MiR-21, MiR-29a, GATA4, and MEF2c Expression Changes in Endothelin-1 and Angiotensin II Cardiac Hypertrophy Stimulated Isl-1+Sca-1+c-kit+ Porcine Cardiac Progenitor Cells In Vitro

 , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
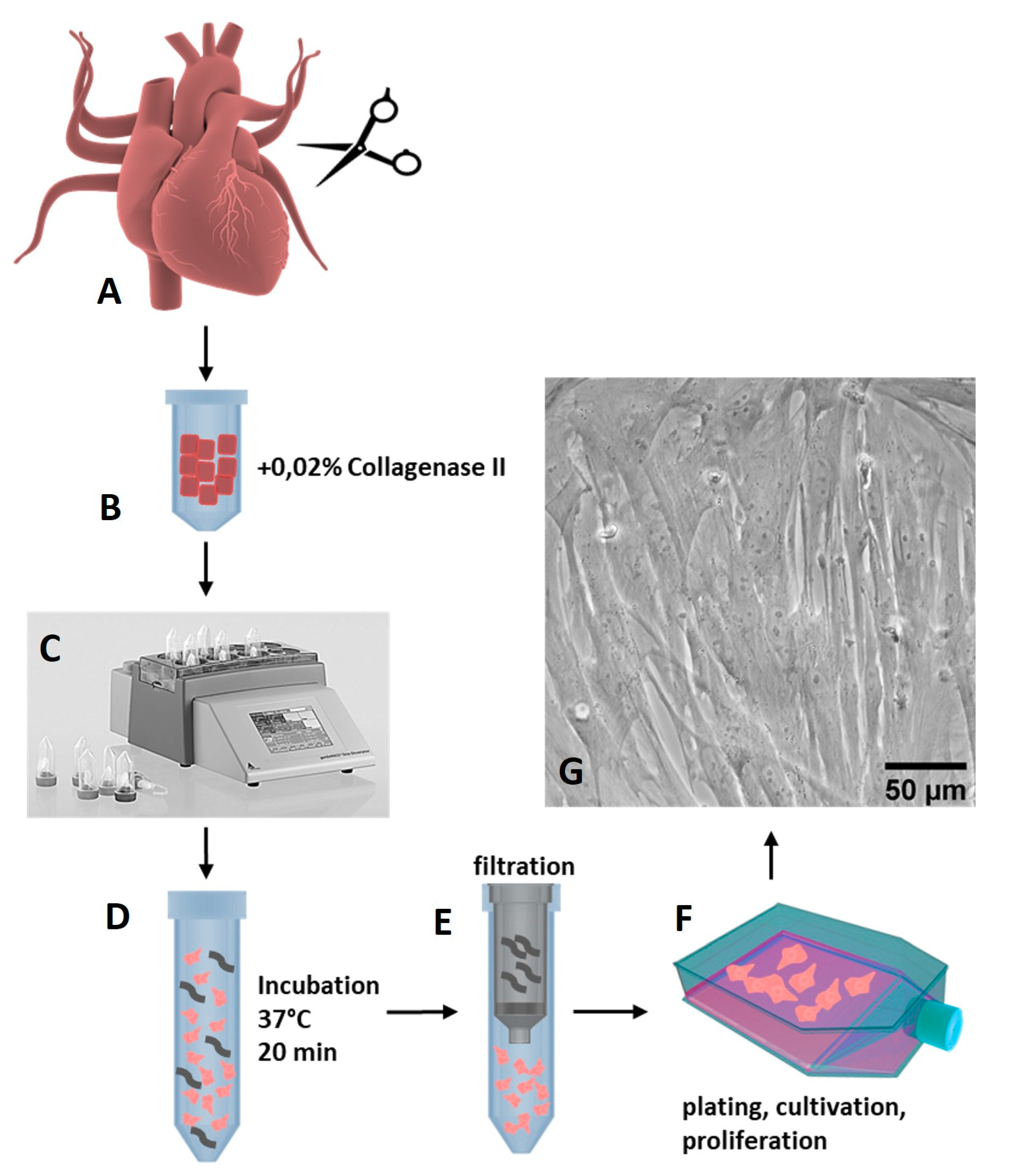
2.1. Isolation of Porcine Cardiac Progenitor Cells from Heart Tissue
2.2. Characterization of pCPCs
2.2.1. Immunofluorescence Staining
2.2.2. Real-Time qPCR
2.2.3. Transfection with pMx-MGT
2.2.4. Induction of Hypertrophy in pCPC
2.3. Measurement of Hypertrophy-Induced Protein Expression and Cell Size
2.4. Quantification of mRNA and miRNA Gene Expression by Real-Time qPCR
2.5. Statistics
3. Results
3.1. Isolation and Characterizaton of Isl1+Sca1+ckit+ pCPC
3.2. Induction of Chemical Hypertrophy with Ang II and ET-1 Results in Hypertrophic Growth of Isl1+Sca1+ckit+ pCPC
3.3. Cardiac Reprogramming of pCPCs with pMx-MGT or Ang II Induced Hypertrophy Alone Leads to Increase of Intracellular MCP-1
3.4. Cx43 is Upregulated in pMx-MGT-Transfected and Ang II Stimulated Isl1+Sca1+ckit+ pCPCs
3.5. Increased Cellular BNP Expression Confirms Cardiac Hypertrophy
3.6. ET-1 and Ang II Induced Hypertrophy Leads to Overexpression of MEF2c and GATA4 in Isl1+Sca1+ckit+ pCPCs
3.7. miR-29a is Upregulated in Response to Ang II Stimulation
3.8. pMx-MGT-Transfected Ang II Stimulated Isl1+Sca1+ckit+ pCPCs Have a Significanly Higher miR-21 Gene Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dorn, G.W.; Robbins, J.; Sugden, P.H. Phenotyping Hypertrophy. Circ. Res. 2003, 92, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Kahan, T.; Bergfeldt, L. Left ventricular hypertrophy in hypertension: Its arrhythmogenic potential. Heart 2005, 91, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Bisping, E.; Wakula, P.; Poteser, M.; Heinzel, F.R. Targeting cardiac hypertrophy: Toward a causal heart failure therapy. J. Cardiovasc. Pharmacol. 2014, 64, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.P.; Davidson, S.M.; Townsend, P.A. Molecular regulation of cardiac hypertrophy. Int. J. Biochem. Cell Biol. 2008, 40, 2023–2039. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Matsa, E.; Burridge, P.W.; Wu, J.C. Human stem cells for modeling heart disease and for drug discovery. Sci. Transl. Med. 2014, 6, 239ps6. [Google Scholar] [CrossRef]
- Perino, M.G.; Yamanaka, S.; Li, J.; Wobus, A.M.; Boheler, K.R. Cardiomyogenic stem and progenitor cell plasticity and the dissection of cardiopoiesis. J. Mol. Cell. Cardiol. 2008, 45, 475–494. [Google Scholar] [CrossRef]
- Engleka, K.A.; Manderfield, L.J.; Brust, R.D.; Li, L.; Cohen, A.; Dymecki, S.M.; Epstein, J.A. Islet1 derivatives in the heart are of both neural crest and second heart field origin. Circ. Res. 2012, 110, 922–926. [Google Scholar] [CrossRef]
- Marketou, M.E.; Parthenakis, F.; Vardas, P.E. Pathological Left Ventricular Hypertrophy and Stem Cells: Current Evidence and New Perspectives. Stem Cells Int. 2016, 2016, 5720758. [Google Scholar] [CrossRef]
