Emerging Role of Cellular Prion Protein in the Maintenance and Expansion of Glioma Stem Cells †

 ,
,  and
and 
{kind=link}
{kind=link}
Abstract
1. Introduction
- (i)
- maintain tumor-initiating activity,
- (ii)
- sustain proliferation and invasiveness,
- (iii)
- acquire multidrug resistance, and
- (iv)
- preserve multipotency and ability to differentiate in non-tumorigenic glioma cells [43,44,45,46]. Finally, as observed in normal tissues, in CSCs PrPC seems to act as receptor or scaffold protein for several extracellular signals dealing with maintenance of self-renewal, adherence, invasiveness, and migration of cells [47].
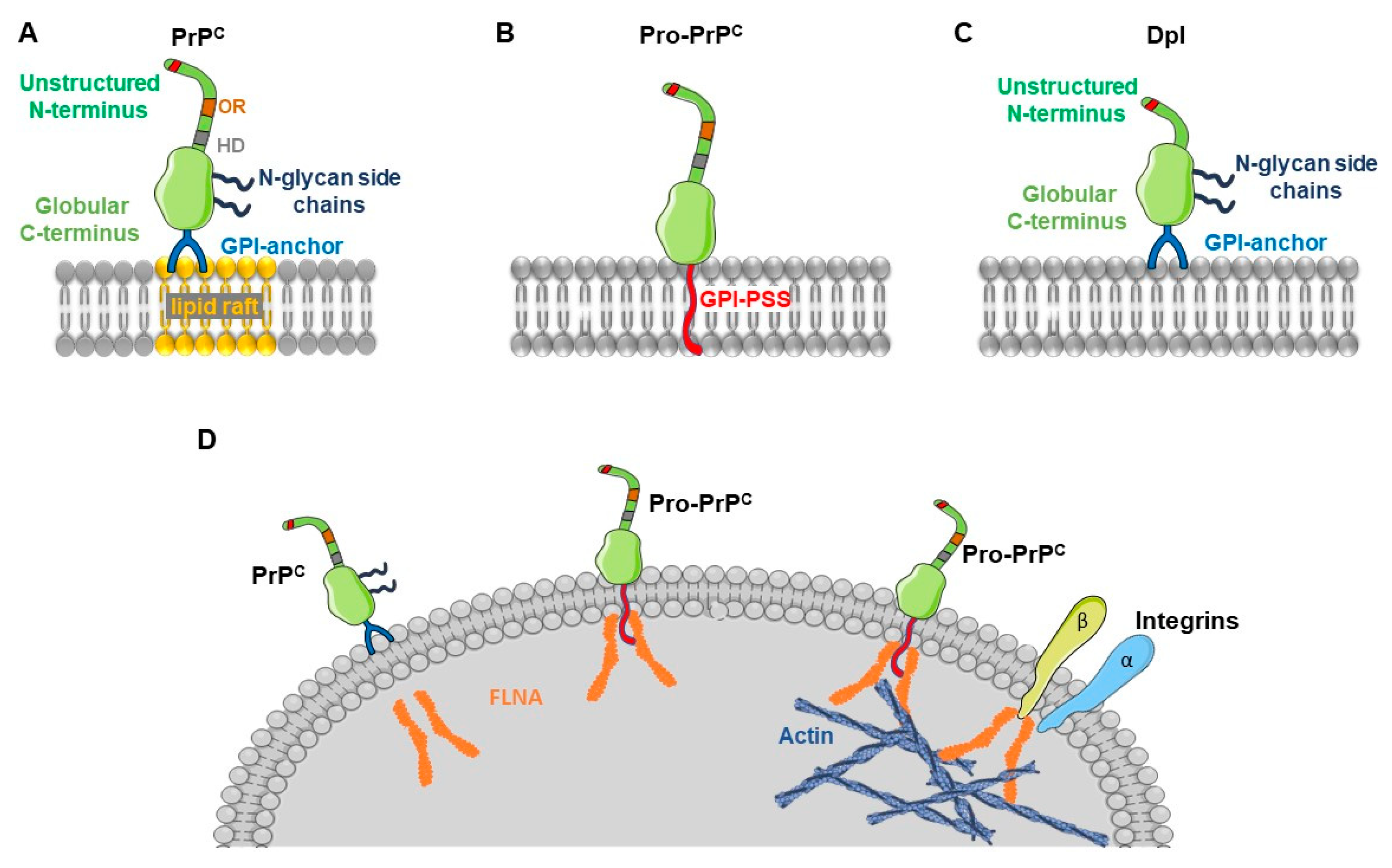
2. The Cellular Prion Protein
3. Physiology of PrPC in the Development and Homeostasis of Normal Tissues
3.1. Role of PrPC in the Development of the Nervous System
3.2. PrPC Stimulates Hematopoiesis
4. PrPC and Cancer Stem Cells in Gliomas and Other Tumors
4.1. PrPC and CD44
4.2. PrPC and Stress-Inducible Protein 1
4.3. PrPC and Laminin
4.4. PrPC and Notch
4.5. PrPC and Wnt Pathway
5. Pro-Prion and Cancer
6. Role of the Prion-Like Protein Doppel in Gliomas and Other Cancers
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, A.; Thellung, S.; Villa, V.; Nizzari, M.; Florio, T. Role of prion protein aggregation in neurotoxicity. Int. J. Mol. Sci. 2012, 13, 8648–8669. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Polymenidou, M. Mammalian prion biology: One century of evolving concepts. Cell 2004, 116, 313–327. [Google Scholar] [CrossRef]
- Martin-Lanneree, S.; Hirsch, T.Z.; Hernandez-Rapp, J.; Halliez, S.; Vilotte, J.L.; Launay, J.M.; Mouillet-Richard, S. PrP(C) from stem cells to cancer. Front. Cell Dev. Biol. 2014, 2, 55. [Google Scholar]
- Blattler, T.; Brandner, S.; Raeber, A.J.; Klein, M.A.; Voigtlander, T.; Weissmann, C.; Aguzzi, A. PrP-expressing tissue required for transfer of scrapie infectivity from spleen to brain. Nature 1997, 389, 69–73. [Google Scholar] [CrossRef]
- Weissmann, C.; Bueler, H.; Fischer, M.; Sailer, A.; Aguzzi, A.; Aguet, M. PrP-deficient mice are resistant to scrapie. Ann. N. Y. Acad. Sci. 1994, 724, 235–240. [Google Scholar] [CrossRef]
- Brandner, S.; Raeber, A.; Sailer, A.; Blattler, T.; Fischer, M.; Weissmann, C.; Aguzzi, A. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc. Natl. Acad. Sci. USA 1996, 93, 13148–13151. [Google Scholar] [CrossRef]
- Bremer, J.; Baumann, F.; Tiberi, C.; Wessig, C.; Fischer, H.; Schwarz, P.; Steele, A.D.; Toyka, K.V.; Nave, K.A.; Weis, J.; et al. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 2010, 13, 310–318. [Google Scholar] [CrossRef]
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef]
- Weissmann, C.; Flechsig, E. PrP knock-out and PrP transgenic mice in prion research. Br. Med. Bull. 2003, 66, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Pericuesta, E.; Ramirez, M.A.; Gutierrez-Adan, A. Prion protein expression regulates embryonic stem cell pluripotency and differentiation. PLoS ONE 2011, 6, e18422. [Google Scholar] [CrossRef] [PubMed]
- Burthem, J.; Urban, B.; Pain, A.; Roberts, D.J. The normal cellular prion protein is strongly expressed by myeloid dendritic cells. Blood 2001, 98, 3733–3738. [Google Scholar] [CrossRef] [PubMed]
- Kubosaki, A.; Yusa, S.; Nasu, Y.; Nishimura, T.; Nakamura, Y.; Saeki, K.; Matsumoto, Y.; Itohara, S.; Onodera, T. Distribution of cellular isoform of prion protein in T lymphocytes and bone marrow, analyzed by wild-type and prion protein gene-deficient mice. Biochem. Biophys. Res. Commun. 2001, 282, 103–107. [Google Scholar] [CrossRef]
- McBride, P.A.; Eikelenboom, P.; Kraal, G.; Fraser, H.; Bruce, M.E. PrP protein is associated with follicular dendritic cells of spleens and lymph nodes in uninfected and scrapie-infected mice. J. Pathol. 1992, 168, 413–418. [Google Scholar] [CrossRef]
- Moleres, F.J.; Velayos, J.L. Expression of PrP(C) in the rat brain and characterization of a subset of cortical neurons. Brain Res. 2005, 1056, 10–21. [Google Scholar] [CrossRef]
- Verghese-Nikolakaki, S.; Michaloudi, H.; Polymenidou, M.; Groschup, M.H.; Papadopoulos, G.C.; Sklaviadis, T. Expression of the prion protein in the rat forebrain--an immunohistochemical study. Neurosci. Lett. 1999, 272, 9–12. [Google Scholar] [CrossRef]
- Ford, M.J.; Burton, L.J.; Morris, R.J.; Hall, S.M. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience 2002, 113, 177–192. [Google Scholar] [CrossRef]
- Manson, J.; West, J.D.; Thomson, V.; McBride, P.; Kaufman, M.H.; Hope, J. The prion protein gene: A role in mouse embryogenesis? Development 1992, 115, 117–122. [Google Scholar]
- McKinley, M.P.; Hay, B.; Lingappa, V.R.; Lieberburg, I.; Prusiner, S.B. Developmental expression of prion protein gene in brain. Dev. Biol. 1987, 121, 105–110. [Google Scholar] [CrossRef]
- Moser, M.; Colello, R.J.; Pott, U.; Oesch, B. Developmental expression of the prion protein gene in glial cells. Neuron 1995, 14, 509–517. [Google Scholar] [CrossRef]
- Steele, A.D.; Emsley, J.G.; Ozdinler, P.H.; Lindquist, S.; Macklis, J.D. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 3416–3421. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, R.; Wong, B.S.; Liu, D.; Pan, T.; Petersen, R.B.; Gambetti, P.; Sy, M.S. Normal cellular prion protein is preferentially expressed on subpopulations of murine hemopoietic cells. J. Immunol. 2001, 166, 3733–3742. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Whittington, M.A.; Sidle, K.C.; Smith, C.J.; Palmer, M.S.; Clarke, A.R.; Jefferys, J.G. Prion protein is necessary for normal synaptic function. Nature 1994, 370, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, L.; Legname, G. Prion protein and aging. Front. Cell Dev. Biol. 2014, 2, 44. [Google Scholar] [CrossRef] [PubMed]
