigel-coated plates) | ROCK inhibitor (Y27632) for 1 day and DMEM/F12/B27-vitamin A + BMP4 + AA + CHIR for 3 days | SSEA1+ MESP1/2+ ISL1+ | Target | -Differentiation into the three cardiac lineages under specific differentiation media -80% efficiency towards CMs, and 90% into SMCs and ECs -Synchronized beating and presence of organized sarcomeric structures | -Both early and late CPC-related markers were co-expressed in the generated CPCs -Repeated passaging leads to a decrease in CPC proliferation rate -Only one iPSC line was tested | [144] | |
| Human iPSCs on inactivated MEFs followed by feeder depletion culture in Matrigel | BMP4 for 3 days and +/− Activin A + bFGF from day 1 until day 3 | DKK1 + VEGF + SB +/− Dorsomorphin/Noggin at day 3 | KDR+ PDGFRα+ | Intermediate | -Low yield of CMs (11%) | -iPSC line variability affects protocol’s efficiency and optimal growth factor concentrations -Presence of the CPC population does not always predict efficient differentiation to CMs | [128] |
| Mouse iPSCs in DMEM with 15% FCS on feeder layers | Differentiation medium with 10% FCS + type IV collagen-coated dishes/OP9 cell sheets for 96–108 h | FLK1+ mesodermal cells co-cultured on OP9 cells + differentiation medium + cyclosporin-A | FLK1+ CXCR4+ VE-cadherin− | Target | -Synchronous beating -Pacemaker and ventricular action potentials -Myofilaments formation with transverse Z-bands -Presence of ion channels (Cav3.2, HCN4 and kir2.1) and intercalated disks | -CPCs were only isolated from mouse iPSCs -Differentiation efficiency was different for various iPSC lines -Incomplete human CM maturation | [133] |
| Human iPSCs on SNL feeder cells and Matrigel-coated plates | Co-culture on END-2 cells + cyclosporin-A at day 8 | Target | |||||
| Human iPSCs on inactivated MEFs with KO-DMEM medium | Serum-free medium (RPMI/B27) + BMP2 + SU5402 for 6 days | OCT4+ SSEA1+ MESP1+ TBX5+ TBX6+ TBX18+ GATA4+ MEF2C+ NKX2.5+ ISL1+ TBX20+ | Target | -Differentiation towards CMs, SMCs and ECs under specific conditions -Arranged sarcomeric organization and gap junctions when CPCs were co-cultured with either fibroblasts + FCS, cardiac fibroblasts + CMs or conditioned medium -Trend towards ventricular CMs | -Only one iPSC line was tested -SSEA1+ CPCs can differentiate into multiple cardiac lineages, like FHF, SHF, epicardium and cardiac neural crest in the presence of FGF signals | [145] | |
| Murine iPSCs on inactivated MEFs | Feeder-free culture on gelatin-coated plates + BIO | IMDM with 15% FCS | FLK1+ MESP1+ NKX2.5+ | Target | -Presence of CM, EC and SMC markers | -Incomplete CM maturation -Functionality of the differentiated cells in in vitro conditions needs further assessment | [146] |
| Human iPSCs on Matrigel-coated plates | E8 medium + ROCK inhibitor for 24 h and RPMI/B27-insulin + CHIR for 48 h/ 4 days | TBX5+ NKX2.5+ CORIN+ HCN4+ GATA4+ | Target | -FHF: mainly differentiates into left ventricular (90%) and some atrial CMs (10%) -Presence of ion channels (Kir2.1) and higher contraction velocity | -4 different CPC populations identified with distinct differentiation potential -Isolation of the CPC populations was performed via a double transgene reporter -Expression of TBX5 and NKX2.5 dynamically changed during differentiation culture, except for the double negative (TBX5−/NKX2.5−) cell population | [147] | |
| TBX5+ NKX2.5− HCN4+ GATA4+ WT1+ TBX18+ KDR+ PECAM1+ | Target | -Epicardial progenitors: contribute to nodal (80%) and some atrial CMs -Formation of tight junctions and expression of the ion channel KCNJ3 -Some potential towards fibroblasts, SMCs and ECs | |||||
| TBX5− NKX2.5+ GATA4+ MEF2C+ ISL1+ | Target | -SHF: differentiation predominantly into atrial (90%) and some nodal and ventricular CMs -Atrial CMs displayed slower beating rates -Some potential towards SMCs and ECs | |||||
| TBX5− NKX2.5− KDR+ PECAM1+ | Target | -Endothelial potential -Formation of tube-like structures under VEGF | |||||