- Santini, M.P.; Forte, E.; Harvey, R.P.; Kovacic, J.C. Developmental origin and lineage plasticity of endogenous cardiac stem cells. Dev. Camb. Engl. 2016, 143, 1242–1258. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.-L.; Liang, X.; Shi, Y.; Chu, P.-H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 Identifies a Cardiac Progenitor Population that Proliferates Prior to Differentiation and Contributes a Majority of Cells to the Heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Kwon, C.; Qian, L.; Cheng, P.; Nigam, V.; Arnold, J.; Srivastava, D. A Regulatory Pathway Involving Notch1/β-Catenin/Isl1 Determines Cardiac Progenitor Cell Fate. Nat. Cell Biol. 2009, 11, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Dodou, E.; Verzi, M.P.; Anderson, J.P.; Xu, S.-M.; Black, B.L. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Dev. Camb. Engl. 2004, 131, 3931–3942. [Google Scholar] [CrossRef] [PubMed]
- Herzig, T.C.; Jobe, S.M.; Aoki, H.; Molkentin, J.D.; Cowley, A.W.; Izumo, S.; Markham, B.E. Angiotensin II type1a receptor gene expression in the heart: AP-1 and GATA-4 participate in the response to pressure overload. Proc. Natl. Acad. Sci. USA 1997, 94, 7543–7548. [Google Scholar] [CrossRef]
- Hasegawa, K.; Lee, S.J.; Jobe, S.M.; Markham, B.E.; Kitsis, R.N. cis-Acting sequences that mediate induction of beta-myosin heavy chain gene expression during left ventricular hypertrophy due to aortic constriction. Circulation 1997, 96, 3943–3953. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Kalvakolanu, D.V.; Markham, B.E. Transcription factor GATA-4 regulates cardiac muscle-specific expression of the alpha-myosin heavy-chain gene. Mol. Cell. Biol. 1994, 14, 4947–4957. [Google Scholar] [CrossRef]
- McGrew, M.J.; Bogdanova, N.; Hasegawa, K.; Hughes, S.H.; Kitsis, R.N.; Rosenthal, N. Distinct gene expression patterns in skeletal and cardiac muscle are dependent on common regulatory sequences in the MLC1/3 locus. Mol. Cell. Biol. 1996, 16, 4524–4534. [Google Scholar] [CrossRef]
- Ip, H.S.; Wilson, D.B.; Heikinheimo, M.; Tang, Z.; Ting, C.N.; Simon, M.C.; Leiden, J.M.; Parmacek, M.S. The GATA-4 transcription factor transactivates the cardiac muscle-specific troponin C promoter-enhancer in nonmuscle cells. Mol. Cell. Biol. 1994, 14, 7517–7526. [Google Scholar] [CrossRef]
- Murphy, A.M.; Thompson, W.R.; Peng, L.F.; Jones, L. Regulation of the rat cardiac troponin I gene by the transcription factor GATA-4. Biochem. J. 1997, 322 Pt 2, 393–401. [Google Scholar] [CrossRef]
- Montgomery, M.O.; Litvin, J. The cardiac-muscle specific enhancer-promoter of slow/cardiac troponin C binds HMG-2. Gene 1997, 187, 159–164. [Google Scholar] [CrossRef]