- Halliez, S.; Passet, B.; Martin-Lanneree, S.; Hernandez-Rapp, J.; Laude, H.; Mouillet-Richard, S.; Vilotte, J.L.; Beringue, V. To develop with or without the prion protein. Front. Cell Dev. Biol. 2014, 2, 58. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Herms, J.W.; Kretzchmar, H.A.; Titz, S.; Keller, B.U. Patch-clamp analysis of synaptic transmission to cerebellar purkinje cells of prion protein knockout mice. Eur. J. Neurosci. 1995, 7, 2508–2512. [Google Scholar] [CrossRef]
- Zhang, C.C.; Steele, A.D.; Lindquist, S.; Lodish, H.F. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc. Natl. Acad. Sci. USA 2006, 103, 2184–2189. [Google Scholar] [CrossRef]
- Prestori, F.; Rossi, P.; Bearzatto, B.; Laine, J.; Necchi, D.; Diwakar, S.; Schiffmann, S.N.; Axelrad, H.; D’Angelo, E. Altered neuron excitability and synaptic plasticity in the cerebellar granular layer of juvenile prion protein knock-out mice with impaired motor control. J. Neurosci. 2008, 28, 7091–7103. [Google Scholar] [CrossRef]
- Linden, R. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front. Mol. Neurosci. 2017, 10, 77. [Google Scholar] [CrossRef]
- Cheng, Y.; Tao, L.; Xu, J.; Li, Q.; Yu, J.; Jin, Y.; Chen, Q.; Xu, Z.; Zou, Q.; Liu, X. CD44/cellular prion protein interact in multidrug resistant breast cancer cells and correlate with responses to neoadjuvant chemotherapy in breast cancer patients. Mol. Carcinog. 2014, 53, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Pan, Y.; Shi, Y.; Guo, C.; Jin, X.; Sun, L.; Liu, N.; Qiao, T.; Fan, D. Overexpression and significance of prion protein in gastric cancer and multidrug-resistant gastric carcinoma cell line SGC7901/ADR. Int. J. Cancer 2005, 113, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhao, L.; Liang, J.; Liu, J.; Shi, Y.; Liu, N.; Zhang, G.; Jin, H.; Gao, J.; Xie, H.; et al. Cellular prion protein promotes invasion and metastasis of gastric cancer. FASEB J. 2006, 20, 1886–1888. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Matarredona, E.R.; Pastor, A.M. Neural Stem Cells of the Subventricular Zone as the Origin of Human Glioblastoma Stem Cells. Therapeutic Implications. Front. Oncol. 2019, 9, 779. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Lindberg, N.; Kastemar, M.; Olofsson, T.; Smits, A.; Uhrbom, L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 2009, 28, 2266–2275. [Google Scholar] [CrossRef]
- Florio, T.; Barbieri, F. The status of the art of human malignant glioma management: The promising role of targeting tumor-initiating cells. Drug Discov. Today 2012, 17, 1103–1110. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.K.; Desai, N.S. Cancer Stem Cells: Acquisition, Characteristics, Therapeutic Implications, Targeting Strategies and Future Prospects. Stem Cell Rev. 2019, 15, 331–355. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, A.; Bajetto, A.; Thellung, S.; Begani, G.; Villa, V.; Nizzari, M.; Pattarozzi, A.; Solari, A.; Gatti, M.; Pagano, A.; et al. Cellular prion protein controls stem cell-like properties of human glioblastoma tumor-initiating cells. Oncotarget 2016, 7, 38638–38657. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Rao, G.; Wang, H.; Li, B.; Tian, W.; Cui, J.; He, L.; Laffin, B.; Tian, X.; Hao, C.; et al. CD44-positive cancer stem cells expressing cellular prion protein contribute to metastatic capacity in colorectal cancer. Cancer Res. 2013, 73, 2682–2694. [Google Scholar] [CrossRef]
- Zhuang, D.; Liu, Y.; Mao, Y.; Gao, L.; Zhang, H.; Luan, S.; Huang, F.; Li, Q. TMZ-induced PrPc/par-4 interaction promotes the survival of human glioma cells. Int. J. Cancer 2012, 130, 309–318. [Google Scholar] [CrossRef]
- Linden, R.; Cordeiro, Y.; Lima, L.M. Allosteric function and dysfunction of the prion protein. Cell Mol. Life Sci. 2012, 69, 1105–1124. [Google Scholar] [CrossRef]
- Santos, T.G.; Lopes, M.H.; Martins, V.R. Targeting prion protein interactions in cancer. Prion 2015, 9, 165–173. [Google Scholar] [CrossRef]
- Stahl, N.; Baldwin, M.A.; Hecker, R.; Pan, K.M.; Burlingame, A.L.; Prusiner, S.B. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 1992, 31, 5043–5053. [Google Scholar] [CrossRef]
- Safar, J.; Prusiner, S.B. Molecular studies of prion diseases. Prog. Brain Res. 1998, 117, 421–434. [Google Scholar]
- Dagher, A.; Zeighami, Y. Testing the Protein Propagation Hypothesis of Parkinson Disease. J. Exp. Neurosci. 2018, 12, 1179069518786715. [Google Scholar] [CrossRef]
- Maniecka, Z.; Polymenidou, M. From nucleation to widespread propagation: A prion-like concept for ALS. Virus Res. 2015, 207, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Morinet, F. Prions: A model of conformational disease? Pathol. Biol. 2014, 62, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.M.P.; Kopito, R.R. Prion-Like Characteristics of Polyglutamine-Containing Proteins. Cold Spring Harb. Perspect. Med. 2018, 8, a024257. [Google Scholar] [CrossRef]
- Watts, J.C.; Prusiner, S.B. beta-Amyloid Prions and the Pathobiology of Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2018, 8, a023507. [Google Scholar] [CrossRef]
- Aulic, S.; Masperone, L.; Narkiewicz, J.; Isopi, E.; Bistaffa, E.; Ambrosetti, E.; Pastore, B.; De Cecco, E.; Scaini, D.; Zago, P.; et al. alpha-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci. Rep. 2017, 7, 10050. [Google Scholar] [CrossRef]
- Freir, D.B.; Nicoll, A.J.; Klyubin, I.; Panico, S.; Mc Donald, J.M.; Risse, E.; Asante, E.A.; Farrow, M.A.; Sessions, R.B.; Saibil, H.R.; et al. Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2011, 2, 336. [Google Scholar] [CrossRef]
- Ondrejcak, T.; Klyubin, I.; Corbett, G.T.; Fraser, G.; Hong, W.; Mably, A.J.; Gardener, M.; Hammersley, J.; Perkinton, M.S.; Billinton, A.; et al. Cellular Prion Protein Mediates the Disruption of Hippocampal Synaptic Plasticity by Soluble Tau In Vivo. J. Neurosci. 2018, 38, 10595–10606. [Google Scholar] [CrossRef]