| Human iPSCs on inactivated MEFs followed by EB suspension culture | BMP4 for 4 days | IWR1/IWP1 for 2 days | NKX2.5+ ISL1+ GATA4+ MEF2C+ | Intermediate | -Low percentage of CMs -Organized sarcomeric structures -Normal calcium transient rhythm | -The CPCs were only identified when using human ESCs -Embryonic action potentials -CPC was an intermediate state during differentiation into CMs | [148] |
| Human iPSCs on MEFs | DMEM/F12 with 20% FBS + AA + EB plating on gelatin-coated dishes at day 7 | MEFs for 24 h and BMP2 + SU5402 for 4 days in RPMI/B27- vitamin A | ISL1+ NKX2.5+ KDR+ MESP1+ TXB20+ GATA4+ | Target | -Differentiation towards myocytes and vascular lineages under specific conditions | -Differentiation trend and CM maturation in vitro were not fully assessed | [149] |
| Human iPSCs on Synthemax-coated plates in E8 medium then mTeSR1/E8 + ROCK inhibitor for 24 h | Albumin-free RPMI + CHIR for 24 h | RPMI + IWP2 for 2 days at day 3 + basal medium at day 5 | ISL1+ NKX2.5+ KDR+ | Intermediate | -Spontaneous contraction and well-organized sarcomere filaments -Development of ventricular action potentials -Spontaneous calcium transients and connexin 43 expression in CMs | -No information about differentiation potential towards ECs and SMCs | [150] |
| Human iPSCs on Matrigel in MEF-CM supplemented with bFGF | RPMI/B27-insulin + Activin A for 24 h + BMP4 and bFGF for 4 days | RPMI/B27- insulin + DKK1 for 2 days | MESP1+ KDR+ ISL1+ NKX2.5+ | Intermediate | -Sarcomere formation -Ventricular and pacemaker action potentials -CM yield varied between 4 and 34% | -Protocol efficiency and CM differentiation and maturation is affected by cell line variability -Incomplete CM maturation -CPC was an intermediate state during differentiation into CMs | [140] |
| Human iPSCs in Geltrex with E8 medium using spheroid culture | RPMI/B27-insulin + CHIR + BMP4 for 48 h | XAV939 for 48 h at day 4 | ISL1+ TBX1+ FGF10+ FGF8+ CXCR4+ (SHF) | Target | -38% efficiency towards CMs -More potential to generate SMCs, ECs and fibroblasts | -No information about the functionality of the differentiated cells -Only one hiPSC line was tested | [151] |
| ISL1+ HCN4+ TBX5+ GATA4+ CXCR4− (FHF) | Target | -62% efficiency towards CMs -Low levels of EC and fibroblast markers | |||||
| Human PSCs on Matrigel/Synthemax-coated plates in mTeSR1/E8 medium with ROCK inhibitor | CHIR in RPMI basal medium for 24 h | IWP2/IWP4 in RPMI basal medium from day 3 to day 5 + LaSR basal or RPMI/Vc/Ins with ROCK inhibitor at day 6 + CHIR for 48 h from day 7 | WT1+ TBX18+ TCF21+ ALDH1A2− KDR+ | Target | -Differentiation towards fibroblasts and SMCs -Fibroblasts and SMCs display fibroid spindle-like shape and a fusiform appearance, respectively -Formation of mature epithelial-like sheets with tight junctions (cobblestone morphology and expression of ZO1 along cell borders) -SMCs display calcium transients and contractibility | -Epicardial progenitor cells are derived from a more multipotent CPC population (PDGFRα+/ ISL1+/NKX2.5+/GATA4+/TBX5+) -Format size of the culture (i.e., 96-well or 6-well plate) affects maturity of the epicardial cells -Different protocols lead to the formation of mesodermal cells expressing distinct markers (PDGFRα+/KDR+ and ISL1+/NKX2.5+) -Epicardial progenitor cells exhibit multiple origins | [152] |
| Albumin-free RPMI + CHIR for 24 h | RPMI + IWP2 for 2 days at day 3 + RPMI/Vc/Ins with ROCK inhibitor for 24 h at day 6 + CHIR in RPMI//Vc/Ins for 48 h at day 7 | Target | [153] | ||||
| Human iPSCs on inactivated MEFs | StemPro-34 medium + BMP4 for 24 h + BMP4, Activin A and bFGF from day 1 until day 3 | StemPro-34 medium + Matrigel-coated plates + BMP4 + CHIR + SB + VEGF for 2 days | Target | [154] | |||
| Human iPSCs in CDM + BSA + Activin A + FGF2 on gelatin-coated plates | CDM + PVA + FGF2 + LY294002 + BMP4 for 36 h and CDM + PVA + FGF2 + BMP4 for 3.5 days | CDM + PVA + BMP4 + WNT3A + RA for 10 days | Target | [155] | |||
| Human iPSCs in E8 medium and monolayer culture on vitronectin-coated plates | S12-insulin medium + CHIR for 24 h | S12-insulin medium + IWR1 for 48 h at day 3 and RA + CHIR between day 5 and 8 | Target | [156] | |||
