Novel Epigenetic Biomarkers in Pregnancy-Related Disorders and Cancers

Abstract
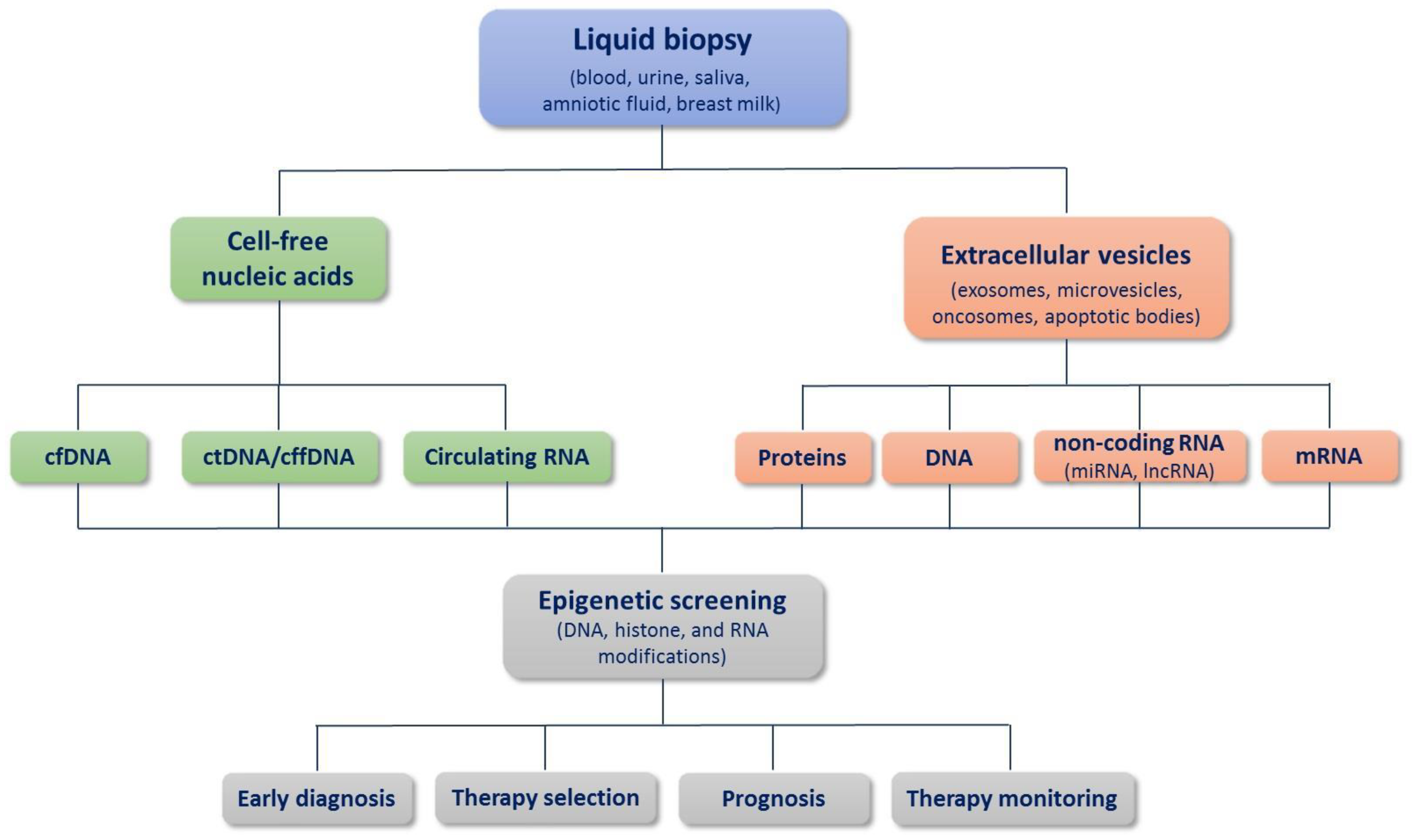
1. Introduction
2. Circulating Cell-Free DNA
2.1. Circulating Cell-Free Tumor DNA
2.2. Epigenetic Alterations in ctDNA
2.3. Circulating Cell-Free Fetal DNA
2.4. Epigenetic Alterations in cffDNA
3. Extracellular Vesicles
3.1. Exosomes
3.1.1. Exosomes in Cancer
3.1.2. Exosomes as Epigenetic Cancer Biomarkers
3.1.3. Placental Exosomes
3.1.4. Placental Exosomes as Epigenetic Biomarkers of Pathological Pregnancies
3.2. Other Extracellular Vesicles and Their Role in Placenta and Cancer
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mandel, P.; Métais, P. Les acides nucléiques du plasma sanguin chez l’homme. CR Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Tan, E.M.; Schur, P.H.; Carr, R.I.; Kunkel, H.G. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J. Clin. Investig. 1966, 45, 1732–1740. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef]
- Sorenson, G.D.; Pribish, D.M.; Valone, F.H.; Memoli, V.A.; Bzik, D.J.; Yao, S.L. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiol. Biomarkers Prev. 1994, 3, 67–71. [Google Scholar]
- Vasioukhin, V.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Stroun, M. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br. J. Haematol. 1994, 86, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Lianidou, E.; Pantel, K. Liquid biopsies. Genes Chromosomes Cancer 2019, 58, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Sisson, B.A.; Uvalic, J.; Kelly, K.; Selvam, P.; Hesse, A.N.; Ananda, G.; Chandok, H.; Bergeron, D.; Holinka, L.; Reddi, H.V. Technical and Regulatory Considerations for Taking Liquid Biopsy to the Clinic: Validation of the JAX PlasmaMonitor(TM) Assay. Biomark. Insights 2019, 14, 1177271919826545. [Google Scholar] [CrossRef]
- Neumann, M.H.D.; Bender, S.; Krahn, T.; Schlange, T. ctDNA and CTCs in Liquid Biopsy-Current Status and Where We Need to Progress. Comput. Struct. Biotechnol. J. 2018, 16, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Page, K.; Shaw, J.A.; Guttery, D.S. The liquid biopsy: Towards standardisation in preparation for prime time. Lancet Oncol. 2019, 20, 758–760. [Google Scholar] [CrossRef]
- Ehrlich, M.; Lacey, M. DNA hypomethylation and hemimethylation in cancer. In Epigenetic Alterations in Oncogenesis; Springer: New York, NY, USA, 2013; pp. 31–56. [Google Scholar]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Romanov, G.A.; Vanyushin, B.F. Methylation of reiterated sequences in mammalian DNAs effects of the tissue type, age, malignancy and hormonal induction. Biochim. Biophys. Acta (BBA)-Nucleic Acids Protein Synth. 1981, 653, 204–218. [Google Scholar] [CrossRef]
- Shinjo, K.; Kondo, Y. Targeting cancer epigenetics: Linking basic biology to clinical medicine. Adv. Drug Deliv. Rev. 2015, 95, 56–64. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Lo, Y.D.; Rainer, T.H.; Chan, L.Y.; Hjelm, N.M.; Cocks, R.A. Plasma DNA as a prognostic marker in trauma patients. Clin. Chem. 2000, 46, 319–323. [Google Scholar]
- Lo, Y.D.; Zhang, J.; Leung, T.N.; Lau, T.K.; Chang, A.M.; Hjelm, N.M. Rapid clearance of fetal DNA from maternal plasma. Am. J. Hum. Genet. 1999, 64, 218–224. [Google Scholar] [CrossRef]
- Zhong, X.Y.; Holzgreve, W.; Hahn, S. The levels of circulatory cell free fetal DNA in maternal plasma are elevated prior to the onset of preeclampsia. Hypertens. Pregnancy 2002, 21, 77–83. [Google Scholar] [CrossRef]
- Wataganara, T.; Bianchi, D.W. Fetal cell-free nucleic acids in the maternal circulation: New clinical applications. Ann. N. Y. Acad. Sci. 2004, 1022, 90–99. [Google Scholar] [CrossRef]
- Leung, T.N.; Zhang, J.; Lau, T.K.; Chan, L.Y.; Lo, Y.D. Increased maternal plasma fetal DNA concentrations in women who eventually develop preeclampsia. Clin. Chem. 2001, 47, 137–139. [Google Scholar]
- Smid, M.; Galbiati, S.; Lojacono, A.; Valsecchi, L.; Platto, C.; Cavoretto, P.; Calza, S.; Ferrari, A.; Ferrari, M.; Cremonesi, L. Correlation of fetal DNA levels in maternal plasma with Doppler status in pathological pregnancies. Prenat. Diagn. 2006, 26, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Alberry, M.S.; Maddocks, D.G.; Hadi, M.A.; Metawi, H.; Hunt, L.P.; Abdel-Fattah, S.A.; Avent, N.D.; Soothill, P.W. Quantification of cell free fetal DNA in maternal plasma in normal pregnancies and in pregnancies with placental dysfunction. Am. J. Obs. Gynecol. 2009, 200. [Google Scholar] [CrossRef] [PubMed]
- Al Nakib, M.; Desbriere, R.; Bonello, N.; Bretelle, F.; Boubli, L.; Gabert, J.; Levy-Mozziconacci, A. Total and fetal cell-free DNA analysis in maternal blood as markers of placental insufficiency in intrauterine growth restriction. Fetal Diagn. Ther. 2009, 26, 24–28. [Google Scholar] [CrossRef]
- Umu, S.U.; Langseth, H.; Bucher-Johannessen, C.; Fromm, B.; Keller, A.; Meese, E.; Lauritzen, M.; Leithaug, M.; Lyle, R.; Rounge, T.B. A comprehensive profile of circulating RNAs in human serum. RNA Biol. 2018, 15, 242–250. [Google Scholar] [CrossRef]
- Fernando, M.R.; Jiang, C.; Krzyzanowski, G.D.; Ryan, W.L. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS ONE 2017, 12, e0183915. [Google Scholar] [CrossRef]
- Armstrong, D.; Wildman, D.E. Extracellular Vesicles and the Promise of Continuous Liquid Biopsies. J. Pathol. Transl. Med. 2018, 52, 1–8. [Google Scholar] [CrossRef]
- Panfoli, I.; Santucci, L.; Bruschi, M.; Petretto, A.; Calzia, D.; Ramenghi, L.A.; Ghiggeri, G.; Candiano, G. Microvesicles as promising biological tools for diagnosis and therapy. Expert Rev. Proteom. 2018, 15, 801–808. [Google Scholar] [CrossRef]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borras, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef]
- Louwen, F.; Muschol-Steinmetz, C.; Reinhard, J.; Reitter, A.; Yuan, J. A lesson for cancer research: Placental microarray gene analysis in preeclampsia. Oncotarget 2012, 3, 759–773. [Google Scholar] [CrossRef]
- Novakovic, B.; Saffery, R. Placental pseudo-malignancy from a DNA methylation perspective: Unanswered questions and future directions. Front. Genet. 2013, 4, 285. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Serman, A.; Simon, F.; Fabijanovic, D.; Serman, L. Epigenetic control of cell invasion-the trophoblast model. Biomol. Concepts 2012, 3, 487–494. [Google Scholar] [CrossRef][Green Version]
- Partl, J.Z.; Fabijanovic, D.; Skrtic, A.; Vranic, S.; Martic, T.N.; Serman, L. Immunohistochemical expression of SFRP1 and SFRP3 proteins in normal and malignant reproductive tissues of rats and humans. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Kreis, N.N.; Friemel, A.; Ritter, A.; Roth, S.; Rolle, U.; Louwen, F.; Yuan, J. Function of p21 (Cip1/Waf1/CDKN1A) in Migration and Invasion of Cancer and Trophoblastic Cells. Cancers 2019, 11, 989. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sui, L.; Qiu, B.; Yin, X.; Liu, J.; Zhang, X. ANXA4 promotes trophoblast invasion via the PI3K/Akt/eNOS pathway in preeclampsia. Am. J. Physiol. Cell Physiol. 2019, 316, C481–C491. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Wang, N.; Lv, H.; Yang, J.; Liu, J.; Li, W.P.; Zhang, C.; Chen, Z.J. The Attenuation of Trophoblast Invasion Caused by the Downregulation of EZH2 Is Involved in the Pathogenesis of Human Recurrent Miscarriage. Mol. Ther. Nucleic Acids 2019, 14, 377–387. [Google Scholar] [CrossRef]
- Xie, D.; Zhu, J.; Liu, Q.; Li, J.; Song, M.; Wang, K.; Zhou, Q.; Jia, Y.; Li, T. Dysregulation of HDAC9 Represses Trophoblast Cell Migration and Invasion Through TIMP3 Activation in Preeclampsia. Am. J. Hypertens 2019, 32, 515–523. [Google Scholar] [CrossRef]
- Raghu, D.; Mobley, R.J.; Shendy, N.A.M.; Perry, C.H.; Abell, A.N. GALNT3 Maintains the Epithelial State in Trophoblast Stem Cells. Cell Rep. 2019, 26. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, K.; Wu, Q.; Jin, L.; Lu, H.; Shi, Y.; Liu, L.; Yang, L.; Lv, L. Let-7 inhibits the migration and invasion of extravillous trophoblast cell via targeting MDM4. Mol. Cell Probes 2019, 45, 48–56. [Google Scholar] [CrossRef]
- Xue, F.; Yang, J.; Li, Q.; Zhou, H. Down-regulation of microRNA-34a-5p promotes trophoblast cell migration and invasion via targetting Smad4. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Ferretti, C.; Bruni, L.; Dangles-Marie, V.; Pecking, A.P.; Bellet, D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum. Reprod. Update 2007, 13, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ozen, M.; Wong, R.J.; Stevenson, D.K. Heme oxygenase-1 in pregnancy and cancer: Similarities in cellular invasion, cytoprotection, angiogenesis, and immunomodulation. Front. Pharm. 2014, 5, 295. [Google Scholar] [CrossRef] [PubMed]
- Piechowski, J. Plausibility of trophoblastic-like regulation of cancer tissue. Cancer Manag. Res. 2019, 11, 5033–5046. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N.; Cicvara-Pecina, T.; Kafka, A. Epithelial-to-mesenchymal transition: Possible role in meningiomas. Front. Biosci. 2012, 4, 889–896. [Google Scholar]