- Pagano, K.; Galante, D.; D’Arrigo, C.; Corsaro, A.; Nizzari, M.; Florio, T.; Molinari, H.; Tomaselli, S.; Ragona, L. Effects of Prion Protein on Abeta42 and Pyroglutamate-Modified AbetapEpsilon3-42 Oligomerization and Toxicity. Mol. Neurobiol. 2019, 56, 1957–1971. [Google Scholar] [CrossRef]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Paradis, E.; Douillard, H.; Koutroumanis, M.; Goodyer, C.; LeBlanc, A. Amyloid beta peptide of Alzheimer’s disease downregulates Bcl-2 and upregulates bax expression in human neurons. J. Neurosci. 1996, 16, 7533–7539. [Google Scholar] [CrossRef]
- Thellung, S.; Corsaro, A.; Villa, V.; Venezia, V.; Nizzari, M.; Bisaglia, M.; Russo, C.; Schettini, G.; Aceto, A.; Florio, T. Amino-terminally truncated prion protein PrP90-231 induces microglial activation in vitro. Ann. N. Y. Acad. Sci. 2007, 1096, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Thellung, S.; Scoti, B.; Corsaro, A.; Villa, V.; Nizzari, M.; Gagliani, M.C.; Porcile, C.; Russo, C.; Pagano, A.; Tacchetti, C.; et al. Pharmacological activation of autophagy favors the clearing of intracellular aggregates of misfolded prion protein peptide to prevent neuronal death. Cell Death Dis. 2018, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Manelli, A.M.; Holmberg, K.H.; Van Eldik, L.J.; Ladu, M.J. Differential effects of oligomeric and fibrillar amyloid-beta 1-42 on astrocyte-mediated inflammation. Neurobiol. Dis. 2005, 18, 459–465. [Google Scholar] [CrossRef]
- Thellung, S.; Corsaro, A.; Villa, V.; Simi, A.; Vella, S.; Pagano, A.; Florio, T. Human PrP90-231-induced cell death is associated with intracellular accumulation of insoluble and protease-resistant macroaggregates and lysosomal dysfunction. Cell Death Dis. 2011, 2, e138. [Google Scholar] [CrossRef]
- Fioriti, L.; Angeretti, N.; Colombo, L.; De Luigi, A.; Colombo, A.; Manzoni, C.; Morbin, M.; Tagliavini, F.; Salmona, M.; Chiesa, R.; et al. Neurotoxic and gliotrophic activity of a synthetic peptide homologous to Gerstmann-Straussler-Scheinker disease amyloid protein. J. Neurosci. 2007, 27, 1576–1583. [Google Scholar] [CrossRef]
- Florio, T.; Grimaldi, M.; Scorziello, A.; Salmona, M.; Bugiani, O.; Tagliavini, F.; Forloni, G.; Schettini, G. Intracellular calcium rise through L-type calcium channels, as molecular mechanism for prion protein fragment 106-126-induced astroglial proliferation. Biochem. Biophys. Res. Commun. 1996, 228, 397–405. [Google Scholar] [CrossRef]
- Forloni, G.; Angeretti, N.; Chiesa, R.; Monzani, E.; Salmona, M.; Bugiani, O.; Tagliavini, F. Neurotoxicity of a prion protein fragment. Nature 1993, 362, 543–546. [Google Scholar] [CrossRef]
- Villa, V.; Thellung, S.; Corsaro, A.; Novelli, F.; Tasso, B.; Colucci-D’Amato, L.; Gatta, E.; Tonelli, M.; Florio, T. Celecoxib Inhibits Prion Protein 90-231-Mediated Pro-inflammatory Responses in Microglial Cells. Mol. Neurobiol. 2016, 53, 57–72. [Google Scholar] [CrossRef]
- Thellung, S.; Gatta, E.; Pellistri, F.; Villa, V.; Corsaro, A.; Nizzari, M.; Robello, M.; Florio, T. Different Molecular Mechanisms Mediate Direct or Glia-Dependent Prion Protein Fragment 90-231 Neurotoxic Effects in Cerebellar Granule Neurons. Neurotox Res. 2017, 32, 381–397. [Google Scholar] [CrossRef]
- Corsaro, A.; Thellung, S.; Chiovitti, K.; Villa, V.; Simi, A.; Raggi, F.; Paludi, D.; Russo, C.; Aceto, A.; Florio, T. Dual modulation of ERK1/2 and p38 MAP kinase activities induced by minocycline reverses the neurotoxic effects of the prion protein fragment 90-231. Neurotox Res. 2009, 15, 138–154. [Google Scholar] [CrossRef]
- Thellung, S.; Villa, V.; Corsaro, A.; Arena, S.; Millo, E.; Damonte, G.; Benatti, U.; Tagliavini, F.; Florio, T.; Schettini, G. p38 MAP kinase mediates the cell death induced by PrP106-126 in the SH-SY5Y neuroblastoma cells. Neurobiol. Dis. 2002, 9, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Villa, V.; Thellung, S.; Bajetto, A.; Gatta, E.; Robello, M.; Novelli, F.; Tasso, B.; Tonelli, M.; Florio, T. Novel celecoxib analogues inhibit glial production of prostaglandin E2, nitric oxide, and oxygen radicals reverting the neuroinflammatory responses induced by misfolded prion protein fragment 90-231 or lipopolysaccharide. Pharmacol. Res. 2016, 113, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Thellung, S.; Villa, V.; Corsaro, A.; Pellistri, F.; Venezia, V.; Russo, C.; Aceto, A.; Robello, M.; Florio, T. ERK1/2 and p38 MAP kinases control prion protein fragment 90-231-induced astrocyte proliferation and microglia activation. Glia 2007, 55, 1469–1485. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Wu, B.; Le, N.T.T.; Imberdis, T.; Mercer, R.C.C.; Harris, D.A. Prions activate a p38 MAPK synaptotoxic signaling pathway. PLoS Pathog. 2018, 14, e1007283. [Google Scholar] [CrossRef] [PubMed]
- Didonna, A.; Legname, G. Aberrant ERK 1/2 complex activation and localization in scrapie-infected GT1-1 cells. Mol. Neurodegener 2010, 5, 29. [Google Scholar] [CrossRef]
- Nordstrom, E.; Fisone, G.; Kristensson, K. Opposing effects of ERK and p38-JNK MAP kinase pathways on formation of prions in GT1-1 cells. FASEB J. 2009, 23, 613–622. [Google Scholar] [CrossRef]
- Manson, J.C.; Clarke, A.R.; Hooper, M.L.; Aitchison, L.; McConnell, I.; Hope, J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 1994, 8, 121–127. [Google Scholar] [CrossRef]
- Mobley, W.C.; Neve, R.L.; Prusiner, S.B.; McKinley, M.P. Nerve growth factor increases mRNA levels for the prion protein and the beta-amyloid protein precursor in developing hamster brain. Proc. Natl. Acad. Sci. USA 1988, 85, 9811–9815. [Google Scholar] [CrossRef]