- Durocher, D.; Charron, F.; Warren, R.; Schwartz, R.J.; Nemer, M. The cardiac transcription factors Nkx2-5 and GATA-4 are mutual cofactors. EMBO J. 1997, 16, 5687–5696. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Hanford, D.S.; Glembotski, C.C. Regulation of rat brain natriuretic peptide transcription. A potential role for GATA-related transcription factors in myocardial cell gene expression. J. Biol. Chem. 1994, 269, 17772–17775. [Google Scholar] [PubMed]
- Pikkarainen, S.; Tokola, H.; Majalahti-Palviainen, T.; Kerkelä, R.; Hautala, N.; Bhalla, S.S.; Charron, F.; Nemer, M.; Vuolteenaho, O.; Ruskoaho, H. GATA-4 Is a Nuclear Mediator of Mechanical Stretch-activated Hypertrophic Program. J. Biol. Chem. 2003, 278, 23807–23816. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; De Windt, L.J.; Witt, S.A.; Kimball, T.R.; Markham, B.E.; Molkentin, J.D. The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J. Biol. Chem. 2001, 276, 30245–30253. [Google Scholar] [CrossRef]
- Stansfield, W.E.; Ranek, M.; Pendse, A.; Schisler, J.C.; Wang, S.; Pulinilkunnil, T.; Willis, M.S. Chapter 4—The Pathophysiology of Cardiac Hypertrophy and Heart Failure. In Cellular and Molecular Pathobiology of Cardiovascular Disease; Willis, M.S., Homeister, J.W., Stone, J.R., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 51–78. ISBN 978-0-12-405206-2. [Google Scholar]
- Muñoz, J.P.; Collao, A.; Chiong, M.; Maldonado, C.; Adasme, T.; Carrasco, L.; Ocaranza, P.; Bravo, R.; Gonzalez, L.; Díaz-Araya, G.; et al. The transcription factor MEF2C mediates cardiomyocyte hypertrophy induced by IGF-1 signaling. Biochem. Biophys. Res. Commun. 2009, 388, 155–160. [Google Scholar] [CrossRef]
- Wei, J.; Joshi, S.; Speransky, S.; Crowley, C.; Jayathilaka, N.; Lei, X.; Wu, Y.; Gai, D.; Jain, S.; Hoosien, M.; et al. Reversal of pathological cardiac hypertrophy via the MEF2-coregulator interface. JCI Insight 2 2017, 2, 91068. [Google Scholar] [CrossRef]
- Hata, A. Functions of microRNAs in cardiovascular biology and disease. Annu. Rev. Physiol. 2013, 75, 69–93. [Google Scholar] [CrossRef]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.-H.; Tatsuguchi, M.; Huang, Z.-P.; Chen, J.-F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Invest. 2009, 119, 2772–2786. [Google Scholar] [CrossRef]
- Lin, Z.; Murtaza, I.; Wang, K.; Jiao, J.; Gao, J.; Li, P.-F. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 12103–12108. [Google Scholar] [CrossRef]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.-P.; Wu, J.; Wang, X.; Sartor, M.A.; Jones, K.; Qian, J.; Nicolaou, P.; Pritchard, T.J.; Fan, G.-C. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation 2009, 119, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Van Laake, L.W.; Huang, Y.; Liu, S.; Wendland, M.F.; Srivastava, D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J. Exp. Med. 2011, 208, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Rane, S.; He, M.; Sayed, D.; Vashistha, H.; Malhotra, A.; Sadoshima, J.; Vatner, D.E.; Vatner, S.F.; Abdellatif, M. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 2009, 104, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Roncarati, R.; Viviani Anselmi, C.; Losi, M.A.; Papa, L.; Cavarretta, E.; Da Costa Martins, P.; Contaldi, C.; Saccani Jotti, G.; Franzone, A.; Galastri, L.; et al. Circulating miR-29a, Among Other Up-Regulated MicroRNAs, Is the Only Biomarker for Both Hypertrophy and Fibrosis in Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 920–927. [Google Scholar] [CrossRef]
- Shi, J.-Y.; Chen, C.; Xu, X.; Lu, Q. miR-29a promotes pathological cardiac hypertrophy by targeting the PTEN/AKT/mTOR signalling pathway and suppressing autophagy. Acta Physiol. Oxf. Engl. 2019, 2019, e13323. [Google Scholar] [CrossRef]
- Adiarto, S.; Heiden, S.; Vignon-Zellweger, N.; Nakayama, K.; Yagi, K.; Yanagisawa, M.; Emoto, N. ET-1 from endothelial cells is required for complete angiotensin II-induced cardiac fibrosis and hypertrophy. Life Sci. 2012, 91, 651–657. [Google Scholar] [CrossRef]
- Moreau, P.; d’Uscio, L.V.; Shaw, S.; Takase, H.; Barton, M.; Lüscher, T.F. Angiotensin II Increases Tissue Endothelin and Induces Vascular Hypertrophy. Circulation 1997, 96, 1593–1597. [Google Scholar] [CrossRef]
- Irukayama-Tomobe, Y.; Miyauchi, T.; Sakai, S.; Kasuya, Y.; Ogata, T.; Takanashi, M.; Iemitsu, M.; Sudo, T.; Goto, K.; Yamaguchi, I. Endothelin-1–Induced Cardiac Hypertrophy Is Inhibited by Activation of Peroxisome Proliferator–Activated Receptor-α Partly Via Blockade of c-Jun NH2-Terminal Kinase Pathway. Circulation 2004, 109, 904–910. [Google Scholar] [CrossRef]
- Bupha-Intr, T.; Haizlip, K.M.; Janssen, P.M.L. Role of Endothelin in the Induction of Cardiac Hypertrophy In Vitro. PLoS ONE 2012, 7, e43179. [Google Scholar] [CrossRef]
- Gray, M.O.; Long, C.S.; Kalinyak, J.E.; Li, H.T.; Karliner, J.S. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc. Res. 1998, 40, 352–363. [Google Scholar] [CrossRef]
- Suzuki, Y.J. Cell signalling pathways for the regulation of GATA4 transcription factor: Implications for cell growth and apoptosis. Cell. Signal. 2011, 23, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Hautala, N.; Tokola, H.; Luodonpää, M.; Puhakka, J.; Romppanen, H.; Vuolteenaho, O.; Ruskoaho, H. Pressure overload increases GATA4 binding activity via endothelin-1. Circulation 2001, 103, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Hasegawa, K.; Kaburagi, S.; Kakita, T.; Wada, H.; Yanazume, T.; Sasayama, S. Phosphorylation of GATA-4 is involved in alpha 1-adrenergic agonist-responsive transcription of the endothelin-1 gene in cardiac myocytes. J. Biol. Chem. 2000, 275, 13721–13726. [Google Scholar] [CrossRef]