| Murine iPSCs in inactivated MEFs in SCM | SCM-LIF + AA at day 2 | Puromycin at day 6 for 3 days | NKX2.5+ c-KIT+ FLK1+ SCA1+ | Target | -Differentiation potential towards ventricular CMs, SMCs and ECs -Sarcomeric organization and intracellular coupling observed | -Presence of CPCs expressing different sets of markers -Application of a plasmid system for CPC enrichment | [157] |
| Human iPSCs on MEFs followed by suspension culture in ESC culture medium | Gelatin-or human laminin211-coated plates + IMDM-serum and CHIR + BIO for 3 days | KY02111 +/− XAV939 or IWP2 from day 3 until day 9 | NKX2.5+ GATA4+ | Intermediate | -Predominantly ventricular CMs and 16% pacemaker cells -Spontaneous beating, sarcomere myofilaments, Z-bands, ion channels (HERG and KCNQ1) intercalated disks observed | -Mechanism of canonical WNT inhibition by KY02111 not fully understood -Protocol efficiency is affected by the presence of serum and cytokines -No differentiation into SMCs and ECs | [131] |
| Human iPSCs in E8 medium on Synthemax/Matrigel-coated plates | CDM3 medium (RPMI basal medium + AA + rice-derived RHA) + CHIR for 2 days | CDM3 medium + WNT-C59 for 48 h at day 2 | MESP1+ KDR+ ISL1+ GATA4+ NKX2.5+ TBX5+ MEF2C+ | Intermediate | -Formation of atrial, ventricular and nodal CMs | -Presence of unspecified CMs, without a defined subtype -Incomplete CM maturation -No differentiation into SMCs and ECs -CPC was an intermediate state during differentiation into CMs | [137] |
| Human iPSCs in mTeSR1 + ROCK inhibitor on Matrigel/Synthemax | Pre-treatment with CHIR/BIO for 3 days | RPMI/B27-insulin + Activin A for 24 h + BMP4 for 4 days | ISL1+ NKX2.5+ | Intermediate | -High yield of CMs -Normal sarcomere organization with transverse Z-bands -Presence of intercalated disks -Maturation trend towards ventricular CMs (80–90%) Some atrial-like action potential (10%) and absence of nodal-like potentials -Some formation of SMCs | -Optimal BMP4 concentration varies with different cell lines -Heterogenous activation of the canonical WNT signaling upon CHIR treatment in transgenic iPSC lines -Requirement of long periods of time (>60 days) to reach advanced CM maturity -Greater efficiency observed in studies using transgenic models | [126,130] |
| Transgenic iPSC lines carrying lentiviral integrated β-catenin shRNA | CHIR in RPMI/B27-insulin for 24 h | Doxycycline at 36 h post-CHIR addition | ISL1+ NKX2.5+ TBX5+ WT1+ | Intermediate | |||
| Non-transgenic hiPSC lines | IWP4 or IWP2 at day 3 | Not reported | - | [130] | |||
| IWP2 at day 3 | ISL1+ NKX2.5+ | Intermediate | [126] | ||||
| Human iPSCs on vitronectin-coated plates in mTeSR1 + ROCK inhibitor for 24 h | RPMI/B27-insulin + ISX-9 for 7 days | NKX2.5+ GATA4+ ISL1+ MEF2C+ | Target | -Differentiation potential towards CMs, ECs, and SMCs in vitro and in vivo -CMs displayed myofilaments, mitochondria and glycogen particles -Formation of tube-like structures and LDL-uptake in ECs -ECs, and SMCs formed vascular structures in vivo | -The exact mechanisms by which ISX-9 induces the expression of cardiac transcription factors is unclear -No reports about electric coupling between generated CMs and endogenous CMs in vivo -No information about the electrophysiology of CMs | [136] | |
| Human iPSCs on Matrigel in mTeSR1 + ROCK inhibitor | CHIR in RPMI/B27-insulin for 24 h + bFGF | IWP2 from day 3 to day 5 | MESP1+ T+ GATA4+ ISL1+ NKX2.5+ TBX1+ HAND2+ at day 2–3 & KDR+ PDGFRα+ at day 4–5 | Intermediate | -Formation of SHF-derived CPCs -Differentiation trend into fibroblasts, which exhibited characteristics of fetal ventricular fibroblasts | -Stage-specific progenitors were generated with this protocol -Differentiation potential was limited to fibroblasts -The fibroblasts generated might represent just one of the populations of cardiac fibroblasts present in the native heart -Only one hiPSC line was tested (line variability effects need further assessment) | [158] |
| Human iPSCs in feeder-free (Geltrex) monolayer culture | RPMI + PVA + BMP4 + FGF2 for 2 days | RPMI-insulin + 20% FBS/human serum for 2 days | MESP1+ ISL1+ NKX2.5+ | Intermediate | -Robust contraction -Striated sarcomeres and gap junction formation -High yield of CMs (64–89%) -Presence of physiological calcium transients and functional electrical coupling -Differentiation trend into ventricular CMs | -FBS is undefined -Incomplete CM maturation -CPC was an intermediate state during differentiation into CMs | [122] |