- Horiuchi, M.; Yamazaki, N.; Ikeda, T.; Ishiguro, N.; Shinagawa, M. A cellular form of prion protein (PrPC) exists in many non-neuronal tissues of sheep. J. Gen. Virol. 1995, 76, 2583–2587. [Google Scholar] [CrossRef]
- Dodelet, V.C.; Cashman, N.R. Prion protein expression in human leukocyte differentiation. Blood 1998, 91, 1556–1561. [Google Scholar] [CrossRef]
- Lee, Y.J.; Baskakov, I.V. The cellular form of the prion protein guides the differentiation of human embryonic stem cells into neuron-, oligodendrocyte-, and astrocyte-committed lineages. Prion 2014, 8, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Peralta, O.A.; Huckle, W.R.; Eyestone, W.H. Expression and knockdown of cellular prion protein (PrPC) in differentiating mouse embryonic stem cells. Differentiation 2011, 81, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Tabar, V.; Panagiotakos, G.; Greenberg, E.D.; Chan, B.K.; Sadelain, M.; Gutin, P.H.; Studer, L. Migration and differentiation of neural precursors derived from human embryonic stem cells in the rat brain. Nat. Biotechnol. 2005, 23, 601–606. [Google Scholar] [CrossRef]
- Bove-Fenderson, E.; Urano, R.; Straub, J.E.; Harris, D.A. Cellular prion protein targets amyloid-beta fibril ends via its C-terminal domain to prevent elongation. J. Biol. Chem. 2017, 292, 16858–16871. [Google Scholar] [CrossRef]
- De Cecco, E.; Legname, G. The role of the prion protein in the internalization of alpha-synuclein amyloids. Prion 2018, 12, 23–27. [Google Scholar] [CrossRef]
- Martins, V.R.; Beraldo, F.H.; Hajj, G.N.; Lopes, M.H.; Lee, K.S.; Prado, M.A.; Linden, R. Prion protein: Orchestrating neurotrophic activities. Curr. Issues Mol. Biol. 2010, 12, 63–86. [Google Scholar]
- Nieznanski, K. Interactions of prion protein with intracellular proteins: So many partners and no consequences? Cell. Mol. Neurobiol. 2010, 30, 653–666. [Google Scholar] [CrossRef]
- Gardiner, N.J. Integrins and the extracellular matrix: Key mediators of development and regeneration of the sensory nervous system. Dev. Neurobiol. 2011, 71, 1054–1072. [Google Scholar] [CrossRef]
- Mercurio, A.M. Laminin receptors: Achieving specificity through cooperation. Trends. Cell Biol. 1995, 5, 419–423. [Google Scholar] [CrossRef]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Forlenza, O.V.; Cabral, A.L.; Veiga, S.S.; Juliano, M.A.; Roesler, R.; Walz, R.; Minetti, A.; et al. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. Mol. Brain Res. 2000, 76, 85–92. [Google Scholar] [CrossRef]
- Loubet, D.; Dakowski, C.; Pietri, M.; Pradines, E.; Bernard, S.; Callebert, J.; Ardila-Osorio, H.; Mouillet-Richard, S.; Launay, J.M.; Kellermann, O.; et al. Neuritogenesis: The prion protein controls beta1 integrin signaling activity. FASEB J. 2012, 26, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Martins, V.R.; Jay, D.G.; Brentani, R.R. Laminin-induced PC-12 cell differentiation is inhibited following laser inactivation of cellular prion protein. FEBS Lett. 2000, 482, 257–260. [Google Scholar] [CrossRef]
- Hajj, G.N.; Lopes, M.H.; Mercadante, A.F.; Veiga, S.S.; da Silveira, R.B.; Santos, T.G.; Ribeiro, K.C.; Juliano, M.A.; Jacchieri, S.G.; Zanata, S.M.; et al. Cellular prion protein interaction with vitronectin supports axonal growth and is compensated by integrins. J. Cell Sci. 2007, 120, 1915–1926. [Google Scholar] [CrossRef] [PubMed]
- Hundt, C.; Peyrin, J.M.; Haik, S.; Gauczynski, S.; Leucht, C.; Rieger, R.; Riley, M.L.; Deslys, J.P.; Dormont, D.; Lasmezas, C.I.; et al. Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. Embo. J. 2001, 20, 5876–5886. [Google Scholar] [CrossRef]
- Rieger, R.; Edenhofer, F.; Lasmezas, C.I.; Weiss, S. The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat. Med. 1997, 3, 1383–1388. [Google Scholar] [CrossRef]
- Gauczynski, S.; Peyrin, J.M.; Haik, S.; Leucht, C.; Hundt, C.; Rieger, R.; Krasemann, S.; Deslys, J.P.; Dormont, D.; Lasmezas, C.I.; et al. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. Embo. J. 2001, 20, 5863–5875. [Google Scholar] [CrossRef]
- Crossin, K.L.; Krushel, L.A. Cellular signaling by neural cell adhesion molecules of the immunoglobulin superfamily. Dev. Dyn. 2000, 218, 260–279. [Google Scholar] [CrossRef]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225. [Google Scholar] [CrossRef]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef]
- Slapsak, U.; Salzano, G.; Amin, L.; Abskharon, R.N.; Ilc, G.; Zupancic, B.; Biljan, I.; Plavec, J.; Giachin, G.; Legname, G. The N Terminus of the Prion Protein Mediates Functional Interactions with the Neuronal Cell Adhesion Molecule (NCAM) Fibronectin Domain. J. Biol. Chem. 2016, 291, 21857–21868. [Google Scholar] [CrossRef]
- Mehrabian, M.; Brethour, D.; Wang, H.; Xi, Z.; Rogaeva, E.; Schmitt-Ulms, G. The Prion Protein Controls Polysialylation of Neural Cell Adhesion Molecule 1 during Cellular Morphogenesis. PLoS ONE 2015, 10, e0133741. [Google Scholar] [CrossRef] [PubMed]
- Prodromidou, K.; Papastefanaki, F.; Sklaviadis, T.; Matsas, R. Functional cross-talk between the cellular prion protein and the neural cell adhesion molecule is critical for neuronal differentiation of neural stem/precursor cells. Stem Cells 2014, 32, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.L.; Lehmann, S.; Launay, J.M.; Kellermann, O. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.; Cabral, A.L.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. Embo. J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef]
- Landemberger, M.C.; de Oliveira, G.P.; Machado, C.F.; Gollob, K.J.; Martins, V.R. Loss of STI1-mediated neuronal survival and differentiation in disease-associated mutations of prion protein. J. Neurochem. 2018, 145, 409–416. [Google Scholar] [CrossRef]