- Knofler, M.; Haider, S.; Saleh, L.; Pollheimer, J.; Gamage, T.; James, J. Human placenta and trophoblast development: Key molecular mechanisms and model systems. Cell Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Davies, J.E.; Pollheimer, J.; Yong, H.E.; Kokkinos, M.I.; Kalionis, B.; Knofler, M.; Murthi, P. Epithelial-mesenchymal transition during extravillous trophoblast differentiation. Cell Adhes. Migr. 2016, 10, 310–321. [Google Scholar] [CrossRef]
- Gamage, T.; Schierding, W.; Hurley, D.; Tsai, P.; Ludgate, J.L.; Bhoothpur, C.; Chamley, L.W.; Weeks, R.J.; Macaulay, E.C.; James, J.L. The role of DNA methylation in human trophoblast differentiation. Epigenetics 2018, 13, 1154–1173. [Google Scholar] [CrossRef]
- Pollheimer, J.; Vondra, S.; Baltayeva, J.; Beristain, A.G.; Knofler, M. Regulation of Placental Extravillous Trophoblasts by the Maternal Uterine Environment. Front. Immunol. 2018, 9, 2597. [Google Scholar] [CrossRef]
- Novakovic, B.; Stunnenberg, H.G. I Remember You: Epigenetic Priming in Epithelial Stem Cells. Immunity 2017, 47, 1019–1021. [Google Scholar] [CrossRef]
- Casadio, V.; Salvi, S.; Martignano, F.; Gunelli, R.; Ravaioli, S.; Calistri, D. Cell-Free DNA Integrity Analysis in Urine Samples. J. Vis. Exp. 2017, 119, e55049. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.Y.; Leung, T.N.; Chan, K.C.; Tai, H.L.; Lau, T.K.; Wong, E.M.; Lo, Y.M. Serial analysis of fetal DNA concentrations in maternal plasma in late pregnancy. Clin. Chem. 2003, 49, 678–680. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chan, M.H.; Chow, K.M.; Chan, A.T.; Leung, C.B.; Chan, L.Y.; Chow, K.C.; Lam, C.W.; Lo, Y.D. Quantitative analysis of pleural fluid cell-free DNA as a tool for the classification of pleural effusions. Clin. Chem. 2003, 49, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef]
- Mouliere, F.; Thierry, A.R. The importance of examining the proportion of circulating DNA originating from tumor, microenvironment and normal cells in colorectal cancer patients. Expert Opin. Biol. Ther. 2012, 12 (Suppl. 1), S209–S215. [Google Scholar] [CrossRef]
- Laktionov, P.P.; Tamkovich, S.N.; Rykova, E.Y.; Bryzgunova, O.E.; Starikov, A.V.; Kuznetsova, N.P.; Sumarokov, S.V.; Kolomiets, S.A.; Sevostianova, N.V.; Vlassov, V.V. Extracellular circulating nucleic acids in human plasma in health and disease. Nucleosides Nucleotides Nucleic Acids 2004, 23, 879–883. [Google Scholar] [CrossRef]
- Rykova, E.Y.; Morozkin, E.S.; Ponomaryova, A.A.; Loseva, E.M.; Zaporozhchenko, I.A.; Cherdyntseva, N.V.; Vlassov, V.V.; Laktionov, P.P. Cell-free and cell-bound circulating nucleic acid complexes: Mechanisms of generation, concentration and content. Expert Opin. Biol. Ther. 2012, 12, S141–S153. [Google Scholar] [CrossRef]
- Mittra, I.; Nair, N.K.; Mishra, P.K. Nucleic acids in circulation: Are they harmful to the host? J. Biosci. 2012, 37, 301–312. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef]
- Jiang, P.; Chan, C.W.; Chan, K.C.; Cheng, S.H.; Wong, J.; Wong, V.W.; Wong, G.L.; Chan, S.L.; Mok, T.S.; Chan, H.L.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Filho, E.M.; Ikuta, N.; Simon, D.; Regner, A.P. Prognostic value of circulating DNA levels in critically ill and trauma patients. Rev. Bras. Ter. Intensiva 2014, 26, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernandez-Delgado, I.; Latorre-Pellicer, A.; Acin-Perez, R.; Martin-Cofreces, N.B.; Jaso-Tamame, A.L.; Iborra, S.; Jorge, I.; et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun. 2018, 9, 2658. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Wagner, D.D. NETosis: A new factor in tumor progression and cancer-associated thrombosis. Semin. Thromb. Hemost. 2014, 40, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Wong, S.L.; Martinod, K.; Gallant, M.; Cabral, J.E.; Wang, Y.; Wagner, D.D. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 2016, 5, e1134073. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Han, Y.; Ren, H.; Chen, C.; He, D.; Zhou, L.; Eisner, G.M.; Asico, L.D.; Jose, P.A.; Zeng, C. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell Biol. 2013, 5, 227–238. [Google Scholar] [CrossRef]
- Mittra, I.; Khare, N.K.; Raghuram, G.V.; Chaubal, R.; Khambatti, F.; Gupta, D.; Gaikwad, A.; Prasannan, P.; Singh, A.; Iyer, A.; et al. Circulating nucleic acids damage DNA of healthy cells by integrating into their genomes. J. Biosci. 2015, 40, 91–111. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.-D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Minciacchi, V.; Zijlstra, A.; Rubin, M.A.; Di Vizio, D. Extracellular vesicles for liquid biopsy in prostate cancer: Where are we and where are we headed? Prostate Cancer Prostatic Dis. 2017, 20, 251. [Google Scholar] [CrossRef]
- Su, Y.-H.; Wang, M.; Brenner, D.E.; Ng, A.; Melkonyan, H.; Umansky, S.; Syngal, S.; Block, T.M. Human urine contains small, 150 to 250 nucleotide-sized, soluble DNA derived from the circulation and may be useful in the detection of colorectal cancer. J. Mol. Diagn. 2004, 6, 101–107. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.; Liang, H.; Wei, F.; Akin, D.; Feng, Z.; Yan, Q.; Li, Y.; Zhen, Y.; Xu, L.; Dong, G.; et al. Evaluation of a novel saliva-based epidermal growth factor receptor mutation detection for lung cancer: A pilot study. Thorac. Cancer 2016, 7, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Soh, J.; Toyooka, S.; Ichihara, S.; Suehisa, H.; Kobayashi, N.; Ito, S.; Yamane, M.; Aoe, M.; Sano, Y.; Kiura, K.; et al. EGFR mutation status in pleural fluid predicts tumor responsiveness and resistance to gefitinib. Lung Cancer 2007, 56, 445–448. [Google Scholar] [CrossRef]
- Pan, W.; Gu, W.; Nagpal, S.; Gephart, M.H.; Quake, S.R. Brain tumor mutations detected in cerebral spinal fluid. Clin. Chem. 2015, 61, 514–522. [Google Scholar] [CrossRef]
- Hayes, B.; Murphy, C.; Crawley, A.; O’Kennedy, R. Developments in Point-of-Care Diagnostic Technology for Cancer Detection. Diagnostics 2018, 8, 39. [Google Scholar] [CrossRef]
- Howlader, N.; Cronin, K.A.; Kurian, A.W.; Andridge, R. Differences in Breast Cancer Survival by Molecular Subtypes in the United States. Cancer Epidemiol. Biomark. Prev. 2018, 27, 619–626. [Google Scholar] [CrossRef]
- Overman, M.J.; Modak, J.; Kopetz, S.; Murthy, R.; Yao, J.C.; Hicks, M.E.; Abbruzzese, J.L.; Tam, A.L. Use of research biopsies in clinical trials: Are risks and benefits adequately discussed? J. Clin. Oncol. 2013, 31, 17. [Google Scholar] [CrossRef]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef]
- Gerlinger, M.; Catto, J.W.; Orntoft, T.F.; Real, F.X.; Zwarthoff, E.C.; Swanton, C. Intratumour heterogeneity in urologic cancers: From molecular evidence to clinical implications. Eur. Urol. 2015, 67, 729–737. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [PubMed]
- Allred, D.C.; Harvey, J.M.; Berardo, M.; Clark, G.M. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod. Pathol. 1998, 11, 155–168. [Google Scholar] [PubMed]
- Volik, S.; Alcaide, M.; Morin, R.D.; Collins, C. Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol. Cancer Res. 2016, 14, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Holdenrieder, S.; Pagliaro, L.; Morgenstern, D.; Dayyani, F. Clinically meaningful use of blood tumor markers in oncology. Biomed. Res. Int. 2016, 2016, 9795269. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra392. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, ra133–ra302. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Kim, H.H.; Han, S.U.; Kim, M.C.; Hyung, W.J.; Kim, W.; Lee, H.J.; Ryu, S.W.; Cho, G.S.; Song, K.Y.; Ryu, S.Y. Long-term results of laparoscopic gastrectomy for gastric cancer: A large-scale case-control and case-matched Korean multicenter study. J. Clin. Oncol. 2014, 32, 627–633. [Google Scholar] [CrossRef]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef]
- Roschewski, M.; Dunleavy, K.; Pittaluga, S.; Moorhead, M.; Pepin, F.; Kong, K.; Shovlin, M.; Jaffe, E.S.; Staudt, L.M.; Lai, C.; et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: A correlative biomarker study. Lancet Oncol. 2015, 16, 541–549. [Google Scholar] [CrossRef]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [PubMed]
- Wimberger, P.; Roth, C.; Pantel, K.; Kasimir-Bauer, S.; Kimmig, R.; Schwarzenbach, H. Impact of platinum-based chemotherapy on circulating nucleic acid levels, protease activities in blood and disseminated tumor cells in bone marrow of ovarian cancer patients. Int. J. Cancer 2011, 128, 2572–2580. [Google Scholar] [CrossRef] [PubMed]
- Catarino, R.; Ferreira, M.M.; Rodrigues, H.; Coelho, A.; Nogal, A.; Sousa, A.; Medeiros, R. Quantification of free circulating tumor DNA as a diagnostic marker for breast cancer. DNA Cell Biol. 2008, 27, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef]