| RPMI-insulin + 20% HSA + AA for 2 days | Intermediate | ||||||
| RPMI-insulin + 20% HSA + AA for 2 days | Intermediate | ||||||
| CPC Property | MiRNA Involved | Target Protein/Pathway | Mechanism | Ref. | |
|---|---|---|---|---|---|
| Proliferation | miR-21 | PTEN | Inhibit negative regulators of cell proliferation | [224] | |
| miR-218 | SFRP2 | ||||
| miR-548c | MEIS1 | ||||
| miR-509 | |||||
| miR-23b | |||||
| miR-204 | ATF2 | Repress proliferation-related transcription factors and induces differentiation | [225] | ||
| miR-1 | HDAC4 | ||||
| HAND2 | |||||
| miR-200b | GATA4 | ||||
| miR-17-92 cluster | Not reported | Increases proliferation rate | [219] | ||
| Differentiation | CMs | miR-133 | NELFA | Suppresses cardiogenesis | [226] |
| miR-218 | SFRP2 | Inhibits a negative regulator of cell proliferation | [227] | ||
| miR-142 | MEF2C | Suppresses CM formation | [228] | ||
| miR-1 | DLL1 | Increases NKX2.5 and Myogenin expression | [229] | ||
| miR-499 | ROD1 | Suppresses inhibitory factors of cardiac differentiation | [224,230] | ||
| SOX6 | |||||
| miR-708 | N-RAS | [231] | |||
| miR-322-503 cluster | CELF1 | [220] | |||
| SMCs | miR-22 | EVI1 | Inhibits negative regulators of SMC marker gene expression and of SMC transcription factors | [232] | |
| miR-29a | YY1 | [233] | |||
| miR-669a | MYOD | Increases CPC differentiation potential by preventing skeletal myogenesis | [234] | ||
| miR-669q | |||||
| Migration | miR-206 | TIMP3 | Suppresses a metalloproteinase inhibitor | [235] | |
| miR-21 | PTEN | Promotes migration of SCA1+ CPCs (not fully clear) | [236] | ||
| Apoptosis | miR-21 | BIM | Inhibit apoptotic activators | [237] | |
| PDCD4 | |||||
| miR-24 | BIM | ||||
| miR-221 | |||||
| Necrotic Cell Death | miR-155 | RIP1 | Inhibits necrosis activators | [238] | |
| Vascular Remodeling | miR-221 | c-KIT eNOS | Inhibit endothelial cell migration and proliferation | [239] | |
| miR-222 | |||||
| Cell Repolarization | miR-1 | KCNE1 KCNQ1 | Reduce potassium current in hyperglycemia conditions | [240] | |
| miR-133 | |||||
| Scaffold Biomaterial | Experimental Design | Outcome | Limitations | Ref. |
|---|---|---|---|---|
| Fibrin patch | SSEA1+ and ISL1+ hESCs-CPCs mixed in fibrinogen, and scaffolds were then transplanted into myocardial infarction rats | -Improved contractility and decrease in adverse ventricular remodeling -Increased angiogenesis and attenuation of fibrosis | -Poor long-term cell engraftment -Functional improvements resulted from paracrine signaling | [247] |
| Same process as above, except the scaffolds were delivered surgically on the infarct area of a 68-year-old patient suffering from severe heart failure | -No observation of ventricular arrhythmias -Decreased in adverse ventricular remodeling | -Presence of T-cell response 3 months post-implantation -Absence of neovascularization in patch-treated area | [248] | |
| mESCs were primed with BMP2 for 36 h and seeded into fibrin matrices The constructs were then implanted onto normal or infarcted rat left ventricles | -Efficient cell engraftment -Attenuation of left ventricle dilation -Promotion of neovascularization | -Rapid inflammation-driven degradation of scaffolds -Unclear whether neovascularization was due to in situ cell differentiation or endogenous EC recruitment | [249] | |
| Polyethylene glycol diacrylate woodpile (PEGDa-Wp) and PEGDa hydrogel. | Human adult LIN−/SCA1+ CPCs were seeded in a PEGDa hydrogel and the mixture was then cultured onto a PEGDa-Wp | -Benefits on cell assembly and alignment -Induction of cell spatial-ordered multilayer organization and differentiation towards a CM phenotype | -Incomplete maturation of CMs -No differentiation into SMCs and ECs -No in vivo testing of the scaffolds | [258] |