- Chiarini, L.B.; Freitas, A.R.; Zanata, S.M.; Brentani, R.R.; Martins, V.R.; Linden, R. Cellular prion protein transduces neuroprotective signals. Embo. J. 2002, 21, 3317–3326. [Google Scholar] [CrossRef]
- Lopes, M.H.; Hajj, G.N.; Muras, A.G.; Mancini, G.L.; Castro, R.M.; Ribeiro, K.C.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef]
- Roffe, M.; Beraldo, F.H.; Bester, R.; Nunziante, M.; Bach, C.; Mancini, G.; Gilch, S.; Vorberg, I.; Castilho, B.A.; Martins, V.R.; et al. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. USA 2010, 107, 13147–13152. [Google Scholar] [CrossRef]
- Santos, T.G.; Silva, I.R.; Costa-Silva, B.; Lepique, A.P.; Martins, V.R.; Lopes, M.H. Enhanced neural progenitor/stem cells self-renewal via the interaction of stress-inducible protein 1 with the prion protein. Stem Cells 2011, 29, 1126–1136. [Google Scholar] [CrossRef]
- Irvin, D.K.; Zurcher, S.D.; Nguyen, T.; Weinmaster, G.; Kornblum, H.I. Expression patterns of Notch1, Notch2, and Notch3 suggest multiple functional roles for the Notch-DSL signaling system during brain development. J. Comp. Neurol. 2001, 436, 167–181. [Google Scholar] [CrossRef]
- Lutolf, S.; Radtke, F.; Aguet, M.; Suter, U.; Taylor, V. Notch1 is required for neuronal and glial differentiation in the cerebellum. Development 2002, 129, 373–385. [Google Scholar] [PubMed]
- Martin-Lanneree, S.; Halliez, S.; Hirsch, T.Z.; Hernandez-Rapp, J.; Passet, B.; Tomkiewicz, C.; Villa-Diaz, A.; Torres, J.M.; Launay, J.M.; Beringue, V.; et al. The Cellular Prion Protein Controls Notch Signaling in Neural Stem/Progenitor Cells. Stem Cells 2017, 35, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; McGovern, G.; Goodsir, C.M.; Brown, K.L.; Bruce, M.E. Sites of prion protein accumulation in scrapie-infected mouse spleen revealed by immuno-electron microscopy. J. Pathol. 2000, 191, 323–332. [Google Scholar] [CrossRef]
- Kitamoto, T.; Muramoto, T.; Mohri, S.; Doh-Ura, K.; Tateishi, J. Abnormal isoform of prion protein accumulates in follicular dendritic cells in mice with Creutzfeldt-Jakob disease. J. Virol. 1991, 65, 6292–6295. [Google Scholar]
- Brown, K.L.; Stewart, K.; Ritchie, D.L.; Mabbott, N.A.; Williams, A.; Fraser, H.; Morrison, W.I.; Bruce, M.E. Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat. Med. 1999, 5, 1308–1312. [Google Scholar] [CrossRef]
- O’Rourke, K.I.; Huff, T.P.; Leathers, C.W.; Robinson, M.M.; Gorham, J.R. SCID mouse spleen does not support scrapie agent replication. J. Gen. Virol. 1994, 75, 1511–1514. [Google Scholar] [CrossRef]
- Fraser, H.; Brown, K.L.; Stewart, K.; McConnell, I.; McBride, P.; Williams, A. Replication of scrapie in spleens of SCID mice follows reconstitution with wild-type mouse bone marrow. J. Gen. Virol. 1996, 77, 1935–1940. [Google Scholar] [CrossRef]
- Montrasio, F.; Frigg, R.; Glatzel, M.; Klein, M.A.; Mackay, F.; Aguzzi, A.; Weissmann, C. Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 2000, 288, 1257–1259. [Google Scholar] [CrossRef]
- Zivny, J.H.; Gelderman, M.P.; Xu, F.; Piper, J.; Holada, K.; Simak, J.; Vostal, J.G. Reduced erythroid cell and erythropoietin production in response to acute anemia in prion protein-deficient (Prnp-/-) mice. Blood Cells Mol. Dis. 2008, 40, 302–307. [Google Scholar] [CrossRef]
- Ballerini, C.; Gourdain, P.; Bachy, V.; Blanchard, N.; Levavasseur, E.; Gregoire, S.; Fontes, P.; Aucouturier, P.; Hivroz, C.; Carnaud, C. Functional implication of cellular prion protein in antigen-driven interactions between T cells and dendritic cells. J. Immunol. 2006, 176, 7254–7262. [Google Scholar] [CrossRef]
- de Almeida, C.J.; Chiarini, L.B.; da Silva, J.P.; PM, E.S.; Martins, M.A.; Linden, R. The cellular prion protein modulates phagocytosis and inflammatory response. J. Leukoc. Biol. 2005, 77, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Siberchicot, C.; Gault, N.; Dechamps, N.; Barroca, V.; Aguzzi, A.; Romeo, P.H.; Radicella, J.P.; Bravard, A.; Bernardino-Sgherri, J. Prion protein deficiency impairs hematopoietic stem cells determination and sensitizes myeloid progenitors to irradiation. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Guzman, M.E.; Bigoni-Ordonez, G.D.; Ibanez Hernandez, M.; Ortiz-Sanchez, E. Cancer stem cell impact on clinical oncology. World, J. Stem Cells 2018, 10, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Wurth, R.; Barbieri, F.; Pattarozzi, A.; Gaudenzi, G.; Gatto, F.; Fiaschi, P.; Ravetti, J.L.; Zona, G.; Daga, A.; Persani, L.; et al. Phenotypical and Pharmacological Characterization of Stem-Like Cells in Human Pituitary Adenomas. Mol. Neurobiol. 2017, 54, 4879–4895. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Yuan, X.; Curtin, J.; Xiong, Y.; Liu, G.; Waschsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 2004, 23, 9392–9400. [Google Scholar] [CrossRef]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- D’Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B.M. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers 2019, 11, 469. [Google Scholar] [CrossRef]
- Perus, L.J.M.; Walsh, L.A. Microenvironmental Heterogeneity in Brain Malignancies. Front. Immunol. 2019, 10, 2294. [Google Scholar] [CrossRef]
- Wurth, R.; Bajetto, A.; Harrison, J.K.; Barbieri, F.; Florio, T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front. Cell. Neurosci. 2014, 8, 144. [Google Scholar]
- Aderetti, D.A.; Hira, V.V.V.; Molenaar, R.J.; van Noorden, C.J.F. The hypoxic peri-arteriolar glioma stem cell niche, an integrated concept of five types of niches in human glioblastoma. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Filatova, A.; Acker, T.; Garvalov, B.K. The cancer stem cell niche(s): The crosstalk between glioma stem cells and their microenvironment. Biochim. Biophys. Acta 2013, 1830, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; You, H.; Liu, F.; An, H.; Shi, Y.; Yu, Q.; Fan, D. Differentially expressed gene profiles between multidrug resistant gastric adenocarcinoma cells and their parental cells. Cancer Lett. 2002, 185, 211–218. [Google Scholar] [CrossRef]