- Gormally, E.; Vineis, P.; Matullo, G.; Veglia, F.; Caboux, E.; Le Roux, E.; Peluso, M.; Garte, S.; Guarrera, S.; Munnia, A.; et al. TP53 and KRAS2 mutations in plasma DNA of healthy subjects and subsequent cancer occurrence: A prospective study. Cancer Res. 2006, 66, 6871–6876. [Google Scholar] [CrossRef]
- Nygaard, A.D.; Garm Spindler, K.L.; Pallisgaard, N.; Andersen, R.F.; Jakobsen, A. The prognostic value of KRAS mutated plasma DNA in advanced non-small cell lung cancer. Lung Cancer 2013, 79, 312–317. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.-H.; Barrett, J.C.; Jänne, P.A. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non–small-cell lung cancer. J. Clin. Oncol. 2016, 34, 3375. [Google Scholar] [CrossRef]
- Zhang, W.; Xia, W.; Lv, Z.; Ni, C.; Xin, Y.; Yang, L. Liquid Biopsy for Cancer: Circulating Tumor Cells, Circulating Free DNA or Exosomes? Cell Physiol. Biochem. 2017, 41, 755–768. [Google Scholar] [CrossRef]
- Esposito, A.; Criscitiello, C.; Trapani, D.; Curigliano, G. The emerging role of “Liquid Biopsies,” circulating tumor cells, and circulating cell-free tumor dna in lung cancer diagnosis and identification of resistance mutations. Curr. Oncol. Rep. 2017, 19, 1. [Google Scholar] [CrossRef]
- Gedvilaite, V.; Schveigert, D.; Cicenas, S. Cell-free DNA in non-small cell lung cancer. Acta Med. Litu. 2017, 24, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Emlen, W.; Mannik, M. Effect of DNA size and strandedness on the in vivo clearance and organ localization of DNA. Clin. Exp. Immunol. 1984, 56, 185–192. [Google Scholar] [PubMed]
- Chan, K.C.; Jiang, P.; Chan, C.W.; Sun, K.; Wong, J.; Hui, E.P.; Chan, S.L.; Chan, W.C.; Hui, D.S.; Ng, S.S.; et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 18761–18768. [Google Scholar] [CrossRef] [PubMed]
- Holdhoff, M.; Schmidt, K.; Donehower, R.; Diaz, L.A., Jr. Analysis of circulating tumor DNA to confirm somatic KRAS mutations. J. Natl. Cancer Inst. 2009, 101, 1284–1285. [Google Scholar] [CrossRef]
- Molparia, B.; Nichani, E.; Torkamani, A. Assessment of circulating copy number variant detection for cancer screening. PLoS ONE 2017, 12, e0180647. [Google Scholar] [CrossRef]
- Yu, H.; Shen, Y.; Ge, Q.; He, Y.; Qiao, D.; Ren, M.; Zhang, J. Quantification of maternal serum cell-free fetal DNA in early-onset preeclampsia. Int. J. Mol. Sci. 2013, 14, 7571–7582. [Google Scholar] [CrossRef]
- Cheng, F.; Su, L.; Qian, C. Circulating tumor DNA: A promising biomarker in the liquid biopsy of cancer. Oncotarget 2016, 7, 48832–48841. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Jones, P.A.; Laird, P.W. Cancer epigenetics comes of age. Nat. Genet. 1999, 21, 163–167. [Google Scholar] [CrossRef]
- Perera, P.A.J. Epigenetic changes in health and disease: DNA Methylation in human development, infection and non-communicable diseases. Ceylon J. Sci. 2018, 47, 3–11. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liang, Y.; Li, S.; Zeng, F.; Meng, Y.; Chen, Z.; Liu, S.; Tao, Y.; Yu, F. The interplay of circulating tumor DNA and chromatin modification, therapeutic resistance, and metastasis. Mol. Cancer 2019, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W. CancerLocator: Non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 2017, 18, 53. [Google Scholar] [CrossRef]
- Gai, W.; Ji, L.; Lam, W.J.; Sun, K.; Jiang, P.; Chan, A.W.; Wong, J.; Lai, P.B.; Ng, S.S.; Ma, B.B. Liver-and colon-specific DNA methylation markers in plasma for investigation of colorectal cancers with or without liver metastases. Clin. Chem. 2018, 64, 1239–1249. [Google Scholar] [CrossRef]
- Song, L.; Jia, J.; Peng, X.; Xiao, W.; Li, Y. The performance of the SEPT9 gene methylation assay and a comparison with other CRC screening tests: A meta-analysis. Sci. Rep. 2017, 7, 3032. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, P.M.; Liu, R.B. Advance in plasma SEPT9 gene methylation assay for colorectal cancer early detection. World J. Gastrointest. Oncol. 2018, 10, 15–22. [Google Scholar] [CrossRef]
- Garrigou, S.; Perkins, G.; Garlan, F.; Normand, C.; Didelot, A.; Le Corre, D.; Peyvandi, S.; Mulot, C.; Niarra, R.; Aucouturier, P. A study of hypermethylated circulating tumor DNA as a universal colorectal cancer biomarker. Clin. Chem. 2016, 62, 1129–1139. [Google Scholar] [CrossRef]
- Murray, D.H.; Symonds, E.L.; Young, G.P.; Byrne, S.; Rabbitt, P.; Roy, A.; Cornthwaite, K.; Karapetis, C.S.; Pedersen, S.K. Relationship between post-surgery detection of methylated circulating tumor DNA with risk of residual disease and recurrence-free survival. J. Cancer Res. Clin. Oncol. 2018, 144, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Ibanez de Caceres, I.; Battagli, C.; Esteller, M.; Herman, J.G.; Dulaimi, E.; Edelson, M.I.; Bergman, C.; Ehya, H.; Eisenberg, B.L.; Cairns, P. Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res. 2004, 64, 6476–6481. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Koukaki, T.; Karamitrousis, E.; Nena, E.; Tsamardinos, I.; Kolios, G.; Lianidou, E. Circulating cell-free DNA in breast cancer: Size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 2019, 38, 3387–3401. [Google Scholar] [CrossRef] [PubMed]
- Salta, S.; P. Nunes, S.; Fontes-Sousa, M.; Lopes, P.; Freitas, M.; Caldas, M.; Antunes, L.; Castro, F.; Antunes, P.; Palma de Sousa, S.; et al. A DNA Methylation-Based Test for Breast Cancer Detection in Circulating Cell-Free DNA. J. Clin. Med. 2018, 7, 420. [Google Scholar] [CrossRef]
- Ooki, A.; Maleki, Z.; Tsay, J.J.; Goparaju, C.; Brait, M.; Turaga, N.; Nam, H.S.; Rom, W.N.; Pass, H.I.; Sidransky, D.; et al. A Panel of Novel Detection and Prognostic Methylated DNA Markers in Primary Non-Small Cell Lung Cancer and Serum DNA. Clin. Cancer Res. 2017, 23, 7141–7152. [Google Scholar] [CrossRef]
- Yang, Z.; Qi, W.; Sun, L.; Zhou, H.; Zhou, B.; Hu, Y. DNA methylation analysis of selected genes for the detection of early-stage lung cancer using circulating cell-free DNA. Adv. Clin. Exp. Med. 2019, 28, 361–366. [Google Scholar] [CrossRef]
- Xu, W.; Lu, J.; Zhao, Q.; Wu, J.; Sun, J.; Han, B.; Zhao, X.; Kang, Y. Genome-Wide Plasma Cell-Free DNA Methylation Profiling Identifies Potential Biomarkers for Lung Cancer. Dis. Markers 2019, 2019, 4108474. [Google Scholar] [CrossRef]
- Hendriks, R.J.; Dijkstra, S.; Smit, F.P.; Vandersmissen, J.; Van de Voorde, H.; Mulders, P.F.; van Oort, I.M.; Van Criekinge, W.; Schalken, J.A. Epigenetic markers in circulating cell-free DNA as prognostic markers for survival of castration-resistant prostate cancer patients. Prostate 2018, 78, 336–342. [Google Scholar] [CrossRef]
- Wasenang, W.; Chaiyarit, P.; Proungvitaya, S.; Limpaiboon, T. Serum cell-free DNA methylation of OPCML and HOXD9 as a biomarker that may aid in differential diagnosis between cholangiocarcinoma and other biliary diseases. Clin. Epigenet. 2019, 11, 39. [Google Scholar] [CrossRef]
- Ramirez, J.L.; Rosell, R.; Taron, M.; Sanchez-Ronco, M.; Alberola, V.; de las Penas, R.; Sanchez, J.M.; Moran, T.; Camps, C.; Massuti, B. 14-3-3σ Methylation in pretreatment serum circulating DNA of cisplatin-plus-gemcitabine-treated advanced Non–small-cell lung cancer patients predicts survival: The Spanish Lung Cancer Group. J. Clin. Oncol. 2005, 23, 9105–9112. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.; Peixoto, A.; Pinto, P.; Pinheiro, M.; Teixeira, M.R. Potential clinical applications of circulating cell-free DNA in ovarian cancer patients. Expert Rev. Mol. Med. 2018, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, X.; Gao, C.; Xing, Y.; Qi, Z.; Liu, R.; Wang, Y.; Zhang, X.; Yang, Y.G.; Li, X.; et al. 5-Hydroxymethylome in Circulating Cell-free DNA as A Potential Biomarker for Non-small-cell Lung Cancer. Genom. Proteom. Bioinform. 2018, 16, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, L.; Shu, X.; Lu, Y.; Shu, X.-O.; Cai, Q.; Beeghly-Fadiel, A.; Li, B.; Ye, F.; Berchuck, A. Genetic data from nearly 63,000 women of European descent predicts DNA methylation biomarkers and epithelial ovarian cancer risk. Cancer Res. 2019, 79, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, A.; Sottile, J.; Sanchez-Tejada, L.; Fajardo, C.; Camara, R.; Lamas, C.; Barbera, V.M.; Pico, A. DNA Methylation of Tumor Suppressor Genes in Pituitary Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2019, 104, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Kardum, V.; Karin, V.; Glibo, M.; Skrtic, A.; Martic, T.N.; Ibisevic, N.; Skenderi, F.; Vranic, S.; Serman, L. Methylation-associated silencing of SFRP1 gene in high-grade serous ovarian carcinomas. Ann. Diagn. Pathol. 2017, 31, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Hosoda, K.; Nishizawa, N.; Katoh, H.; Watanabe, M. Epigenetic biomarkers of promoter DNA methylation in the new era of cancer treatment. Cancer Sci. 2018, 109, 3695. [Google Scholar] [CrossRef]
- Kel, A.; Boyarskikh, U.; Stegmaier, P.; Leskov, L.S.; Sokolov, A.V.; Yevshin, I.; Mandrik, N.; Stelmashenko, D.; Koschmann, J.; Kel-Margoulis, O.; et al. Walking pathways with positive feedback loops reveal DNA methylation biomarkers of colorectal cancer. BMC Bioinform. 2019, 20, 119. [Google Scholar] [CrossRef]