- Kockskämper, J.; von Lewinski, D.; Khafaga, M.; Elgner, A.; Grimm, M.; Eschenhagen, T.; Gottlieb, P.A.; Sachs, F.; Pieske, B. The slow force response to stretch in atrial and ventricular myocardium from human heart: Functional relevance and subcellular mechanisms. Prog. Biophys. Mol. Biol. 2008, 97, 250–267. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Xiao, W. MicroRNA-26a protects against cardiac hypertrophy via inhibiting GATA4 in rat model and cultured cardiomyocytes. Mol. Med. Rep. 2016, 14, 2860–2866. [Google Scholar] [CrossRef]
- Lu, D.; Wang, J.; Li, J.; Guan, F.; Zhang, X.; Dong, W.; Liu, N.; Gao, S.; Zhang, L. Meox1 accelerates myocardial hypertrophic decompensation through Gata4. Cardiovasc. Res. 2018, 114, 300–311. [Google Scholar] [CrossRef]
- Ovchinnikova, E.; Hoes, M.; Ustyantsev, K.; Bomer, N.; de Jong, T.V.; van der Mei, H.; Berezikov, E.; van der Meer, P. Modeling Human Cardiac Hypertrophy in Stem Cell-Derived Cardiomyocytes. Stem Cell Rep. 2018, 10, 794–807. [Google Scholar] [CrossRef]
- Aggarwal, P.; Turner, A.; Matter, A.; Kattman, S.J.; Stoddard, A.; Lorier, R.; Swanson, B.J.; Arnett, D.K.; Broeckel, U. RNA expression profiling of human iPSC-derived cardiomyocytes in a cardiac hypertrophy model. PLoS ONE 2014, 9, e108051. [Google Scholar] [CrossRef]
- Milani-Nejad, N.; Janssen, P.M.L. Small and large animal models in cardiac contraction research: Advantages and disadvantages. Pharmacol. Ther. 2014, 141, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Z.; Yin, C.; Asfour, H.; Chen, O.; Li, Y.; Bursac, N.; Liu, J.; Qian, L. Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ. Res. 2015, 116, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, I.A.; Christoffels, V.M. Regulation of expression of atrial and brain natriuretic peptide, biomarkers for heart development and disease. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2013, 1832, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Kerkelä, R.; Ulvila, J.; Magga, J. Natriuretic Peptides in the Regulation of Cardiovascular Physiology and Metabolic Events. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2015, 4, e002423. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef]
- Urbanek, K.; Torella, D.; Sheikh, F.; De Angelis, A.; Nurzynska, D.; Silvestri, F.; Beltrami, C.A.; Bussani, R.; Beltrami, A.P.; Quaini, F.; et al. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 8692–8697. [Google Scholar] [CrossRef]
- Davis, D.R.; Zhang, Y.; Smith, R.R.; Cheng, K.; Terrovitis, J.; Malliaras, K.; Li, T.-S.; White, A.; Makkar, R.; Marbán, E. Validation of the Cardiosphere Method to Culture Cardiac Progenitor Cells from Myocardial Tissue. PLoS ONE 2009, 4, e7195. [Google Scholar] [CrossRef]
- Bu, L.; Jiang, X.; Martin-Puig, S.; Caron, L.; Zhu, S.; Shao, Y.; Roberts, D.J.; Huang, P.L.; Domian, I.J.; Chien, K.R. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 2009, 460, 113–117. [Google Scholar] [CrossRef]