- Diarra-Mehrpour, M.; Arrabal, S.; Jalil, A.; Pinson, X.; Gaudin, C.; Pietu, G.; Pitaval, A.; Ripoche, H.; Eloit, M.; Dormont, D.; et al. Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res. 2004, 64, 719–727. [Google Scholar] [CrossRef]
- Li, Q.Q.; Cao, X.X.; Xu, J.D.; Chen, Q.; Wang, W.J.; Tang, F.; Chen, Z.Q.; Liu, X.P.; Xu, Z.D. The role of P-glycoprotein/cellular prion protein interaction in multidrug-resistant breast cancer cells treated with paclitaxel. Cell Mol. Life Sci. 2009, 66, 504–515. [Google Scholar] [CrossRef]
- Comincini, S.; Facoetti, A.; Del Vecchio, I.; Peoc’h, K.; Laplanche, J.L.; Magrassi, L.; Ceroni, M.; Ferretti, L.; Nano, R. Differential expression of the prion-like protein doppel gene (PRND) in astrocytomas: A new molecular marker potentially involved in tumor progression. Anticancer Res. 2004, 24, 1507–1517. [Google Scholar]
- Iglesia, R.P.; Prado, M.B.; Cruz, L.; Martins, V.R.; Santos, T.G.; Lopes, M.H. Engagement of cellular prion protein with the co-chaperone Hsp70/90 organizing protein regulates the proliferation of glioblastoma stem-like cells. Stem Cell Res. 2017, 8, 76. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Kakeya, T.; Yamazaki, T.; Takekida, K.; Nakamura, N.; Matsuda, H.; Takatori, K.; Tanimura, A.; Tanamoto, K.; Sawada, J. G1-dependent prion protein expression in human glioblastoma cell line T98G. Biol. Pharm. Bull. 2002, 25, 728–733. [Google Scholar] [CrossRef][Green Version]
- Hinton, C.; Antony, H.; Hashimi, S.M.; Munn, A.; Wei, M.Q. Significance of prion and prion-like proteins in cancer development, progression and multi-drug resistance. Curr. Cancer Drug Targets 2013, 13, 895–904. [Google Scholar] [CrossRef]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar]
- Johannessen, T.C.; Bjerkvig, R.; Tysnes, B.B. DNA repair and cancer stem-like cells--potential partners in glioma drug resistance? Cancer Treat. Rev. 2008, 34, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Cheema, S.K.; Mishra, S.K.; Rangnekar, V.M.; Tari, A.M.; Kumar, R.; Lopez-Berestein, G. Par-4 transcriptionally regulates Bcl-2 through a WT1-binding site on the bcl-2 promoter. J. Biol. Chem. 2003, 278, 19995–20005. [Google Scholar] [CrossRef] [PubMed]
- Huet, S.; Groux, H.; Caillou, B.; Valentin, H.; Prieur, A.M.; Bernard, A. CD44 contributes to T cell activation. J. Immunol 1989, 143, 798–801. [Google Scholar] [PubMed]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 cell adhesion molecules. Mol. Pathol. 1999, 52, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Ishi, Y.; Motegi, H.; Okamoto, M.; Ou, Y.; Chen, J.; Yamaguchi, S. Overexpression of CD44 is associated with a poor prognosis in grade II/III gliomas. J. Neurooncol. 2019, 145, 201–210. [Google Scholar] [CrossRef]
- Dong, Q.; Li, Q.; Wang, M.; Hu, J.; Dai, J.; Niu, L.; Yuan, G.; Pan, Y. Elevated CD44 expression predicts poor prognosis in patients with low-grade glioma. Oncol. Lett. 2019, 18, 3698–3704. [Google Scholar] [CrossRef]
- Guadagno, E.; Borrelli, G.; Califano, M.; Cali, G.; Solari, D.; Del Basso De Caro, M. Immunohistochemical expression of stem cell markers CD44 and nestin in glioblastomas: Evaluation of their prognostic significance. Pathol. Res. Pr. 2016, 212, 825–832. [Google Scholar] [CrossRef]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 2018, 5387041. [Google Scholar] [CrossRef]
- Johansson, E.; Grassi, E.S.; Pantazopoulou, V.; Tong, B.; Lindgren, D.; Berg, T.J.; Pietras, E.J.; Axelson, H.; Pietras, A. CD44 Interacts with HIF-2alpha to Modulate the Hypoxic Phenotype of Perinecrotic and Perivascular Glioma Cells. Cell Rep. 2017, 20, 1641–1653. [Google Scholar] [CrossRef]
- Wang, H.H.; Liao, C.C.; Chow, N.H.; Huang, L.L.; Chuang, J.I.; Wei, K.C.; Shin, J.W. Whether CD44 is an applicable marker for glioma stem cells. Am. J. Transl. Res. 2017, 9, 4785–4806. [Google Scholar]
- Johnson, B.D.; Schumacher, R.J.; Ross, E.D.; Toft, D.O. Hop modulates Hsp70/Hsp90 interactions in protein folding. J. Biol. Chem. 1998, 273, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Kravats, A.N.; Hoskins, J.R.; Reidy, M.; Johnson, J.L.; Doyle, S.M.; Genest, O.; Masison, D.C.; Wickner, S. Functional and physical interaction between yeast Hsp90 and Hsp70. Proc. Natl. Acad. Sci. USA 2018, 115, E2210–E2219. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.H.; Santos, T.G.; Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Cunha, I.W.; Wasilewska-Sampaio, A.P.; Costa-Silva, B.; Marchi, F.A.; Bleggi-Torres, L.F.; Sanematsu, P.I.; et al. Disruption of prion protein-HOP engagement impairs glioblastoma growth and cognitive decline and improves overall survival. Oncogene 2015, 34, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Baindur-Hudson, S.; Edkins, A.L.; Blatch, G.L. Hsp70/Hsp90 organising protein (hop): Beyond interactions with chaperones and prion proteins. Subcell Biochem. 2015, 78, 69–90. [Google Scholar]
- Erlich, R.B.; Kahn, S.A.; Lima, F.R.; Muras, A.G.; Martins, R.A.; Linden, R.; Chiarini, L.B.; Martins, V.R.; Moura Neto, V. STI1 promotes glioma proliferation through MAPK and PI3K pathways. Glia 2007, 55, 1690–1698. [Google Scholar] [CrossRef]
- Fonseca, A.C.; Romao, L.; Amaral, R.F.; Assad Kahn, S.; Lobo, D.; Martins, S.; Marcondes de Souza, J.; Moura-Neto, V.; Lima, F.R. Microglial stress inducible protein 1 promotes proliferation and migration in human glioblastoma cells. Neuroscience 2012, 200, 130–141. [Google Scholar] [CrossRef]