- Gezer, U.; Holdenrieder, S. Post-translational histone modifications in circulating nucleosomes as new biomarkers in colorectal cancer. In Vivo 2014, 28, 287–292. [Google Scholar]
- Cameron, E.E.; Bachman, K.E.; Myohanen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.-F.; Druez, A.; Faugeras, L.; Martinet, J.-P.; Géhénot, M.; Josseaux, E.; Herzog, M.; Micallef, J.; George, F.; Delos, M. Circulating nucleosomes as new blood-based biomarkers for detection of colorectal cancer. Clin. Epigenet. 2017, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Gezer, U.; Yörüker, E.; Keskin, M.; Kulle, C.; Dharuman, Y.; Holdenrieder, S. Histone methylation marks on circulating nucleosomes as novel blood-based biomarker in colorectal cancer. Int. J. Mol. Sci. 2015, 16, 29654–29662. [Google Scholar] [CrossRef] [PubMed]
- Bauden, M.; Pamart, D.; Ansari, D.; Herzog, M.; Eccleston, M.; Micallef, J.; Andersson, B.; Andersson, R. Circulating nucleosomes as epigenetic biomarkers in pancreatic cancer. Clin. Epigenet. 2015, 7, 106. [Google Scholar] [CrossRef]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA: Apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Hahn, S.; Huppertz, B.; Holzgreve, W. Fetal cells and cell free fetal nucleic acids in maternal blood: New tools to study abnormal placentation? Placenta 2005, 26, 515–526. [Google Scholar] [CrossRef]
- Faas, B.H.; de Ligt, J.; Janssen, I.; Eggink, A.J.; Wijnberger, L.D.; van Vugt, J.M.; Vissers, L.; Geurts van Kessel, A. Non-invasive prenatal diagnosis of fetal aneuploidies using massively parallel sequencing-by-ligation and evidence that cell-free fetal DNA in the maternal plasma originates from cytotrophoblastic cells. Expert Opin. Biol. Ther. 2012, 12, S19–S26. [Google Scholar] [CrossRef]
- Chan, K.A.; Ding, C.; Gerovassili, A.; Yeung, S.W.; Chiu, R.W.; Leung, T.N.; Lau, T.K.; Chim, S.S.; Chung, G.T.; Nicolaides, K.H. Hypermethylated RASSF1A in maternal plasma: A universal fetal DNA marker that improves the reliability of noninvasive prenatal diagnosis. Clin. Chem. 2006, 52, 2211–2218. [Google Scholar] [CrossRef]
- Huppertz, B.; Kingdom, J.C. Apoptosis in the trophoblast--role of apoptosis in placental morphogenesis. J. Soc. Gynecol. Investig. 2004, 11, 353–362. [Google Scholar] [CrossRef]
- Litton, C.; Stone, J.; Eddleman, K.; Lee, M.J. Noninvasive prenatal diagnosis: Past, present, and future. Mt. Sinai J. Med. 2009, 76, 521–528. [Google Scholar] [CrossRef]
- Sarzynska-Nowacka, U.; Kosinski, P.; Wielgos, M. Is there a future for cell-free fetal dna tests in screening for preeclampsia? Ginekol. Pol. 2019, 90, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Canick, J.A.; Palomaki, G.E.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E. The impact of maternal plasma DNA fetal fraction on next generation sequencing tests for common fetal aneuploidies. Prenat. Diagn. 2013, 33, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Lui, Y.Y.; Chik, K.W.; Chiu, R.W.; Ho, C.Y.; Lam, C.W.; Lo, Y.M. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [PubMed]
- Rolnik, D.L.; da Silva Costa, F.; Lee, T.J.; Schmid, M.; McLennan, A.C. Association between fetal fraction on cell-free DNA testing and first-trimester markers for pre-eclampsia. Ultrasound Obstet. Gynecol. 2018, 52, 722–727. [Google Scholar] [CrossRef]
- Wright, D.; Wright, A.; Nicolaides, K.H. A unified approach to risk assessment for fetal aneuploidies. Ultrasound Obstet. Gynecol. 2015, 45, 48–54. [Google Scholar] [CrossRef]
- Ashoor, G.; Syngelaki, A.; Poon, L.; Rezende, J.; Nicolaides, K. Fetal fraction in maternal plasma cell-free DNA at 11–13 weeks’ gestation: Relation to maternal and fetal characteristics. Ultrasound Obstet. Gynecol. 2013, 41, 26–32. [Google Scholar] [CrossRef]
- Revello, R.; Sarno, L.; Ispas, A.; Akolekar, R.; Nicolaides, K. Screening for trisomies by cell-free DNA testing of maternal blood: Consequences of a failed result. Ultrasound Obstet. Gynecol. 2016, 47, 698–704. [Google Scholar] [CrossRef]
- Wataganara, T.; Gratacos, E.; Jani, J.; Becker, J.; Lewi, L.; Sullivan, L.M.; Bianchi, D.W.; Deprest, J.A. Persistent elevation of cell-free fetal DNA levels in maternal plasma after selective laser coagulation of chorionic plate anastomoses in severe midgestational twin-twin transfusion syndrome. Am. J. Obs. Gynecol. 2005, 192, 604–609. [Google Scholar] [CrossRef]
- Sifakis, S.; Koukou, Z.; Spandidos, D.A. Cell-free fetal DNA and pregnancy-related complications (review). Mol. Med. Rep. 2015, 11, 2367–2372. [Google Scholar] [CrossRef]
- Bréchot, P.P.; Mouawia, H.; Saker, A. Diagnostic prénatal non invasif de la mucoviscidose. Arch. Pédiatr. 2011, 18, 111–118. [Google Scholar]
- Bustamante-Aragones, A.; Trujillo-Tiebas, M.J.; Gallego-Merlo, J.; Rodriguez de Alba, M.; Gonzalez-Gonzalez, C.; Cantalapiedra, D.; Ayuso, C.; Ramos, C. Prenatal diagnosis of Huntington disease in maternal plasma: Direct and indirect study. Eur. J. Neurol. 2008, 15, 1338–1344. [Google Scholar] [CrossRef] [PubMed]
- Chitty, L.S.; Chatelain, P.; Wolffenbuttel, K.P.; Aigrain, Y. Prenatal management of disorders of sex development. J. Pediatr. Urol. 2012, 8, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Chitty, L.S.; Mason, S.; Barrett, A.N.; McKay, F.; Lench, N.; Daley, R.; Jenkins, L.A. Non-invasive prenatal diagnosis of achondroplasia and thanatophoric dysplasia: Next-generation sequencing allows for a safer, more accurate, and comprehensive approach. Prenat. Diagn. 2015, 35, 656–662. [Google Scholar] [CrossRef] [PubMed]
- González-González, M.C.; Garcia-Hoyos, M.; Trujillo-Tiebas, M.J.; Aragonés, A.B.; De Alba, M.R.; Alvarez, D.D.; Diaz-Recasens, J.; Ayuso, C.; Ramos, C. Improvement in strategies for the non-invasive prenatal diagnosis of Huntington disease. J. Assist. Reprod. Genet. 2008, 25, 477–481. [Google Scholar] [CrossRef]
- Lench, N.; Barrett, A.; Fielding, S.; McKay, F.; Hill, M.; Jenkins, L.; White, H.; Chitty, L.S. The clinical implementation of non-invasive prenatal diagnosis for single-gene disorders: Challenges and progress made. Prenat. Diagn. 2013, 33, 555–562. [Google Scholar] [CrossRef]
- Hill, M.; Finning, K.; Martin, P.; Hogg, J.; Meaney, C.; Norbury, G.; Daniels, G.; Chitty, L. Non-invasive prenatal determination of fetal sex: Translating research into clinical practice. Clin. Genet. 2011, 80, 68–75. [Google Scholar] [CrossRef]
- Akolekar, R.; Beta, J.; Picciarelli, G.; Ogilvie, C.; D’Antonio, F. Procedure-related risk of miscarriage following amniocentesis and chorionic villus sampling: A systematic review and meta-analysis. Ultrasound Obs. Gynecol. 2015, 45, 16–26. [Google Scholar] [CrossRef]
- Tabor, A.; Alfirevic, Z. Update on procedure-related risks for prenatal diagnosis techniques. Fetal Diagn. Ther. 2010, 27, 1–7. [Google Scholar] [CrossRef]
- Norton, M.; Jacobsson, B.; Swamy, G.; Laurent, L.; Ranzini, A.; Brar, H.; Tomlinson, M.; Pereira, L.; Spitz, J.; Holleman, D. Non-invasive examination of Trisomy using directed cell-free DNA analysis: The NEXT study. Prenat. Diagn. 2014, 34, e2. [Google Scholar]
- Lo, Y.M.; Tein, M.S.; Lau, T.K.; Haines, C.J.; Leung, T.N.; Poon, P.M.; Wainscoat, J.S.; Johnson, P.J.; Chang, A.M.; Hjelm, N.M. Quantitative analysis of fetal DNA in maternal plasma and serum: Implications for noninvasive prenatal diagnosis. Am. J. Hum. Genet. 1998, 62, 768–775. [Google Scholar] [CrossRef]
- Gourvas, V.; Dalpa, E.; Konstantinidou, A.; Vrachnis, N.; Spandidos, D.A.; Sifakis, S. Angiogenic factors in placentas from pregnancies complicated by fetal growth restriction. Mol. Med. Rep. 2012, 6, 23–27. [Google Scholar] [PubMed]
- Sekizawa, A.; Sugito, Y.; Iwasaki, M.; Watanabe, A.; Jimbo, M.; Hoshi, S.; Saito, H.; Okai, T. Cell-free fetal DNA is increased in plasma of women with hyperemesis gravidarum. Clin. Chem. 2001, 47, 2164–2165. [Google Scholar] [PubMed]
- Duley, L. The global impact of pre-eclampsia and eclampsia. Semin. Perinatol. 2009, 33, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Damsky, C.H.; Fisher, S.J. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J. Clin. Investig. 1997, 99, 2152–2164. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L. Latest advances in understanding preeclampsia. Science 2005, 308, 1592–1594. [Google Scholar] [CrossRef]
- Lau, T.-W.; Leung, T.N.; Chan, L.Y.; Lau, T.K.; Chan, K.A.; Tam, W.H.; Lo, Y.D. Fetal DNA clearance from maternal plasma is impaired in preeclampsia. Clin. Chem. 2002, 48, 2141–2146. [Google Scholar]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.-H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef]
- Levine, R.J.; Qian, C.; LeShane, E.S.; Kai, F.Y.; England, L.J.; Schisterman, E.F.; Wataganara, T.; Romero, R.; Bianchi, D.W. Two-stage elevation of cell-free fetal DNA in maternal sera before onset of preeclampsia. Am. J. Obstet. Gynecol. 2004, 190, 707–713. [Google Scholar] [CrossRef]