| Poly(l-lactic acid) Nanofibres | mESC-derived ISL1+/GATA4+ CPCs were seeded onto nanofibres After 7 days of in vitro differentiation, the scaffolds were implanted subcutaneously in the dorsal area of athymic nude mice | -Enhancement of cell attachment, extension and differentiation in vitro -Improvement of cell survival, integration and commitment to the three cardiac lineages in vivo -Induction of angiogenesis in vivo | -Poor in vitro differentiation into ECs -Unclear whether neovascularization was due to paracrine factors or CPC-derived SMCs and ECs | [257] |
| Tissue Printing using Sodium Alginate | Human SCA1+ CPCs were mixed with alginate matrixes, including an RGD-modified alginate, which were then used to print porous and non-porous scaffolds | -Porosity preserved viability and proliferation and increased cardiac commitment of CPCs -CPCs migrated from the construct and formed tubular-like structures | -Incomplete maturation of the differentiated cells -No in vivo testing of the scaffolds | [250] |
| Porcine- and human-derived myocardial matrices | Human SCA1+ CPCs were seeded onto porcine and human ECM Scaffolds were injected into the left ventricular free wall of healthy hearts of Sprague Dawley rats | -Porcine-derived ECM was more efficient at promoting CPC differentiation, whereas human-derived ECM promoted CPC proliferation | -Variation in ECM properties due to distinct decellularised methods used, patient-to-patient variability and tissue age | [259] |
| 3D-printed hyaluronic acid/gelatin-based matrix | Human SCA1+ CPCs were printed together with the matrix The cell-loaded patches were transplanted in myocardial infarction mice | -Reduction of adverse remodeling and fibrosis -Long-term CPC survival and engraftment -Formation of vessel-like structures within the scaffold in vivo | -Absence of neovascularization in the infarcted region -Incomplete maturation of CMs in vivo | [251] |
| Collagen/Matrigel hydrogels | Human SCA1+ CPCs were encapsulated in collagen/Matrigel hydrogels which were cultured in either stress-free or unidirectional constrained conditions | -Enhanced cardiac differentiation and matrix remodeling -Constrained hydrogels stabilized CPC viability, attachment and proliferation -Static strain stimulated actin fiber formation and cell alignment | -Differentiation trend towards CMs -Incomplete maturation of CMs -No CPC differentiation into SMCs and ECs -No in vivo testing | [260] |
| Decellularised porcine ventricular ECM | Human Foetal and adult SCA1+ CPCs were resuspended in porcine myocardial matrix and collagen type I solutions The cell/matrix mixtures were injected into the left ventricular wall of Sprague Dawley rats | -The myocardial matrix improved CPCs adhesion, survival, proliferation and cardiac commitment both in vitro and in vivo -Foetal CPCs survived better than adult CPCs in vivo | -Rats were euthanized 30 min post-implantation, preventing assessment of long-term effects on cell survival, migration and cardiac function | [261] |
| Same procedure as above, exceptions: use of adult rat c-KIT+ CPCs and no in vivo implantation | -The cardiac ECM improved cardiac commitment, cell survival, proliferation and adhesion | -Differentiation trend towards CMs. -Low differentiation efficiency towards ECs and SMCs | [262] | |
| Whole decellularised mouse heart | hiPSC- and hESC-derived KDR+/c-KIT− CPCs were seeded into a whole decellularised mouse heart The repopulated hearts were perfused with VEGF and DKK1 or VEGF and bFGF | -Efficient control of in situ iPSC-CPC differentiation -Advanced CM maturation -Development of vessel-like structures and spontaneous contraction for both iPSC-and ESC-CPC constructs | -Scattered regions of uncoupled cells -Insufficient mechanical force generation and incomplete electrical synchronization of the constructs | [252] |
| FLT1 (VEGFR1)+/PDGFRα+ hESC-CPCs were seeded onto decellularised mice hearts, which were implanted subcutaneously into SCID mice | -In situ generation of CMs, SMCs and ECs -Formation of a vascular network and higher expression of CM markers in vivo | -In vivo differentiated ECs were not ubiquitously distributed in the decellularised scaffold -Absence of beating populations | [263] | |