- Lima, F.R.; Arantes, C.P.; Muras, A.G.; Nomizo, R.; Brentani, R.R.; Martins, V.R. Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J. Neurochem. 2007, 103, 2164–2176. [Google Scholar] [CrossRef]
- Carvalho da Fonseca, A.C.; Wang, H.; Fan, H.; Chen, X.; Zhang, I.; Zhang, L.; Lima, F.R.; Badie, B. Increased expression of stress inducible protein 1 in glioma-associated microglia/macrophages. J. Neuroimmunol. 2014, 274, 71–77. [Google Scholar] [CrossRef]
- Susantad, T.; Smith, D.R. siRNA-mediated silencing of the 37/67-kDa high affinity laminin receptor in Hep3B cells induces apoptosis. Cell Mol. Biol. Lett. 2008, 13, 452–464. [Google Scholar] [CrossRef]
- Wu, H.; Li, J.; Xu, D.; Jv, D.; Meng, X.; Qiao, P.; Cui, T.; Shi, B. The 37-kDa laminin receptor precursor regulates the malignancy of human glioma cells. Cell Biochem. Funct. 2016, 34, 516–521. [Google Scholar] [CrossRef]
- Yin, S.M.; Sy, M.S.; Yang, H.Y.; Tien, P. Interaction of Doppel with the full-length laminin receptor precursor protein. Arch. Biochem. Biophys. 2004, 428, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Wang, W.; Wu, Q.; Lu, Y.; Su, T.; Gu, N.; Li, K.; Wang, J.; Du, R.; Zhao, X.; et al. MGr1-Antigen/37 kDa laminin receptor precursor promotes cellular prion protein induced multi-drug-resistance of gastric cancer. Oncotarget 2017, 8, 71630–71641. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Imayoshi, I.; Sakamoto, M.; Yamaguchi, M.; Mori, K.; Kageyama, R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J. Neurosci. 2010, 30, 3489–3498. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Purow, B.W.; Haque, R.M.; Noel, M.W.; Su, Q.; Burdick, M.J.; Lee, J.; Sundaresan, T.; Pastorino, S.; Park, J.K.; Mikolaenko, I.; et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005, 65, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hou, H.; Li, M.; Yang, Y.; Sun, L. Anticancer effect of eupatilin on glioma cells through inhibition of the Notch-1 signaling pathway. Mol. Med. Rep. 2016, 13, 1141–1146. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, S.; Huang, D.; Cui, M.; Hu, H.; Zhang, L.; Wang, W.; Parameswaran, N.; Jackson, M.; Osborne, B.; et al. Cellular Prion Protein Mediates Pancreatic Cancer Cell Survival and Invasion through Association with and Enhanced Signaling of Notch1. Am. J. Pathol. 2016, 186, 2945–2956. [Google Scholar] [CrossRef][Green Version]
- Zhang, X.P.; Zheng, G.; Zou, L.; Liu, H.L.; Hou, L.H.; Zhou, P.; Yin, D.D.; Zheng, Q.J.; Liang, L.; Zhang, S.Z.; et al. Notch activation promotes cell proliferation and the formation of neural stem cell-like colonies in human glioma cells. Mol. Cell Biochem. 2008, 307, 101–108. [Google Scholar] [CrossRef]
- Han, N.; Hu, G.; Shi, L.; Long, G.; Yang, L.; Xi, Q.; Guo, Q.; Wang, J.; Dong, Z.; Zhang, M. Notch1 ablation radiosensitizes glioblastoma cells. Oncotarget 2017, 8, 88059–88068. [Google Scholar] [CrossRef]
- Kanamori, M.; Kawaguchi, T.; Nigro, J.M.; Feuerstein, B.G.; Berger, M.S.; Miele, L.; Pieper, R.O. Contribution of Notch signaling activation to human glioblastoma multiforme. J. Neurosurg. 2007, 106, 417–427. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Matsui, W.; Khaki, L.; Stearns, D.; Chun, J.; Li, Y.M.; Eberhart, C.G. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006, 66, 7445–7452. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, K.; Nedergaard, M.K.; Villingshoj, M.; Borup, R.; Broholm, H.; Kjaer, A.; Poulsen, H.S.; Stockhausen, M.T. Inhibition of Notch signaling alters the phenotype of orthotopic tumors formed from glioblastoma multiforme neurosphere cells but does not hamper intracranial tumor growth regardless of endogene Notch pathway signature. Cancer Biol. 2014, 15, 862–877. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, K.; Villingshoj, M.; Poulsen, H.S.; Stockhausen, M.T. Level of Notch activation determines the effect on growth and stem cell-like features in glioblastoma multiforme neurosphere cultures. Cancer Biol. 2013, 14, 625–637. [Google Scholar] [CrossRef]
- Hirsch, T.Z.; Martin-Lanneree, S.; Reine, F.; Hernandez-Rapp, J.; Herzog, L.; Dron, M.; Privat, N.; Passet, B.; Halliez, S.; Villa-Diaz, A.; et al. Epigenetic Control of the Notch and Eph Signaling Pathways by the Prion Protein: Implications for Prion Diseases. Mol. Neurobiol. 2019, 56, 2159–2173. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Varnat, F.; Zacchetti, G.; Ruiz i Altaba, A. Hedgehog pathway activity is required for the lethality and intestinal phenotypes of mice with hyperactive Wnt signaling. Mech. Dev. 2010, 127, 73–81. [Google Scholar] [CrossRef]
- He, X.C.; Zhang, J.; Tong, W.G.; Tawfik, O.; Ross, J.; Scoville, D.H.; Tian, Q.; Zeng, X.; He, X.; Wiedemann, L.M.; et al. signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat. Genet. 2004, 36, 1117–1121. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt signaling. Cold Spring Harb Perspect Biol. 2012, 4, a011163. [Google Scholar] [CrossRef] [PubMed]
- Tompa, M.; Kalovits, F.; Nagy, A.; Kalman, B. Contribution of the Wnt Pathway to Defining Biology of Glioblastoma. Neuromol. Med. 2018, 20, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Besnier, L.S.; Cardot, P.; Da Rocha, B.; Simon, A.; Loew, D.; Klein, C.; Riveau, B.; Lacasa, M.; Clair, C.; Rousset, M.; et al. The cellular prion protein PrPc is a partner of the Wnt pathway in intestinal epithelial cells. Mol. Biol. Cell 2015, 26, 3313–3328. [Google Scholar] [CrossRef] [PubMed]
- Rousset, M.; Leturque, A.; Thenet, S. The nucleo-junctional interplay of the cellular prion protein: A new partner in cancer-related signaling pathways? Prion 2016, 10, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, T.; Fisher, S.; Olofsson, S.; Endo, T.; Groth, D.; Tarentino, A.; Borchelt, D.R.; Teplow, D.; Hood, L.; Burlingame, A.; et al. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch. Biochem. Biophys. 1989, 274, 1–13. [Google Scholar] [CrossRef]