- Poon, L.C.; Nicolaides, K.H. Early prediction of preeclampsia. Obstet. Gynecol. Int. 2014, 2014, 297397. [Google Scholar] [CrossRef]
- Rolnik, D.; O’gorman, N.; Fiolna, M.; Van Den Boom, D.; Nicolaides, K.; Poon, L. Maternal plasma cell-free DNA in the prediction of pre-eclampsia. Ultrasound Obstet. Gynecol. 2015, 45, 106–111. [Google Scholar] [CrossRef]
- Thurik, F.F.; Lamain-de Ruiter, M.; Javadi, A.; Kwee, A.; Woortmeijer, H.; Page-Christiaens, G.C.; Franx, A.; van der Schoot, C.E.; Koster, M.P. Absolute first trimester cell-free DNA levels and their associations with adverse pregnancy outcomes. Prenat. Diagn. 2016, 36, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Simons, F.E.; Schatz, M. Anaphylaxis during pregnancy. J. Allergy Clin. Immunol. 2012, 130, 597–606. [Google Scholar] [CrossRef]
- Farina, A.; LeShane, E.S.; Romero, R.; Gomez, R.; Chaiworapongsa, T.; Rizzo, N.; Bianchi, D.W. High levels of fetal cell-free DNA in maternal serum: A risk factor for spontaneous preterm delivery. Am. J. Obs. Gynecol. 2005, 193, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, R.; Rao, A.J. Trophoblast ‘pseudo-tumorigenesis’: Significance and contributory factors. Reprod. Biol. Endocrinol. 2004, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Zukerberg, L.R.; Ngwu, C.; Harlow, E.; Lees, J.A. In vivo association of E2F and DP family proteins. Mol. Cell Biol. 1995, 15, 2536–2546. [Google Scholar] [CrossRef] [PubMed]
- Liggett, W.H., Jr.; Sidransky, D. Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar] [CrossRef]
- Van der Weyden, L.; Adams, D.J. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim. Biophys. Acta 2007, 1776, 58–85. [Google Scholar] [CrossRef]
- Lee, J.H.; Kang, M.J.; Han, H.Y.; Lee, M.G.; Jeong, S.I.; Ryu, B.K.; Ha, T.K.; Her, N.G.; Han, J.; Park, S.J.; et al. Epigenetic alteration of PRKCDBP in colorectal cancers and its implication in tumor cell resistance to TNFalpha-induced apoptosis. Clin. Cancer Res. 2011, 17, 7551–7562. [Google Scholar] [CrossRef]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef]
- Rahat, B.; Thakur, S.; Hamid, A.; Bagga, R.; Kaur, J. Association of aberrant methylation at promoter regions of tumor suppressor genes with placental pathologies. Epigenomics 2016, 8, 767–787. [Google Scholar] [CrossRef]
- Papantoniou, N.; Bagiokos, V.; Agiannitopoulos, K.; Kolialexi, A.; Destouni, A.; Tounta, G.; Kanavakis, E.; Antsaklis, A.; Mavrou, A. RASSF1A in maternal plasma as a molecular marker of preeclampsia. Prenat. Diagn. 2013, 33, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, S.Y.; Park, S.Y.; Ahn, H.K.; Chung, J.H.; Ryu, H.M. Association of fetal-derived hypermethylated RASSF1A concentration in placenta-mediated pregnancy complications. Placenta 2013, 34, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, H.J.; Park, S.Y.; Han, Y.J.; Choi, J.S.; Ryu, H.M. Early prediction of hypertensive disorders of pregnancy using cell-free fetal DNA, cell-free total DNA, and biochemical markers. Fetal Diagn. Ther. 2016, 40, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Rahat, B.; Hamid, A.; Ahmad Najar, R.; Bagga, R.; Kaur, J. Epigenetic mechanisms regulate placental c-myc and hTERT in normal and pathological pregnancies; c-myc as a novel fetal DNA epigenetic marker for pre-eclampsia. Mol. Hum. Reprod. 2014, 20, 1026–1040. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.D.; Liu, L.; Zhao, H.R.; Hou, Q.F.; Yan, J.B.; Shi, W.L.; Guo, Q.N.; Wang, L.; Liao, S.X.; Zhu, B.F. Detection of fetal epigenetic biomarkers through genome-wide DNA methylation study for non-invasive prenatal diagnosis. Mol. Med. Rep. 2017, 15, 3989–3998. [Google Scholar] [CrossRef] [PubMed]
- ACOG. Committee Opinion No. 545: Noninvasive prenatal testing for fetal aneuploidy. Obstet. Gynecol. 2012, 120, 1532–1534. [Google Scholar] [CrossRef]
- Benn, P.; Borrell, A.; Cuckle, H.; Dugoff, L.; Gross, S.; Johnson, J.a.; Maymon, R.; Odibo, A.; Schielen, P.; Spencer, K. Prenatal Detection of Down Syndrome using Massively Parallel Sequencing (MPS): A rapid response statement from a committee on behalf of the Board of the International Society for Prenatal Diagnosis, 24 October 2011. Prenat. Diagn. 2012, 32, 1–2. [Google Scholar] [CrossRef]
- Salomon, L.; Alfirevic, Z.; Audibert, F.; Kagan, K.; Paladini, D.; Yeo, G.; Raine-Fenning, N.; Committee, I.C.S. ISUOG consensus statement on the impact of non-invasive prenatal testing (NIPT) on prenatal ultrasound practice. Ultrasound Obstet. Gynecol. 2014, 44, 122–123. [Google Scholar] [CrossRef]
- Futch, T.; Spinosa, J.; Bhatt, S.; de Feo, E.; Rava, R.P.; Sehnert, A.J. Initial clinical laboratory experience in noninvasive prenatal testing for fetal aneuploidy from maternal plasma DNA samples. Prenat. Diagn. 2013, 33, 569–574. [Google Scholar] [CrossRef]
- Wang, J.C.; Sahoo, T.; Schonberg, S.; Kopita, K.A.; Ross, L.; Patek, K.; Strom, C.M. Discordant noninvasive prenatal testing and cytogenetic results: A study of 109 consecutive cases. Genet. Med. 2015, 17, 234–236. [Google Scholar] [CrossRef]
- Pan, M.; Li, F.T.; Li, Y.; Jiang, F.M.; Li, D.Z.; Lau, T.K.; Liao, C. Discordant results between fetal karyotyping and non-invasive prenatal testing by maternal plasma sequencing in a case of uniparental disomy 21 due to trisomic rescue. Prenat. Diagn. 2013, 33, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Verweij, E.; De Boer, M.; Oepkes, D. Non-invasive prenatal testing for trisomy 13: More harm than good? Ultrasound Obstet. Gynecol. 2014, 44, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.; Tian, F.; Zhang, J.; Song, Z.; Wu, Y.; Han, X.; Hu, W.; Ma, D.; Cram, D. Maternal mosaicism is a significant contributor to discordant sex chromosomal aneuploidies associated with noninvasive prenatal testing. Clin. Chem. 2014, 60, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.M.; Hardisty, E.; Devers, P.; Kaiser-Rogers, K.; Hayden, M.A.; Goodnight, W.; Vora, N.L. Discordant noninvasive prenatal testing results in a patient subsequently diagnosed with metastatic disease. Prenat. Diagn. 2013, 33, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Everett, T.R.; Chitty, L.S. Cell-free fetal DNA: The new tool in fetal medicine. Ultrasound Obs. Gynecol. 2015, 45, 499–507. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581. [Google Scholar] [CrossRef]
- Mincheva-Nilsson, L.; Baranov, V. The role of placental exosomes in reproduction. Am. J. Reprod. Immunol. 2010, 63, 520–533. [Google Scholar] [CrossRef]
- Gupta, A.; Pulliam, L. Exosomes as mediators of neuroinflammation. J. Neuroinflamm. 2014, 11, 68. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef]
- Behbahani, G.D.; Khani, S.; Hosseini, H.M.; Abbaszadeh-Goudarzi, K.; Nazeri, S. The role of exosomes contents on genetic and epigenetic alterations of recipient cancer cells. Iran. J. Basic Med. Sci. 2016, 19, 1031–1039. [Google Scholar] [PubMed]
- Stahl, P.D.; Raposo, G. Extracellular Vesicles: Exosomes and Microvesicles, Integrators of Homeostasis. Physiology 2019, 34, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Arenaccio, C.; Federico, M. The Multifaceted Functions of Exosomes in Health and Disease: An Overview. Adv. Exp. Med. Biol. 2017, 998, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Salomon, C.; Rice, G.E. Role of Exosomes in Placental Homeostasis and Pregnancy Disorders. Prog. Mol. Biol. Transl. Sci. 2017, 145, 163–179. [Google Scholar] [CrossRef]
- Tannetta, D.; Collett, G.; Vatish, M.; Redman, C.; Sargent, I. Syncytiotrophoblast extracellular vesicles–Circulating biopsies reflecting placental health. Placenta 2017, 52, 134–138. [Google Scholar] [CrossRef]
- Monguio-Tortajada, M.; Moron-Font, M.; Gamez-Valero, A.; Carreras-Planella, L.; Borras, F.E.; Franquesa, M. Extracellular-Vesicle Isolation from Different Biological Fluids by Size-Exclusion Chromatography. Curr. Protoc. Stem Cell Biol. 2019, 49, e82. [Google Scholar] [CrossRef]
- Lane, R.E.; Korbie, D.; Hill, M.M.; Trau, M. Extracellular vesicles as circulating cancer biomarkers: Opportunities and challenges. Clin. Transl. Med. 2018, 7, 14. [Google Scholar] [CrossRef]
- Gyorgy, B.; Paloczi, K.; Kovacs, A.; Barabas, E.; Beko, G.; Varnai, K.; Pallinger, E.; Szabo-Taylor, K.; Szabo, T.G.; Kiss, A.A.; et al. Improved circulating microparticle analysis in acid-citrate dextrose (ACD) anticoagulant tube. Thromb. Res. 2014, 133, 285–292. [Google Scholar] [CrossRef]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From garbage bins to promising therapeutic targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, X.; Shi, H.; Wu, L.; Qian, H.; Xu, W. Exosomes in cancer: Small particle, big player. J. Hematol. Oncol. 2015, 8, 83. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Tumor-derived exosomes and their role in cancer progression. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; Volume 74, pp. 103–141. [Google Scholar]