- Valente, M.; Nascimento, D.S.; Cumano, A.; Pinto-do-Ó, P. Sca-1+ Cardiac Progenitor Cells and Heart-Making: A Critical Synopsis. Stem Cells Dev. 2014, 23, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Mauretti, A.; Spaans, S.; Bax, N.A.M.; Sahlgren, C.; Bouten, C.V.C. Cardiac Progenitor Cells and the Interplay with Their Microenvironment. Stem Cells Int. 2017, 2017, 7471582. [Google Scholar] [CrossRef] [PubMed]
- Bearzi, C.; Rota, M.; Hosoda, T.; Tillmanns, J.; Nascimbene, A.; De Angelis, A.; Yasuzawa-Amano, S.; Trofimova, I.; Siggins, R.W.; LeCapitaine, N.; et al. Human cardiac stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 14068–14073. [Google Scholar] [CrossRef] [PubMed]
- Al-Maqtari, T.; Hong, K.U.; Vajravelu, B.N.; Moktar, A.; Cao, P.; Moore, J.B.; Bolli, R. Transcription factor-induced activation of cardiac gene expression in human c-kit+ cardiac progenitor cells. PLoS ONE 2017, 12, e0174242. [Google Scholar] [CrossRef]
- Ong, S.-B.; Lee, W.H.; Shao, N.-Y.; Ismail, N.I.; Katwadi, K.; Lim, M.-M.; Kwek, X.-Y.; Michel, N.A.; Li, J.; Newson, J.; et al. Calpain Inhibition Restores Autophagy and Prevents Mitochondrial Fragmentation in a Human iPSC Model of Diabetic Endotheliopathy. Stem Cell Rep. 2019, 12, 597–610. [Google Scholar] [CrossRef]
- Hu, J.; Verzi, M.P.; Robinson, A.S.; Tang, P.L.-F.; Hua, L.L.; Xu, S.-M.; Kwok, P.-Y.; Black, B.L. Endothelin signaling activates Mef2c expression in the neural crest through a MEF2C-dependent positive-feedback transcriptional pathway. Dev. Camb. Engl. 2015, 142, 2775–2780. [Google Scholar] [CrossRef]
- Yelamanchili, S.V.; Chaudhuri, A.D.; Chen, L.-N.; Xiong, H.; Fox, H.S. MicroRNA-21 dysregulates the expression of MEF2C in neurons in monkey and human SIV/HIV neurological disease. Cell Death Dis. 2010, 1, e77. [Google Scholar] [CrossRef]
- Wang, J.; Duan, L.; Gao, Y.; Zhou, S.; Liu, Y.; wei, S.; An, S.; Liu, J.; Tian, L.; Wang, S. Angiotensin II receptor blocker valsartan ameliorates cardiac fibrosis partly by inhibiting miR-21 expression in diabetic nephropathy mice. Mol. Cell. Endocrinol. 2018, 472, 149–158. [Google Scholar] [CrossRef]
- Adam, O.; Löhfelm, B.; Thum, T.; Gupta, S.K.; Puhl, S.-L.; Schäfers, H.-J.; Böhm, M.; Laufs, U. Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res. Cardiol. 2012, 107, 278. [Google Scholar] [CrossRef]
- Ning, Z.-W.; Luo, X.-Y.; Wang, G.-Z.; Li, Y.; Pan, M.-X.; Yang, R.-Q.; Ling, X.-G.; Huang, S.; Ma, X.-X.; Jin, S.-Y.; et al. MicroRNA-21 mediates angiotensin II-induced liver fibrosis by activating NLRP3 Inflammasome/IL-1β axis via targeting Smad7 and Spry1. Antioxid. Redox Signal. 2017, 27, 1–20. [Google Scholar] [CrossRef]
- Foglia, M.J.; Poss, K.D. Building and re-building the heart by cardiomyocyte proliferation. Development 2016, 143, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Yutzey, K.E. Cardiomyocyte Proliferation. Circ. Res. 2017, 120, 627–629. [Google Scholar] [CrossRef] [PubMed]
- Soonpaa, M.H.; Kim, K.K.; Pajak, L.; Franklin, M.; Field, L.J. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 1996, 271, H2183–H2189. [Google Scholar] [CrossRef] [PubMed]