- Li, C.; Yu, S.; Nakamura, F.; Pentikainen, O.T.; Singh, N.; Yin, S.; Xin, W.; Sy, M.S. Pro-prion binds filamin A, facilitating its interaction with integrin beta1, and contributes to melanomagenesis. J. Biol. Chem. 2010, 285, 30328–30339. [Google Scholar] [CrossRef]
- Li, C.; Yu, S.; Nakamura, F.; Yin, S.; Xu, J.; Petrolla, A.A.; Singh, N.; Tartakoff, A.; Abbott, D.W.; Xin, W.; et al. Binding of pro-prion to filamin A disrupts cytoskeleton and correlates with poor prognosis in pancreatic cancer. J. Clin. Investig. 2009, 119, 2725–2736. [Google Scholar] [CrossRef]
- Li, C.; Xin, W.; Sy, M.S. Binding of pro-prion to filamin A: By design or an unfortunate blunder. Oncogene 2010, 29, 5329–5345. [Google Scholar] [CrossRef][Green Version]
- Kim, H.; McCulloch, C.A. Filamin A mediates interactions between cytoskeletal proteins that control cell adhesion. FEBS Lett. 2011, 585, 18–22. [Google Scholar] [CrossRef]
- Sy, M.S.; Li, C.; Yu, S.; Xin, W. The fatal attraction between pro-prion and filamin A: Prion as a marker in human cancers. Biomark Med. 2010, 4, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gao, Z.; Hu, L.; Wu, G.; Yang, X.; Zhang, L.; Zhu, Y.; Wong, B.S.; Xin, W.; Sy, M.S.; et al. Glycosylphosphatidylinositol Anchor Modification Machinery Deficiency Is Responsible for the Formation of Pro-Prion Protein (PrP) in BxPC-3 Protein and Increases Cancer Cell Motility. J. Biol. Chem. 2016, 291, 3905–3917. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Wang, W.; Xie, Z.; Wong, B.S.; Li, R.; Petersen, R.B.; Sy, M.S.; Chen, S.G. Expression and structural characterization of the recombinant human doppel protein. Biochemistry 2000, 39, 13575–13583. [Google Scholar] [CrossRef] [PubMed]
- Silverman, G.L.; Qin, K.; Moore, R.C.; Yang, Y.; Mastrangelo, P.; Tremblay, P.; Prusiner, S.B.; Cohen, F.E.; Westaway, D. Doppel is an N-glycosylated, glycosylphosphatidylinositol-anchored protein. Expression in testis and ectopic production in the brains of Prnp(0/0) mice predisposed to Purkinje cell loss. J. Biol. Chem. 2000, 275, 26834–26841. [Google Scholar] [PubMed]
- Behrens, A.; Genoud, N.; Naumann, H.; Rulicke, T.; Janett, F.; Heppner, F.L.; Ledermann, B.; Aguzzi, A. Absence of the prion protein homologue Doppel causes male sterility. Embo. J. 2002, 21, 3652–3658. [Google Scholar] [CrossRef]
- Moore, R.C.; Lee, I.Y.; Silverman, G.L.; Harrison, P.M.; Strome, R.; Heinrich, C.; Karunaratne, A.; Pasternak, S.H.; Chishti, M.A.; Liang, Y.; et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J. Mol. Biol. 1999, 292, 797–817. [Google Scholar] [CrossRef]
- Al-Hilal, T.A.; Chung, S.W.; Choi, J.U.; Alam, F.; Park, J.; Kim, S.W.; Kim, S.Y.; Ahsan, F.; Kim, I.S.; Byun, Y. Targeting prion-like protein doppel selectively suppresses tumor angiogenesis. J. Clin. Investig. 2016, 126, 1251–1266. [Google Scholar] [CrossRef]
- Travaglino, E.; Comincini, S.; Benatti, C.; Azzalin, A.; Nano, R.; Rosti, V.; Ferretti, L.; Invernizzi, R. Overexpression of the Doppel protein in acute myeloid leukaemias and myelodysplastic syndromes. Br. J. Haematol. 2005, 128, 877–884. [Google Scholar] [CrossRef]
- Comincini, S.; Ferrara, V.; Arias, A.; Malovini, A.; Azzalin, A.; Ferretti, L.; Benericetti, E.; Cardarelli, M.; Gerosa, M.; Passarin, M.G.; et al. Diagnostic value of PRND gene expression profiles in astrocytomas: Relationship to tumor grades of malignancy. Oncol. Rep. 2007, 17, 989–996. [Google Scholar] [CrossRef]
- Azzalin, A.; Sbalchiero, E.; Barbieri, G.; Palumbo, S.; Muzzini, C.; Comincini, S. The doppel (Dpl) protein influences in vitro migration capability in astrocytoma-derived cells. Cell Oncol. 2008, 30, 491–501. [Google Scholar]
- Comincini, S.; Chiarelli, L.R.; Zelini, P.; Del Vecchio, I.; Azzalin, A.; Arias, A.; Ferrara, V.; Rognoni, P.; Dipoto, A.; Nano, R.; et al. Nuclear mRNA retention and aberrant doppel protein expression in human astrocytic tumor cells. Oncol. Rep. 2006, 16, 1325–1332. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sbalchiero, E.; Azzalin, A.; Palumbo, S.; Barbieri, G.; Arias, A.; Simonelli, L.; Ferretti, L.; Comincini, S. Altered cellular distribution and sub-cellular sorting of doppel (Dpl) protein in human astrocytoma cell lines. Cell Oncol. 2008, 30, 337–347. [Google Scholar] [PubMed]
- Rognoni, P.; Chiarelli, L.R.; Comincini, S.; Azzalin, A.; Miracco, C.; Valentini, G. Biochemical signatures of doppel protein in human astrocytomas to support prediction in tumor malignancy. J. Biomed. Biotechnol. 2010, 2010, 301067. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thellung, S.; Corsaro, A.; Bosio, A.G.; Zambito, M.; Barbieri, F.; Mazzanti, M.; Florio, T. Emerging Role of Cellular Prion Protein in the Maintenance and Expansion of Glioma Stem Cells. Cells 2019, 8, 1458. https://doi.org/10.3390/cells8111458
Thellung S, Corsaro A, Bosio AG, Zambito M, Barbieri F, Mazzanti M, Florio T. Emerging Role of Cellular Prion Protein in the Maintenance and Expansion of Glioma Stem Cells. Cells. 2019; 8(11):1458. https://doi.org/10.3390/cells8111458
Chicago/Turabian StyleThellung, Stefano, Alessandro Corsaro, Alessia G. Bosio, Martina Zambito, Federica Barbieri, Michele Mazzanti, and Tullio Florio. 2019. "Emerging Role of Cellular Prion Protein in the Maintenance and Expansion of Glioma Stem Cells" Cells 8, no. 11: 1458. https://doi.org/10.3390/cells8111458
APA StyleThellung, S., Corsaro, A., Bosio, A. G., Zambito, M., Barbieri, F., Mazzanti, M., & Florio, T. (2019). Emerging Role of Cellular Prion Protein in the Maintenance and Expansion of Glioma Stem Cells. Cells, 8(11), 1458. https://doi.org/10.3390/cells8111458