- Greening, D.W.; Gopal, S.K.; Xu, R.; Simpson, R.J.; Chen, W. Exosomes and their roles in immune regulation and cancer. Semin. Cell. Dev. Biol. 2015, 40, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Bhome, R.; Del Vecchio, F.; Lee, G.H.; Bullock, M.D.; Primrose, J.N.; Sayan, A.E.; Mirnezami, A.H. Exosomal microRNAs (exomiRs): Small molecules with a big role in cancer. Cancer Lett. 2018, 420, 228–235. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genom. 2013, 14, 319. [Google Scholar] [CrossRef]
- Rabinowits, G.; Gercel-Taylor, C.; Day, J.M.; Taylor, D.D.; Kloecker, G.H. Exosomal microRNA: A diagnostic marker for lung cancer. Clin. Lung Cancer 2009, 10, 42–46. [Google Scholar] [CrossRef]
- Taylor, D.D.; Gercel-Taylor, C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008, 110, 13–21. [Google Scholar] [CrossRef]
- Eichelser, C.; Stuckrath, I.; Muller, V.; Milde-Langosch, K.; Wikman, H.; Pantel, K.; Schwarzenbach, H. Increased serum levels of circulating exosomal microRNA-373 in receptor-negative breast cancer patients. Oncotarget 2014, 5, 9650–9663. [Google Scholar] [CrossRef]
- Ogata-Kawata, H.; Izumiya, M.; Kurioka, D.; Honma, Y.; Yamada, Y.; Furuta, K.; Gunji, T.; Ohta, H.; Okamoto, H.; Sonoda, H. Circulating exosomal microRNAs as biomarkers of colon cancer. PLoS ONE 2014, 9, e92921. [Google Scholar] [CrossRef]
- Endzelins, E.; Berger, A.; Melne, V.; Bajo-Santos, C.; Sobolevska, K.; Abols, A.; Rodriguez, M.; Santare, D.; Rudnickiha, A.; Lietuvietis, V.; et al. Detection of circulating miRNAs: Comparative analysis of extracellular vesicle-incorporated miRNAs and cell-free miRNAs in whole plasma of prostate cancer patients. BMC Cancer 2017, 17, 730. [Google Scholar] [CrossRef]
- Cheng, L.; Sharples, R.A.; Scicluna, B.J.; Hill, A.F. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Pochampally, R.; Watabe, K.; Lu, Z.; Mo, Y.Y. Exosome-mediated transfer of miR-10b promotes cell invasion in breast cancer. Mol. Cancer 2014, 13, 256. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.; Hamar, P.; Guo, C.; Basar, E.; Perdigao-Henriques, R.; Balaj, L.; Lieberman, J. miR-200-containing extracellular vesicles promote breast cancer cell metastasis. J. Clin. Investig. 2014, 124, 5109–5128. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.Y.; Li, M.X.; Pan, W.L.; Chen, Y.; Li, M.M.; Pang, J.X.; Zheng, L.; Chen, J.X.; Duan, W.J. In Situ Detection of Plasma Exosomal MicroRNA-1246 for Breast Cancer Diagnostics by a Au Nanoflare Probe. ACS Appl. Mater. Interfaces 2018, 10, 39478–39486. [Google Scholar] [CrossRef]
- Liao, J.; Liu, R.; Shi, Y.J.; Yin, L.H.; Pu, Y.P. Exosome-shuttling microRNA-21 promotes cell migration and invasion-targeting PDCD4 in esophageal cancer. Int. J. Oncol. 2016, 48, 2567–2579. [Google Scholar] [CrossRef]
- Liu, M.X.; Liao, J.; Xie, M.; Gao, Z.K.; Wang, X.H.; Zhang, Y.; Shang, M.H.; Yin, L.H.; Pu, Y.P.; Liu, R. miR-93-5p Transferred by Exosomes Promotes the Proliferation of Esophageal Cancer Cells via Intercellular Communication by Targeting PTEN. Biomed. Environ. Sci. 2018, 31, 171–185. [Google Scholar] [CrossRef]
- Sakha, S.; Muramatsu, T.; Ueda, K.; Inazawa, J. Exosomal microRNA miR-1246 induces cell motility and invasion through the regulation of DENND2D in oral squamous cell carcinoma. Sci. Rep. 2016, 6, 38750. [Google Scholar] [CrossRef]
- Lan, F.; Qing, Q.; Pan, Q.; Hu, M.; Yu, H.; Yue, X. Serum exosomal miR-301a as a potential diagnostic and prognostic biomarker for human glioma. Cell Oncol. 2018, 41, 25–33. [Google Scholar] [CrossRef]
- Cai, Q.; Zhu, A.; Gong, L. Exosomes of glioma cells deliver miR-148a to promote proliferation and metastasis of glioblastoma via targeting CADM1. Bull. Cancer 2018, 105, 643–651. [Google Scholar] [CrossRef]
- Qu, Z.; Wu, J.; Wu, J.; Ji, A.; Qiang, G.; Jiang, Y.; Jiang, C.; Ding, Y. Exosomal miR-665 as a novel minimally invasive biomarker for hepatocellular carcinoma diagnosis and prognosis. Oncotarget 2017, 8, 80666–80678. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Hu, J.; Zhou, K.; Chen, F.; Wang, Z.; Liao, B.; Dai, Z.; Cao, Y.; Fan, J.; Zhou, J. Serum exosomal miR-125b is a novel prognostic marker for hepatocellular carcinoma. Onco Targets 2017, 10, 3843–3851. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.J.; Fang, J.H.; Yang, X.J.; Zhang, C.; Yuan, Y.; Zheng, L.; Zhuang, S.M. Hepatocellular Carcinoma Cell-Secreted Exosomal MicroRNA-210 Promotes Angiogenesis In Vitro and In Vivo. Mol. Ther. Nucleic Acids 2018, 11, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Wang, X.; Zhao, Y.; Hu, R.; Qin, L. Exosomal miR-93 promotes proliferation and invasion in hepatocellular carcinoma by directly inhibiting TIMP2/TP53INP1/CDKN1A. Biochem. Biophys. Res. Commun. 2018, 502, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.H.; Zhang, Z.J.; Shang, L.R.; Luo, Y.W.; Lin, Y.F.; Yuan, Y.; Zhuang, S.M. Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology 2018, 68, 1459–1475. [Google Scholar] [CrossRef]
- Gong, L.; Bao, Q.; Hu, C.; Wang, J.; Zhou, Q.; Wei, L.; Tong, L.; Zhang, W.; Shen, Y. Exosomal miR-675 from metastatic osteosarcoma promotes cell migration and invasion by targeting CALN1. Biochem. Biophys. Res. Commun. 2018, 500, 170–176. [Google Scholar] [CrossRef]
- Feng, M.; Zhao, J.; Wang, L.; Liu, J. Upregulated Expression of Serum Exosomal microRNAs as Diagnostic Biomarkers of Lung Adenocarcinoma. Ann. Clin. Lab. Sci. 2018, 48, 712–718. [Google Scholar]
- Zhang, X.; Sai, B.; Wang, F.; Wang, L.; Wang, Y.; Zheng, L.; Li, G.; Tang, J.; Xiang, J. Hypoxic BMSC-derived exosomal miRNAs promote metastasis of lung cancer cells via STAT3-induced EMT. Mol. Cancer 2019, 18, 40. [Google Scholar] [CrossRef]
- Yoshimura, A.; Sawada, K.; Nakamura, K.; Kinose, Y.; Nakatsuka, E.; Kobayashi, M.; Miyamoto, M.; Ishida, K.; Matsumoto, Y.; Kodama, M. Exosomal miR-99a-5p is elevated in sera of ovarian cancer patients and promotes cancer cell invasion by increasing fibronectin and vitronectin expression in neighboring peritoneal mesothelial cells. BMC Cancer 2018, 18, 1065. [Google Scholar] [CrossRef]
- Su, Y.Y.; Sun, L.; Guo, Z.R.; Li, J.C.; Bai, T.T.; Cai, X.X.; Li, W.H.; Zhu, Y.F. Upregulated expression of serum exosomal miR-375 and miR-1307 enhance the diagnostic power of CA125 for ovarian cancer. J. Ovarian Res. 2019, 12, 6. [Google Scholar] [CrossRef]
- Yan, S.; Jiang, Y.; Liang, C.; Cheng, M.; Jin, C.; Duan, Q.; Xu, D.; Yang, L.; Zhang, X.; Ren, B.; et al. Exosomal miR-6803-5p as potential diagnostic and prognostic marker in colorectal cancer. J. Cell Biochem. 2018, 119, 4113–4119. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pan, B.; Sun, L.; Chen, X.; Zeng, K.; Hu, X.; Xu, T.; Xu, M.; Wang, S. Circulating Exosomal miR-27a and miR-130a Act as Novel Diagnostic and Prognostic Biomarkers of Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2018, 27, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wu, X.; Xia, M.; Wu, F.; Ding, J.; Jiao, Y.; Zhan, Q.; An, F. Upregulated exosomic miR23b3p plays regulatory roles in the progression of pancreatic cancer. Oncol. Rep. 2017, 38, 2182–2188. [Google Scholar] [CrossRef]
- Goto, T.; Fujiya, M.; Konishi, H.; Sasajima, J.; Fujibayashi, S.; Hayashi, A.; Utsumi, T.; Sato, H.; Iwama, T.; Ijiri, M.; et al. An elevated expression of serum exosomal microRNA-191, -21, -451a of pancreatic neoplasm is considered to be efficient diagnostic marker. BMC Cancer 2018, 18, 116. [Google Scholar] [CrossRef]
- Yang, H.; Fu, H.; Wang, B.; Zhang, X.; Mao, J.; Li, X.; Wang, M.; Sun, Z.; Qian, H.; Xu, W. Exosomal miR-423-5p targets SUFU to promote cancer growth and metastasis and serves as a novel marker for gastric cancer. Mol. Carcinog. 2018, 57, 1223–1236. [Google Scholar] [CrossRef]
- Kumata, Y.; Iinuma, H.; Suzuki, Y.; Tsukahara, D.; Midorikawa, H.; Igarashi, Y.; Soeda, N.; Kiyokawa, T.; Horikawa, M.; Fukushima, R. Exosomeencapsulated microRNA23b as a minimally invasive liquid biomarker for the prediction of recurrence and prognosis of gastric cancer patients in each tumor stage. Oncol. Rep. 2018, 40, 319–330. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, H.; Ge, S.; Ning, T.; Bai, M.; Li, J.; Li, S.; Sun, W.; Deng, T.; Zhang, L.; et al. Exosome-Derived miR-130a Activates Angiogenesis in Gastric Cancer by Targeting C-MYB in Vascular Endothelial Cells. Mol. Ther. 2018, 26, 2466–2475. [Google Scholar] [CrossRef]
- Wang, J.; Guan, X.; Zhang, Y.; Ge, S.; Zhang, L.; Li, H.; Wang, X.; Liu, R.; Ning, T.; Deng, T.; et al. Exosomal miR-27a Derived from Gastric Cancer Cells Regulates the Transformation of Fibroblasts into Cancer-Associated Fibroblasts. Cell Physiol. Biochem. 2018, 49, 869–883. [Google Scholar] [CrossRef]