| Whole decellularised rat heart | hESC-derived KDR+/PDGFRα+ CPCs were expanded in a stirred-suspension bioreactor and seeded onto perfusion-decellularised Wistar rat hearts containing immobilized bFGF | -Improved CPC retention, proliferation and cardiac differentiation potential -Spontaneous and synchronous contractions -Advanced CM maturation | -Growth factor immobilization prevents spatiotemporal control -No in vivo testing | [264] |
| Whole decellularised human heart | Human adult c-KIT+ CPCs from human cardiac biopsies were cultured onto perfused-decellularised heart ventricles | -Increased CPC growth and stimulated differentiation towards cardiac lineages in vitro | -Poor CPC infiltration into the matrix -No electrical signal propagation. -No in vivo testing | [265] |
| Rat and pig collagen matrix and decellularised left ventricle ECM | iPSC-CPCs were cultured on rat or pig collagen matrices and decellularised ECM CPCs were also co-cultured with ECs and CMs | -Enhanced expression of contractile protein gene expression -Cell communication was observed in co-cultures | -No results reported on CPC proliferation and differentiation -No information about the CPC markers | [253] |
| 3D-bioprinted patch containing decellularised porcine ventricular ECM | Bioinks composed of decellularised ECM, human neonatal c-KIT+ CPCs and gelatin methacrylate were used to print patches, which were implanted onto the epicardial surface of the right ventricle of Sprague Dawley rat hearts | -Good CPC retention and viability in the scaffolds -Enhanced cardiogenic differentiation and angiogenic potential -Presence of vascularization in the patches in vivo | -Main purpose of the patch was to improve the paracrine release from the CPCs -No influence in SMC differentiation | [266] |
| Foetal and adult rat decellularised ventricle ECM | Immortalized adult mouse LIN−/SCA1+ CPCs were seeded onto embryonic, neonatal and adult rat ECM | -Good CPC retention, motility and viability -Remodeling of the supporting ECM -Enhanced production of cardiac repair factors | -No evidence of CPC differentiation -No in vivo testing | [267] |
| Decellularised murine embryonic heart | Day 5 and 9 mESC-CPCs were then seeded onto the decellularised scaffolds | -Day 5 progenitors formed spontaneously beating constructs in the scaffolds | -Mixed cell population isolated -Not all cell populations led to functional maturation | [268] |
| Decellularised human pericardium-derived microporous scaffold | Human SCA1+ CPCs were seeded onto 3D microporous pericardium scaffolds, which were then implanted subcutaneously into Wistar rats | -Improved CPC migration, survival, proliferation and differentiation -Reduction of immunological response and enhanced angiogenesis | -No influence in CPC differentiation towards SMCs | [269] |
| Self-assembling peptide nanofibers | Adult LIN−/c-KIT+ rat CPCs were seeded onto IGF1-tethered nanofibres CPCs and scaffolds were injected into myocardial infarction rats | -Enhanced CPC survival, proliferation and differentiation into CMs -Improved angiogenesis, recruitment of resident CPCs and attenuation of ventricle dilation | -Growth factor immobilization prevents spatiotemporal control -Newly formed CMs were derived from resident CPCs -CPCs were not cultured on the scaffolds prior to implantation | [254] |
| Adult mouse SCA1+ CPCs were mixed with Puramatrix® complex and injected into the border area of the myocardium in myocardial infarction mice | -Reduction of the infarct area and attenuation of ventricular dilation. -Enhanced neovascularization | -No CPC differentiation towards ECs -Functional improvements resulted from paracrine signaling -Poor CPC engraftment | [255] | |
| RDG-modified collagen and porous gelatin solid foam | Human adult CS-CPC were grown as secondary CSs, which were seeded onto the scaffolds | -Enhanced cell migration and ECM production -Increased CPC cardiogenic potential, cell retention and adherence | -Cardiac commitment trend towards CMs -Distinct scaffold morphologies promoted different biological processes | [270] |