- Aiello Robert, J.; Bourassa Patricia-Ann, K.; Lindsey, S.; Weng, W.; Natoli, E.; Rollins, B.J.; Milos, P.M. Monocyte Chemoattractant Protein-1 Accelerates Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1518–1525. [Google Scholar] [CrossRef]
- Behr Thomas, M.; Wang, X.; Aiyar, N.; Coatney, R.W.; Li, X.; Koster, P.; Angermann, C.E.; Ohlstein, E.; Feuerstein, G.Z.; Winaver, J. Monocyte Chemoattractant Protein-1 is upregulated in rats with volume-overload congestive heart failure. Circulation 2000, 102, 1315–1322. [Google Scholar] [CrossRef]
- Niu, J.; Kolattukudy, P.E. Role of MCP-1 in cardiovascular disease: Molecular mechanisms and clinical implications. Clin. Sci. 2009, 117, 95–109. [Google Scholar] [CrossRef]
- Iwai, M.; Mogi, M.; Horiuchi, M. Role of NAD(P)H oxidase and its regulation in chronic hypertension. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2006, 29, 743–744. [Google Scholar] [CrossRef][Green Version]
- Harrison, D.G.; Gongora, M.C. Oxidative stress and hypertension. Med. Clin. N. Am. 2009, 93, 621–635. [Google Scholar] [CrossRef]
- Harrison, D.G.; Guzik, T.J.; Lob, H.; Madhur, M.; Marvar, P.J.; Thabet, S.; Vinh, A.; Weyand, C. Inflammation, Immunity and Hypertension. Hypertension 2011, 57, 132–140. [Google Scholar] [CrossRef]
- Matsuda, S.; Umemoto, S.; Yoshimura, K.; Itoh, S.; Murata, T.; Fukai, T.; Matsuzaki, M. Angiotensin II Activates MCP-1 and Induces Cardiac Hypertrophy and Dysfunction via Toll-like Receptor 4. J. Atheroscler. Thromb. 2015, 22, 833–844. [Google Scholar] [CrossRef]
- Lo Cecilia, W. Role of Gap Junctions in Cardiac Conduction and Development. Circ. Res. 2000, 87, 346–348. [Google Scholar]
- Manias, J.L.; Plante, I.; Gong, X.-Q.; Shao, Q.; Churko, J.; Bai, D.; Laird, D.W. Fate of connexin43 in cardiac tissue harbouring a disease-linked connexin43 mutant. Cardiovasc. Res. 2008, 80, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Kostin, S.; Dammer, S.; Hein, S.; Klovekorn, W.P.; Bauer, E.P.; Schaper, J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc. Res. 2004, 62, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, B.E.J.; Jongsma, H.J.; Bierhuizen, M.F.A. Regulation of myocardial connexins during hypertrophic remodelling. Eur. Heart J. 2004, 25, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Bupha-Intr, T.; Haizlip, K.M.; Janssen, P.M.L. Temporal changes in expression of connexin 43 after load-induced hypertrophy in vitro. Am. J. Physiol.-Heart Circ. Physiol. 2009, 296, H806–H814. [Google Scholar] [CrossRef]
- Cao, L.; Chen, Y.; Lu, L.; Liu, Y.; Wang, Y.; Fan, J.; Yin, Y. Angiotensin II upregulates fibroblast-myofibroblast transition through Cx43-dependent CaMKII and TGF-β1 signaling in neonatal rat cardiac fibroblasts. Acta Biochim. Biophys. Sin. 2018, 50, 843–852. [Google Scholar] [CrossRef]