- Jiang, L.; Deng, T.; Wang, D.; Xiao, Y. Elevated Serum Exosomal miR-125b Level as a Potential Marker for Poor Prognosis in Intermediate-Risk Acute Myeloid Leukemia. Acta Haematol. 2018, 140, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Tao, Y.W.; Gao, S.; Li, P.; Zheng, J.M.; Zhang, S.E.; Liang, J.; Zhang, Y. Cancer-associated fibroblasts contribute to oral cancer cells proliferation and metastasis via exosome-mediated paracrine miR-34a-5p. EBioMedicine 2018, 36, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, X.; Sun, W.; Yue, S.; Yang, J.; Li, J.; Ma, B.; Wang, J.; Yang, X.; Pu, M. Loss of exosomal miR-320a from cancer-associated fibroblasts contributes to HCC proliferation and metastasis. Cancer Lett. 2017, 397, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Kuranaga, Y.; Kumazaki, M.; Sugito, N.; Yoshikawa, Y.; Takai, T.; Taniguchi, K.; Ito, Y.; Akao, Y. Regulated polarization of tumor-associated macrophages by mir-145 via colorectal cancer–derived extracellular vesicles. J. Immunol. 2017, 199, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.H.; Spieker, T.; Koschmieder, S.; Schaffers, S.; Humberg, J.; Jungen, D.; Bulk, E.; Hascher, A.; Wittmer, D.; Marra, A.; et al. The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J. Thorac. Oncol. 2011, 6, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Liang, W.; Fu, M.; Huang, Z.H.; Li, X.; Zhang, W.; Zhang, P.; Qian, H.; Jiang, P.C.; Xu, W.R.; et al. Exosomes-mediated transfer of long noncoding RNA ZFAS1 promotes gastric cancer progression. J. Cancer Res. Clin. Oncol. 2017, 143, 991–1004. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, P.; Li, J.; Peng, M.; Zhao, X.; Zhang, X.; Chen, K.; Zhang, Y.; Liu, H.; Gan, L. Tumor-derived exosomal lnc-Sox2ot promotes EMT and stemness by acting as a ceRNA in pancreatic ductal adenocarcinoma. Oncogene 2018, 37, 3822. [Google Scholar] [CrossRef]
- Bian, E.B.; Chen, E.F.; Xu, Y.D.; Yang, Z.H.; Tang, F.; Ma, C.C.; Wang, H.L.; Zhao, B. Exosomal lncRNA-ATB activates astrocytes that promote glioma cell invasion. Int. J. Oncol. 2019, 54, 713–721. [Google Scholar] [CrossRef]
- Yoshida, Y.; Yamamoto, H.; Morita, R.; Oikawa, R.; Matsuo, Y.; Maehata, T.; Nosho, K.; Watanabe, Y.; Yasuda, H.; Itoh, F. Detection of DNA methylation of gastric juice-derived exosomes in gastric cancer. Integr. Mol. Med. 2014, 1, 17–21. [Google Scholar]
- Stefanski, A.L.; Martinez, N.; Peterson, L.K.; Callahan, T.J.; Treacy, E.; Luck, M.; Friend, S.F.; Hermesch, A.; Maltepe, E.; Phang, T.; et al. Murine trophoblast-derived and pregnancy-associated exosome-enriched extracellular vesicle microRNAs: Implications for placenta driven effects on maternal physiology. PLoS ONE 2019, 14, e0210675. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Akyol, S.; Gercel-Taylor, C. Pregnancy-associated exosomes and their modulation of T cell signaling. J. Immunol. 2006, 176, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Nguyen, H.P.; Elgass, K.; Simpson, R.J.; Salamonsen, L.A. Human endometrial exosomes contain hormone-specific cargo modulating trophoblast adhesive capacity: Insights into endometrial-embryo interactions. Biol. Reprod. 2016, 94, 38. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Stenqvist, A.-C.; Nagaeva, O.; Kjellberg, L.; Wulff, M.; Baranov, V.; Mincheva-Nilsson, L. Human placenta expresses and secretes NKG2D ligands via exosomes that down-modulate the cognate receptor expression: Evidence for immunosuppressive function. J. Immunol. 2009, 183, 340–351. [Google Scholar] [CrossRef]
- Mincheva-Nilsson, L.; Nagaeva, O.; Chen, T.; Stendahl, U.; Antsiferova, J.; Mogren, I.; Hernestal, J.; Baranov, V. Placenta-derived soluble MHC class I chain-related molecules down-regulate NKG2D receptor on peripheral blood mononuclear cells during human pregnancy: A possible novel immune escape mechanism for fetal survival. J. Immunol. 2006, 176, 3585–3592. [Google Scholar] [CrossRef]
- Stenqvist, A.C.; Nagaeva, O.; Baranov, V.; Mincheva-Nilsson, L. Exosomes secreted by human placenta carry functional Fas ligand and TRAIL molecules and convey apoptosis in activated immune cells, suggesting exosome-mediated immune privilege of the fetus. J. Immunol. 2013, 191, 5515–5523. [Google Scholar] [CrossRef]
- Saadeldin, I.M.; Oh, H.J.; Lee, B.C. Embryonic–maternal cross-talk via exosomes: Potential implications. Stem Cells Cloning Adv. Appl. 2015, 8, 103. [Google Scholar]
- Salomon, C.; Nuzhat, Z.; Dixon, C.L.; Menon, R. Placental exosomes during gestation: Liquid biopsies carrying signals for the regulation of human parturition. Curr. Pharm. Des. 2018, 24, 974–982. [Google Scholar] [CrossRef]
- Salomon, C.; Torres, M.J.; Kobayashi, M.; Scholz-Romero, K.; Sobrevia, L.; Dobierzewska, A.; Illanes, S.E.; Mitchell, M.D.; Rice, G.E. A gestational profile of placental exosomes in maternal plasma and their effects on endothelial cell migration. PLoS ONE 2014, 9, e98667. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L. Circulating microparticles in normal pregnancy and pre-eclampsia. Placenta 2008, 29 (Suppl. A), S73–S77. [Google Scholar] [CrossRef]
- Atay, S.; Gercel-Taylor, C.; Suttles, J.; Mor, G.; Taylor, D.D. Trophoblast-derived exosomes mediate monocyte recruitment and differentiation. Am. J. Reprod. Immunol. 2011, 65, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Dragovic, R.A.; Southcombe, J.H.; Tannetta, D.S.; Redman, C.W.; Sargent, I.L. Multicolor flow cytometry and nanoparticle tracking analysis of extracellular vesicles in the plasma of normal pregnant and pre-eclamptic women. Biol. Reprod. 2013, 89, 151. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Menon, R. Placental exosomes: A proxy to understand pregnancy complications. Am. J. Reprod. Immunol. 2018, 79, e12788. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.D.; Peiris, H.N.; Kobayashi, M.; Koh, Y.Q.; Duncombe, G.; Illanes, S.E.; Rice, G.E.; Salomon, C. Placental exosomes in normal and complicated pregnancy. Am. J. Obs. Gynecol. 2015, 213, S173–S181. [Google Scholar] [CrossRef]
- Morales-Prieto, D.M.; Ospina-Prieto, S.; Chaiwangyen, W.; Schoenleben, M.; Markert, U.R. Pregnancy-associated miRNA-clusters. J. Reprod. Immunol. 2013, 97, 51–61. [Google Scholar] [CrossRef]
- Malnou, E.C.; Umlauf, D.; Mouysset, M.; Cavaille, J. Imprinted MicroRNA Gene Clusters in the Evolution, Development, and Functions of Mammalian Placenta. Front. Genet. 2018, 9, 706. [Google Scholar] [CrossRef]
- Takahashi, H.; Ohkuchi, A.; Kuwata, T.; Usui, R.; Baba, Y.; Suzuki, H.; Chaw Kyi, T.T.; Matsubara, S.; Saito, S.; Takizawa, T. Endogenous and exogenous miR-520c-3p modulates CD44-mediated extravillous trophoblast invasion. Placenta 2017, 50, 25–31. [Google Scholar] [CrossRef]
- Anton, L.; Olarerin-George, A.O.; Hogenesch, J.B.; Elovitz, M.A. Placental expression of miR-517a/b and miR-517c contributes to trophoblast dysfunction and preeclampsia. PLoS ONE 2015, 10, e0122707. [Google Scholar] [CrossRef]
- Ding, J.; Huang, F.; Wu, G.; Han, T.; Xu, F.; Weng, D.; Wu, C.; Zhang, X.; Yao, Y.; Zhu, X. MiR-519d-3p suppresses invasion and migration of trophoblast cells via targeting MMP-2. PLoS ONE 2015, 10, e0120321. [Google Scholar] [CrossRef]
- Jiang, L.; Long, A.; Tan, L.; Hong, M.; Wu, J.; Cai, L.; Li, Q. Elevated microRNA-520g in pre-eclampsia inhibits migration and invasion of trophoblasts. Placenta 2017, 51, 70–75. [Google Scholar] [CrossRef]
- Hromadnikova, I.; Kotlabova, K.; Ondrackova, M.; Pirkova, P.; Kestlerova, A.; Novotna, V.; Hympanova, L.; Krofta, L. Expression profile of C19MC microRNAs in placental tissue in pregnancy-related complications. DNA Cell Biol. 2015, 34, 437–457. [Google Scholar] [CrossRef] [PubMed]
- Hromadnikova, I.; Kotlabova, K.; Ivankova, K.; Krofta, L. First trimester screening of circulating C19MC microRNAs and the evaluation of their potential to predict the onset of preeclampsia and IUGR. PLoS ONE 2017, 12, e0171756. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, A.V.; Gusar, V.A.; Kan, N.E.; Prozorovskaya, K.N.; Karapetyan, A.O.; Bayev, O.R.; Chagovets, V.V.; Kliver, S.F.; Iakovishina, D.Y.; Frankevich, V.E. Identification of potential early biomarkers of preeclampsia. Placenta 2018, 61, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Higashijima, A.; Miura, K.; Mishima, H.; Kinoshita, A.; Jo, O.; Abe, S.; Hasegawa, Y.; Miura, S.; Yamasaki, K.; Yoshida, A. Characterization of placenta-specific microRNAs in fetal growth restriction pregnancy. Prenat. Diagn. 2013, 33, 214–222. [Google Scholar] [CrossRef]
- Hromadnikova, I.; Kotlabova, K.; Ivankova, K.; Krofta, L. Expression profile of C19MC microRNAs in placental tissue of patients with preterm prelabor rupture of membranes and spontaneous preterm birth. Mol. Med. Rep. 2017, 16, 3849–3862. [Google Scholar] [CrossRef]
- Dai, Y.; Qiu, Z.; Diao, Z.; Shen, L.; Xue, P.; Sun, H.; Hu, Y. MicroRNA-155 inhibits proliferation and migration of human extravillous trophoblast derived HTR-8/SVneo cells via down-regulating cyclin D1. Placenta 2012, 33, 824–829. [Google Scholar] [CrossRef]
- Anton, L.; Olarerin-George, A.O.; Schwartz, N.; Srinivas, S.; Bastek, J.; Hogenesch, J.B.; Elovitz, M.A. miR-210 inhibits trophoblast invasion and is a serum biomarker for preeclampsia. Am. J. Pathol. 2013, 183, 1437–1445. [Google Scholar] [CrossRef]