| Degradable Poly(N-isopropylacrylamide) hydrogel | Mouse CDCs were added into hydrogel solutions, with or without collagen and containing different stiffness | -Preservation of CDC proliferation -Stimulation of differentiation into mature cardiac cells in hydrogels with medium stiffness and collagen | -No differentiation into ECs and SMCs -No in vivo testing | [256] |
| Biodegradable gelatin | Human CDCs were seeded onto bFGF immobilized gelatin hydrogels, which were implanted in the epicardium of immunosuppressed myocardial infarction pigs | -Enhanced angiogenesis, cell engraftment -Reduction of the infarct area and attenuation of adverse ventricular remodeling | -Growth factor immobilization prevents spatiotemporal control -No differentiation into ECs and SMCs | [271] |
| Fibrinogen/Matrigel mixture and PDMS molds | NKX2.5+/c-KIT+/either FLK1+ or SCA1+ iPSC-CPCs were mixed in a fibrinogen/Matrigel hydrogel and applied into PDMS molds | -Spontaneous and synchronous contraction -Highly organized sarcomere structures and robust electromechanical connections | -Improper nutrient access within the construct -No differentiation potential towards SMCs and ECs -No in vivo testing | [157] |
| Collagen sponge | CPCs were seeded onto collagen sponges and then transplanted into rat hearts with atrioventricular conduction block | -Enhanced vascularization -Gap junction formation -Differentiation into CMs, conduction cells and ECs | -No information about the functionality of the CPC-derived cells | [272] |
| Cells | Biomaterial/Scaffold | Platform | Stimulation | Electrophysiology | Ref. |
|---|---|---|---|---|---|
| hiPSC-CMs | Graphene substrate | 2D | FET (current pulse with f = 1 Hz) For calcium: voltage ramp from −80 to +60 mV at 20 mV/s | -Enhanced electrophysiological properties: RP = −40.54 ± 1.72 mV AP = 75.24 ± 3.91 mV CV = 5.34 ± 1.60 cm/s ICa2+ density = −9.31 ± 2.35 pA/pF ICa2+,L density = −2.47 ± 0.6 pA/pF Ik density = 46.24 ± 8.45 pA/pF Ikr density = 36.57 ± 5.84 pA/pF Ca2+ transients: Amplitude intensity = 1.69 ± 0.20 u Upstroke velocity 3.09 ± 0.99 u/s Decay velocity (50%) = 0.84 ± 0.29 s | [273] |
| iCell® CMs & hESC-CMs | Reduced graphene oxide (rGO) | 2D | Light: intensity >1 mW/mm2, duration 40-ms-2-Hz light pulses and 3-s step of light | -Optical stimulation on rGO substrates improves CMs electrophysiology -rGO increases AP peaks frequency -On rGO CMs contraction frequency increases with light intensity | [274] |
| Neonatal Sprague Dawley rat vCMs | Electrospun gelatine + PCL nanofibres | 3D | FET (1–3 V, 50-ms-long pulses at 1–2 Hz) | -Electrical stimulation results in regularly spaced spikes (f = 1–2 Hz) with shape and width consistent with CM extracellular signals -NE increases electrical activity and frequency of calcium transients | [275] |
| hiPSC-CMs | PLGA electrospun aligned nanofibres | 3D | Not applied | -Enhanced CM maturity and electrical activity -CM drug (E4031) response showed higher electrophysiological homogeneity -L-ANFs increased FP amplitude, number of electrically active cells, synchronization and anisotropic propagation of the electrical signal | [276] |
| hESC-CMs & hiPSC-CMs | Type I collagen gel template suture (Biowires) | 3D | Electrical field with daily and progressively frequency increase: low frequency ramp-up regimen (from 1 to 3 Hz) or high frequency ramp-up regimen (from 1 to 6 Hz) | -Electrical stimulation enhanced electrical activity frequency -High frequency increased electrophysiological properties, contractile activity, synchronization and CV -High frequency decreased excitation threshold and variability in AP duration -High frequency improved CM response to caffeine and Ca2+ handling properties: IERG = 0.81 ± 0.09 pA/pF IK1 = 1.53 ± 0.25 pA/pF | [277] |
| hESC-CMs | MEA coated with collagen type I + agarose layer | 2D | Anti-arrhythmic and pro-arrhythmic drugs | -Pharmacological stimulation influences CMs electrophysiology -FPD and CT are dependent on the dose of arrhythmogenic drugs: E-4031 & Astemizole increased FPD Flecainide & Terfenadine decreased FPD Flecainide, Astemizole & Terfenadine increased CT and of safe drugs: Verapamil & Lidocine decreased FPD Lidocine slightly increased CT | [278] |