- Dodge, S.M.; Beardslee, M.A.; Darrow, B.J.; Green, K.G.; Beyer, E.C.; Saffitz, J.E. Effects of angiotensin II on expression of the gap junction channel protein connexin43 in neonatal rat ventricular myocytes. J. Am. Coll. Cardiol. 1998, 32, 800–807. [Google Scholar] [CrossRef]
- Spinella, F.; Rosanò, L.; Castro, V.D.; Nicotra, M.R.; Natali, P.G.; Bagnato, A. Endothelin-1 Decreases Gap Junctional Intercellular Communication by Inducing Phosphorylation of Connexin 43 in Human Ovarian Carcinoma Cells. J. Biol. Chem. 2003, 278, 41294–41301. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| ISL-1 | ACATGACGGTGGCTTACAGG | ATGTCACTCTGCAAGGCGAA |
| SCA-1/Ly6a | AGCTCAGGGACTGGAGTGTT | ATCAGGGTAGGGGCAGGTAA |
| KIT (c-kit) | GCCTGTGACTATCTGGGCTC | GCCTTATTCATGCCCGGAGA |
| BNP | CGCAGTAGCATCTTCCAAGTC | ACCTCCTGAGCACATTGCAG |
| ACTB (HK) | TCAACACCCCAGCCATGTAC | CTCCGGAGTCCATCACGATG |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| MEF2c | TAACATGCCGCCATCCGCCC | ATCCTCTCGGTCGCTGCCGT |
| GATA4 | AGAAAACGGAAGCCCAAGAAC | CCACACTGCTGGAGTTGCTG |
| miR-29a | CGGACCTAGCACCATCTGAA | miScript Universal Primer (Qiagen) |
| miR-21 | CGTAGCTAGCTTATCAGACTG | miScript Universal Primer (Qiagen) |
| ACTB (HK) | TCAACACCCCAGCCATGTAC | CTCCGGAGTCCATCACGATG |
| let7a (HK) | GCAGTGAGGTAGTAGGTTGT | miScript Universal Primer (Qiagen) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zlabinger, K.; Spannbauer, A.; Traxler, D.; Gugerell, A.; Lukovic, D.; Winkler, J.; Mester-Tonczar, J.; Podesser, B.; Gyöngyösi, M. MiR-21, MiR-29a, GATA4, and MEF2c Expression Changes in Endothelin-1 and Angiotensin II Cardiac Hypertrophy Stimulated Isl-1+Sca-1+c-kit+ Porcine Cardiac Progenitor Cells In Vitro. Cells 2019, 8, 1416. https://doi.org/10.3390/cells8111416
Zlabinger K, Spannbauer A, Traxler D, Gugerell A, Lukovic D, Winkler J, Mester-Tonczar J, Podesser B, Gyöngyösi M. MiR-21, MiR-29a, GATA4, and MEF2c Expression Changes in Endothelin-1 and Angiotensin II Cardiac Hypertrophy Stimulated Isl-1+Sca-1+c-kit+ Porcine Cardiac Progenitor Cells In Vitro. Cells. 2019; 8(11):1416. https://doi.org/10.3390/cells8111416
Chicago/Turabian StyleZlabinger, Katrin, Andreas Spannbauer, Denise Traxler, Alfred Gugerell, Dominika Lukovic, Johannes Winkler, Julia Mester-Tonczar, Bruno Podesser, and Mariann Gyöngyösi. 2019. "MiR-21, MiR-29a, GATA4, and MEF2c Expression Changes in Endothelin-1 and Angiotensin II Cardiac Hypertrophy Stimulated Isl-1+Sca-1+c-kit+ Porcine Cardiac Progenitor Cells In Vitro" Cells 8, no. 11: 1416. https://doi.org/10.3390/cells8111416
APA StyleZlabinger, K., Spannbauer, A., Traxler, D., Gugerell, A., Lukovic, D., Winkler, J., Mester-Tonczar, J., Podesser, B., & Gyöngyösi, M. (2019). MiR-21, MiR-29a, GATA4, and MEF2c Expression Changes in Endothelin-1 and Angiotensin II Cardiac Hypertrophy Stimulated Isl-1+Sca-1+c-kit+ Porcine Cardiac Progenitor Cells In Vitro. Cells, 8(11), 1416. https://doi.org/10.3390/cells8111416