- Umemura, K.; Ishioka, S.i.; Endo, T.; Ezaka, Y.; Takahashi, M.; Saito, T. Roles of microRNA-34a in the pathogenesis of placenta accreta. J. Obstet. Gynaecol. Res. 2013, 39, 67–74. [Google Scholar] [CrossRef]
- Bai, Y.; Yang, W.; Yang, H.X.; Liao, Q.; Ye, G.; Fu, G.; Ji, L.; Xu, P.; Wang, H.; Li, Y.X.; et al. Downregulated miR-195 detected in preeclamptic placenta affects trophoblast cell invasion via modulating ActRIIA expression. PLoS ONE 2012, 7, e38875. [Google Scholar] [CrossRef]
- Fu, G.; Ye, G.; Nadeem, L.; Ji, L.; Manchanda, T.; Wang, Y.; Zhao, Y.; Qiao, J.; Wang, Y.L.; Lye, S.; et al. MicroRNA-376c impairs transforming growth factor-beta and nodal signaling to promote trophoblast cell proliferation and invasion. Hypertension 2013, 61, 864–872. [Google Scholar] [CrossRef]
- Luo, L.; Ye, G.; Nadeem, L.; Fu, G.; Yang, B.B.; Honarparvar, E.; Dunk, C.; Lye, S.; Peng, C. MicroRNA-378a-5p promotes trophoblast cell survival, migration and invasion by targeting Nodal. J. Cell Sci. 2012, 125, 3124–3132. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef]
- Rackov, G.; Garcia-Romero, N.; Esteban-Rubio, S.; Carrión-Navarro, J.; Belda-Iniesta, C.; Ayuso-Sacido, A. Vesicle-mediated control of cell function: The role of extracellular matrix and microenvironment. Front. Physiol. 2018, 9, 651. [Google Scholar] [CrossRef]
- Qian, Z.; Shen, Q.; Yang, X.; Qiu, Y.; Zhang, W. The role of extracellular vesicles: An epigenetic view of the cancer microenvironment. Biomed. Res. Int. 2015, 2015, 649161. [Google Scholar] [CrossRef]
- Zhu, X.; You, Y.; Li, Q.; Zeng, C.; Fu, F.; Guo, A.; Zhang, H.; Zou, P.; Zhong, Z.; Wang, H. BCR-ABL1–positive microvesicles transform normal hematopoietic transplants through genomic instability: Implications for donor cell leukemia. Leukemia 2014, 28, 1666. [Google Scholar] [CrossRef]
- Baig, S.; Kothandaraman, N.; Manikandan, J.; Rong, L.; Ee, K.H.; Hill, J.; Lai, C.W.; Tan, W.Y.; Yeoh, F.; Kale, A.; et al. Proteomic analysis of human placental syncytiotrophoblast microvesicles in preeclampsia. Clin. Proteom. 2014, 11, 40. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; You, S.; Spinelli, C.; Morley, S.; Zandian, M.; Aspuria, P.J.; Cavallini, L.; Ciardiello, C.; Reis Sobreiro, M.; Morello, M.; et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget 2015, 6, 11327–11341. [Google Scholar] [CrossRef]
- Kim, D.K.; Kang, B.; Kim, O.Y.; Choi, D.S.; Lee, J.; Kim, S.R.; Go, G.; Yoon, Y.J.; Kim, J.H.; Jang, S.C.; et al. EVpedia: An integrated database of high-throughput data for systemic analyses of extracellular vesicles. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef]


{kind=link}
| Cancer | Biomarker | Findings | Refs. |
|---|---|---|---|
| Colorectal cancer (CRC) | SEPT9 | The blood SEPT9 gene methylation assay exhibited better performance in a symptomatic population than in an asymptomatic population when compared with other colorectal cancer screening tests. | [119] |
| SEPT9-reinforced promoter DNA methylation in ctDNA was a significant discriminator between premalignant and malignant alterations in colorectal carcinogenesis. | [120] | ||
| WIF1, NYP | DNA methylation of WIF1 and NPY genes showed that hypermethylation of their gene promoters was detectable in both CRC tissue and corresponding ctDNA. | [121] | |
| BCAT1, IKZF1 | Post-surgery detection of methylated BCAT1 and IKZF1 in ctDNA in CRC patients was related to increased risk of residual disease and subsequently recurrence. | [122] | |
| Ovarian cancer | RASSF1, BRCA1 | Tumor cell-specific hypermethylation of BRCA1 and RASSF1A promoter regions were found in serum and peritoneal fluid from ovarian cancer patients. | [123] |
| Breast cancer | WNT5A, KLK10, MSH2, SOX17, | WNT5A, KLK10, MSH2, and SOX17 genes were found more frequently methylated in ctDNA in all the patient groups than in healthy individuals. More intense methylation of WNT5A gene correlated with greater tumor size, advanced disease, and poor outcome; SOX17 methylation was significantly correlated with the incidence of death, shorter progression-free survival, and overall survival, while KLK10 methylation was significantly correlated with unfavorable clinicopathological characteristics and relapse. | [124] |
| APC, CCND2, FOXA1, PSAT1, RASSF1A, SCGB3A1 | Methylated APC, CCND2, FOXA1, PSAT1, RASSF1A, and SCGB3A1 in ctDNA in breast cancer patients were confirmed as cancer-specific, although with variable performance. A panel combining four of them (APC, FOXA1, RASSF1A, and SCGB3A1) revealed the highest accuracy for disease detection (94%). | [125] | |
| Lung cancer | CDO1, HOXA9, AJAP1, PTGDR, UNCX, MARCH11 | Promoter methylation of a panel of six genes (CDO1, HOXA9, AJAP1, PTGDR, UNCX, and MARCH11) in ctDNA was found to have potential as a diagnostic and prognostic biomarker in non-small-cell lung cancer (NSCLC) patients. | [126] |
| CALCA, HOXA9, RASSF1A, CDKN2A, DLEC1, CDH13, PITX2, WT1 | Methylation levels of CALCA, HOXA9, RASSF1A, CDKN2A, DLEC1, CDH13, PITX2, and WT1 in ctDNA in patients with lung cancer were significantly higher than in the non-lung cancer group. Three of them (RASSF1A, CDKN2A, and DLEC1) were found only in lung cancer patients. | [127] | |
| B3GAT2, BCAR1, HLF, HOPX, HOXD11, MIR1203, MYL9, SLC9A3R2, SYT5, VTRNA1-3 | Using genome-wide plasma cfDNA methylation profiling, ten genes (B3GAT2, BCAR1, HLF, HOPX, HOXD11, MIR1203, MYL9, SLC9A3R2, SYT5, and VTRNA1-3) were identified as potential biomarkers for lung cancer. | [128] | |
| Prostate cancer | GSTP1, APC | Epigenetic markers GSTP1 and APC in circulating cell-free DNA have the potential to serve as prognostic markers for survival of castration-resistant prostate cancer patients. | [129] |
| Cholangiocarcinoma | OPCML, HOXD9 | cfDNA methylation of OPCML and HOXD9 may serve as a differential biomarker of cholangiocarcinoma and other biliary diseases. | [130] |
| Cancer | miRNAs | Target | Refs. |
|---|---|---|---|
| Breast cancer | miR-10b | HOXD10, KLF4 | [235] |
| miR-373 | Unknown | [230] | |
| miR-200 family members | Unknown | [236] | |
| miR-1246 | Unknown | [237] | |
| Esophageal cancer | miR-21 | PDCD4 | [238] |
| miR-93-5p | PTEN | [239] | |
| Oral squamous cell carcinoma | miR-1246 | DENND2D | [240] |
| Glioblastoma | miR-301a | Unknown | [241] |
| miR-148a | CADM1 | [242] | |
| Hepatocellular carcinoma | miR-665 | Unknown | [243] |
| miR-125b | Unknown | [244] | |
| miR-210 | SMAD4, STAT6 | [245] | |
| miR-93 | TP53INP1, TIMP2, CDKN1A | [246] | |
| miR-1247-3p | B4GALT3 | [247] | |
| miR-103 | Unknown | [248] | |
| Osteosarcoma | miR-675 | CALN1 | [249] |
| Lung cancer | miR-17-3p, miR-21, miR-106a, miR-146, miR-155, miR-191, miR-192, miR-203, miR-205, miR-210, miR-212, miR-214 | Unknown | [228] |
| miR-21-5p, miR-126-3p, miR-140-5p | Unknown | [250] | |
| miR-193a-3p, miR-210-3p, miR-5100 | Unknown | [251] | |
| Ovarian cancer | miR-21, miR-141, miR-200a, miR-200b, miR-200c, miR-203, miR-205, miR-214 | Unknown | [229] |
| miR-99a-5p | Unknown | [252] | |
| miR-375, miR-1307 | Unknown | [253] | |
| Colorectal cancer | let-7a, miR-1229, miR-1246, miR-150, miR-21, miR-223, miR-23a | Unknown | [231] |
| miR-6803-5p | Unknown | [254] | |
| miR-27a, miR-130a | Unknown | [255] | |
| miR-25-3p | KLF2, KLF4 | [256] | |
| Prostate cancer | miR-200c-3p, miR-21-5p, let-7a-5p | Unknown | [232] |
| Pancreatic cancer | miR-23b-3p | Unknown | [257] |
| miR-191, miR-21, miR-451a | Unknown | [258] | |
| Gastric cancer | miR-423-5p | SUFU | [259] |
| miR-23b | Unknown | [260] | |
| miR-130a | C-MYB | [261] | |
| miR-27a | Unknown | [262] | |
| Acute myeloid leukemia | miR-125b | Unknown | [263] |
| Biomarker | Cancer | Refs. | |
|---|---|---|---|
| lncRNA | MALAT1 | Non-small-cell lung cancer | [268] |
| ZFAS1 | Gastric cancer | [269] | |
| SOX2OT | Pancreatic ductal adenocarcinoma | [270] | |
| lncRNA-ATB | Glioma | [271] | |
| cfDNA | LINE1 and SOX17 methylation | Gastric cancer | [272] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karin-Kujundzic, V.; Sola, I.M.; Predavec, N.; Potkonjak, A.; Somen, E.; Mioc, P.; Serman, A.; Vranic, S.; Serman, L. Novel Epigenetic Biomarkers in Pregnancy-Related Disorders and Cancers. Cells 2019, 8, 1459. https://doi.org/10.3390/cells8111459
Karin-Kujundzic V, Sola IM, Predavec N, Potkonjak A, Somen E, Mioc P, Serman A, Vranic S, Serman L. Novel Epigenetic Biomarkers in Pregnancy-Related Disorders and Cancers. Cells. 2019; 8(11):1459. https://doi.org/10.3390/cells8111459
Chicago/Turabian StyleKarin-Kujundzic, Valentina, Ida Marija Sola, Nina Predavec, Anamarija Potkonjak, Ema Somen, Pavao Mioc, Alan Serman, Semir Vranic, and Ljiljana Serman. 2019. "Novel Epigenetic Biomarkers in Pregnancy-Related Disorders and Cancers" Cells 8, no. 11: 1459. https://doi.org/10.3390/cells8111459
APA StyleKarin-Kujundzic, V., Sola, I. M., Predavec, N., Potkonjak, A., Somen, E., Mioc, P., Serman, A., Vranic, S., & Serman, L. (2019). Novel Epigenetic Biomarkers in Pregnancy-Related Disorders and Cancers. Cells, 8(11), 1459. https://doi.org/10.3390/cells8111459