| hiPSC-CMs | MEA coated with hydrogel containing fluorescence microbeads | 2D | Electrical: periodic voltage pulses (biphasic square waves with pulse width = 4 ms, f = 0.2 Hz, peak-to-peak amplitude = 4 V) Pharmacological: drug exposure (NE and Blebbistatin) | -Good electrical coupling of CMs (FP = 9–35 µV and CV = 16 cm/s) -Electrical pacing promoted synchronized contraction (f = 11 bpm) -Recorded impedance increased with cell attachment and at each contraction -Blebbistan inhibited beating activity and has no effect on FP -NE increased CV and contraction spikes rate | [279] |
| Clinical Trial Name | Phase | Start/End Date | CPC Type | Delivery of Cells | Biomaterial Added | Results | Ref. |
|---|---|---|---|---|---|---|---|
| CADUCEUS prospective, randomized trial | I | 2009–2012 | CDCs | Direct injection via catheter | none | LVEF unchanged at 12 months Scar size decreased 12.3% at 12 months Regional contractility and systolic wall thickening increased | [283,293] |
| ALCADIA Open-label, non-randomized trial | I | 2010–2013 | CDCs | Direct injection via catheter | Biodegradable gelatin hydrogel sheet containing 200 μg of bFGF planted onto epicardium covering the injection site | LVEF increase 12% at 6 months Scar size decrease 3.3% at 6 months | [285] |
| ALLSTAR Open-label cohort (PI), double-blinded, randomized, placebo-controlled study (PII) | I/II | 2012–2019 | CDCs | Direct injection via catheter | none | Terminated (follow-up activities were ceased) | [294] |
| ESCORT Open-label trial | I | 2013–2018 | ESC-derived ISL1+/CD15+ | Epicardial patch via coronary artery bypass procedure | Fibrin gel patch containing progenitor cells | LVEF increase of 12.5% No arrhythmias, or tumor formation | [287] |
| CAREMI Double blinded, randomized, placebo-controlled trial | I/II | 2014–2016 | CDCs | Direct injection via catheter | none | Infarct size decreased to 15.6% at 12 months LVEF increase of 7.7% at 12 months | [295] |
| DYNAMIC Open-label trial, randomized, double-blinded, placebo-controlled trial | I | 2014–ongoing | CDCs | Direct injection via catheter to multi-vessel areas of heart | none | Ongoing | [296] |
| CONCERT-HF Randomized, double-blinded, placebo-controlled trial | II | 2015–ongoing | c-KIT+ | Direct injection via catheter | none | Ongoing (paused on 29.10.18, re-approved 06.02.2019) | [297] |
| TICAP Open-label trial, non-randomized | I | 2011–2013 | CDCs | Direct injection via catheter | none | RVEF increase of around 8.0% at 18 and 36 months No tumor formation | [288,290] |
| PERSEUS Open-label trial, randomized | II | 2013–2016 | CDCs | Direct injection via catheter | none | LVEF increase of 6.4% at 3 months Reduction in scar size | [289] |
| APOLLON Randomized, single-blinded | III | 2016 & Unknown | CDCs | Direct injection via catheter | none | Unknown status (last update was September 2017) | [291] |
| TICAP-DCM Randomized | I | 2017–ongoing | CDCs | Direct injection via catheter | none | Recruiting | [292] |
| REGRESS-HFpEF Randomized, double-blinded, placebo-controlled trial | II | 2017–ongoing | CDCs | Direct injection via catheter | none | Ongoing | [298] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barreto, S.; Hamel, L.; Schiatti, T.; Yang, Y.; George, V. Cardiac Progenitor Cells from Stem Cells: Learning from Genetics and Biomaterials. Cells 2019, 8, 1536. https://doi.org/10.3390/cells8121536
Barreto S, Hamel L, Schiatti T, Yang Y, George V. Cardiac Progenitor Cells from Stem Cells: Learning from Genetics and Biomaterials. Cells. 2019; 8(12):1536. https://doi.org/10.3390/cells8121536
Chicago/Turabian StyleBarreto, Sara, Leonie Hamel, Teresa Schiatti, Ying Yang, and Vinoj George. 2019. "Cardiac Progenitor Cells from Stem Cells: Learning from Genetics and Biomaterials" Cells 8, no. 12: 1536. https://doi.org/10.3390/cells8121536
APA StyleBarreto, S., Hamel, L., Schiatti, T., Yang, Y., & George, V. (2019). Cardiac Progenitor Cells from Stem Cells: Learning from Genetics and Biomaterials. Cells, 8(12), 1536. https://doi.org/10.3390/cells8121536