Hypoxia and EGF Stimulation Regulate VEGF Expression in Human Glioblastoma Multiforme (GBM) Cells by Differential Regulation of the PI3K/Rho-GTPase and MAPK Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibodies and Reagents
2.3. EGF Stimulation
2.4. Hypoxia Mimicking
2.5. Hypoxia Chamber
2.6. Inhibitors Treatment
2.7. Cell Transfection with Small Interfering RNA (siRNA)
2.8. Western Blot Analysis
2.9. Pull Down Assays
2.10. ELISA
2.11. Statistical Analysis
3. Results
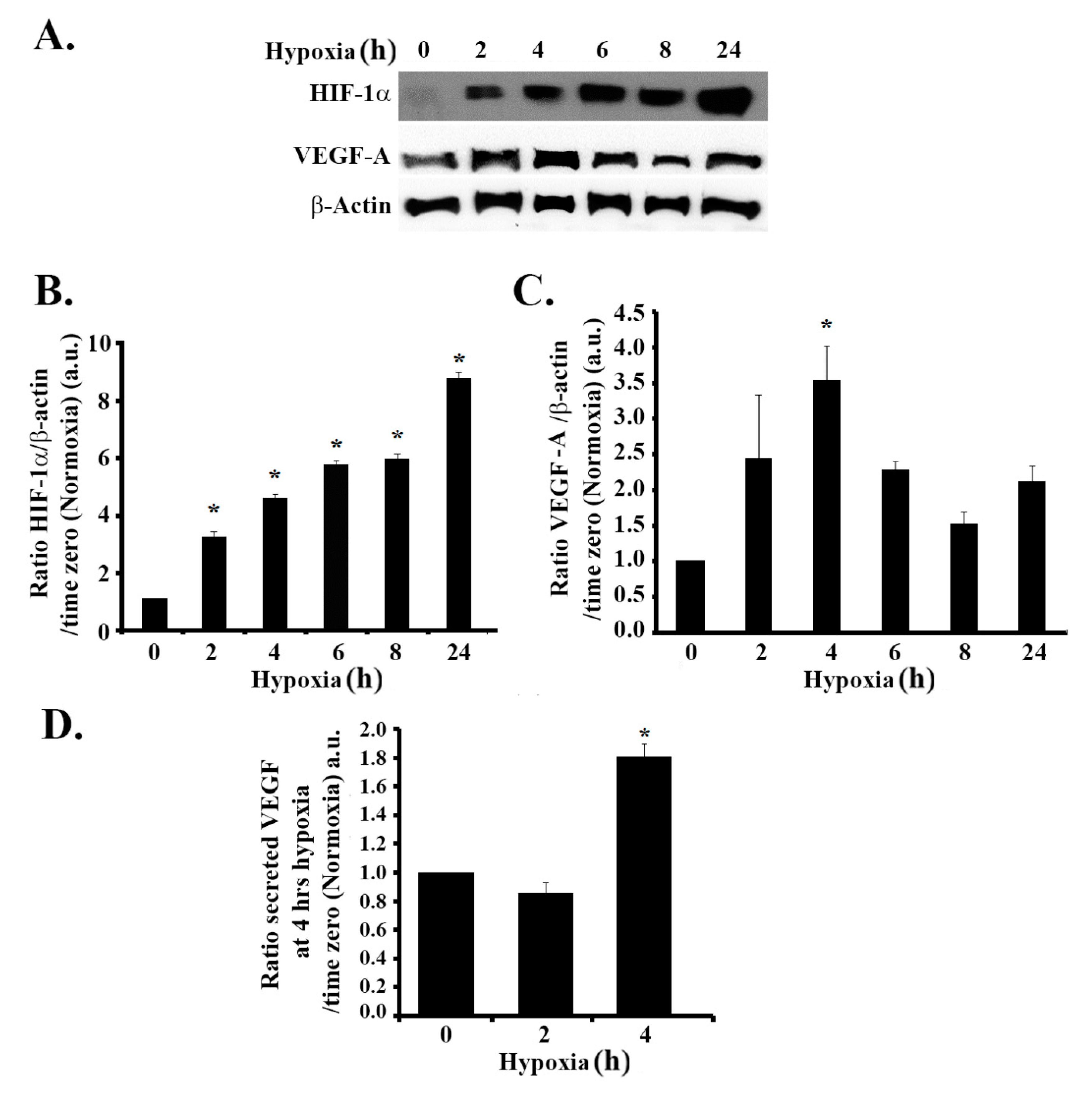
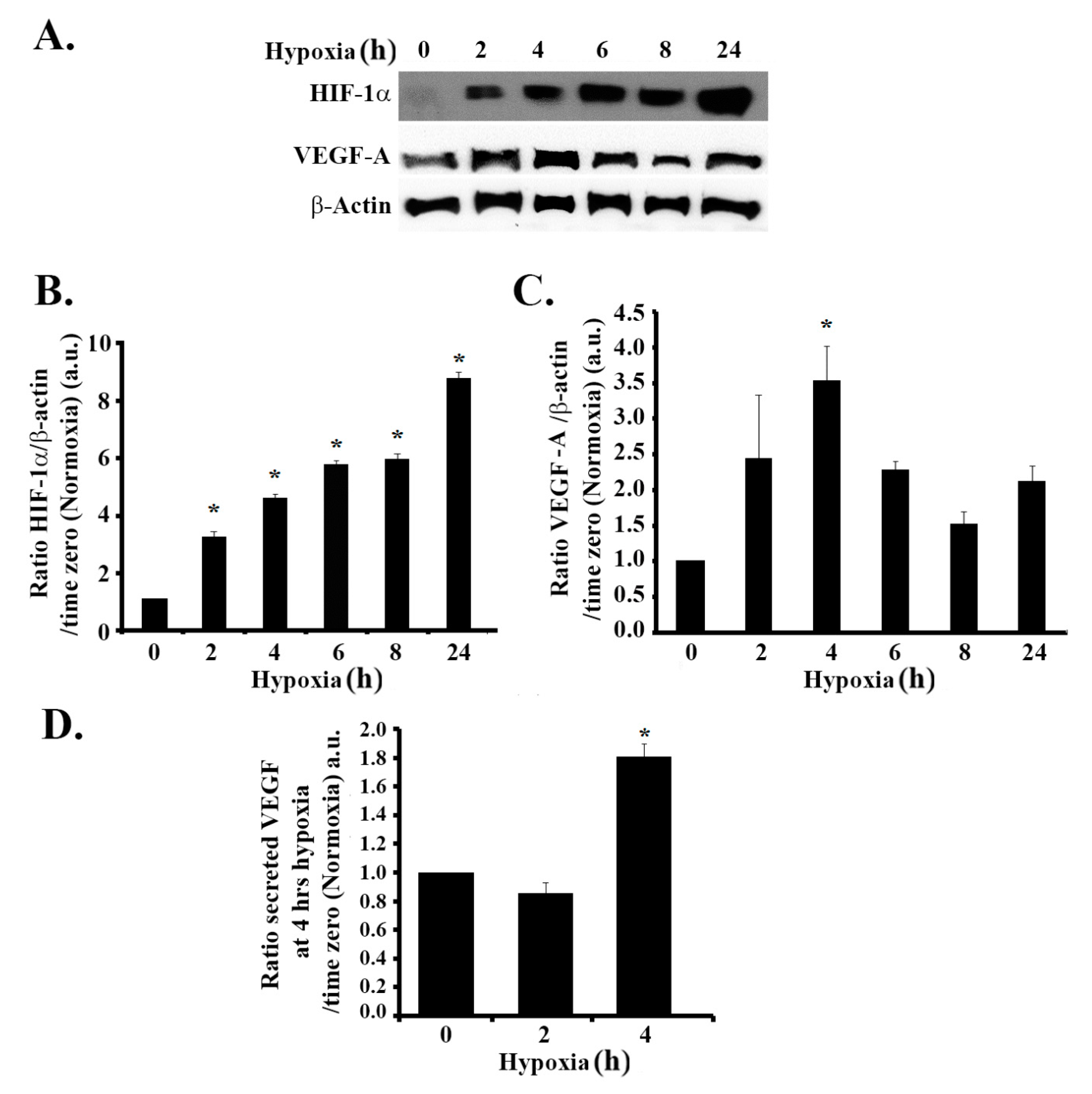
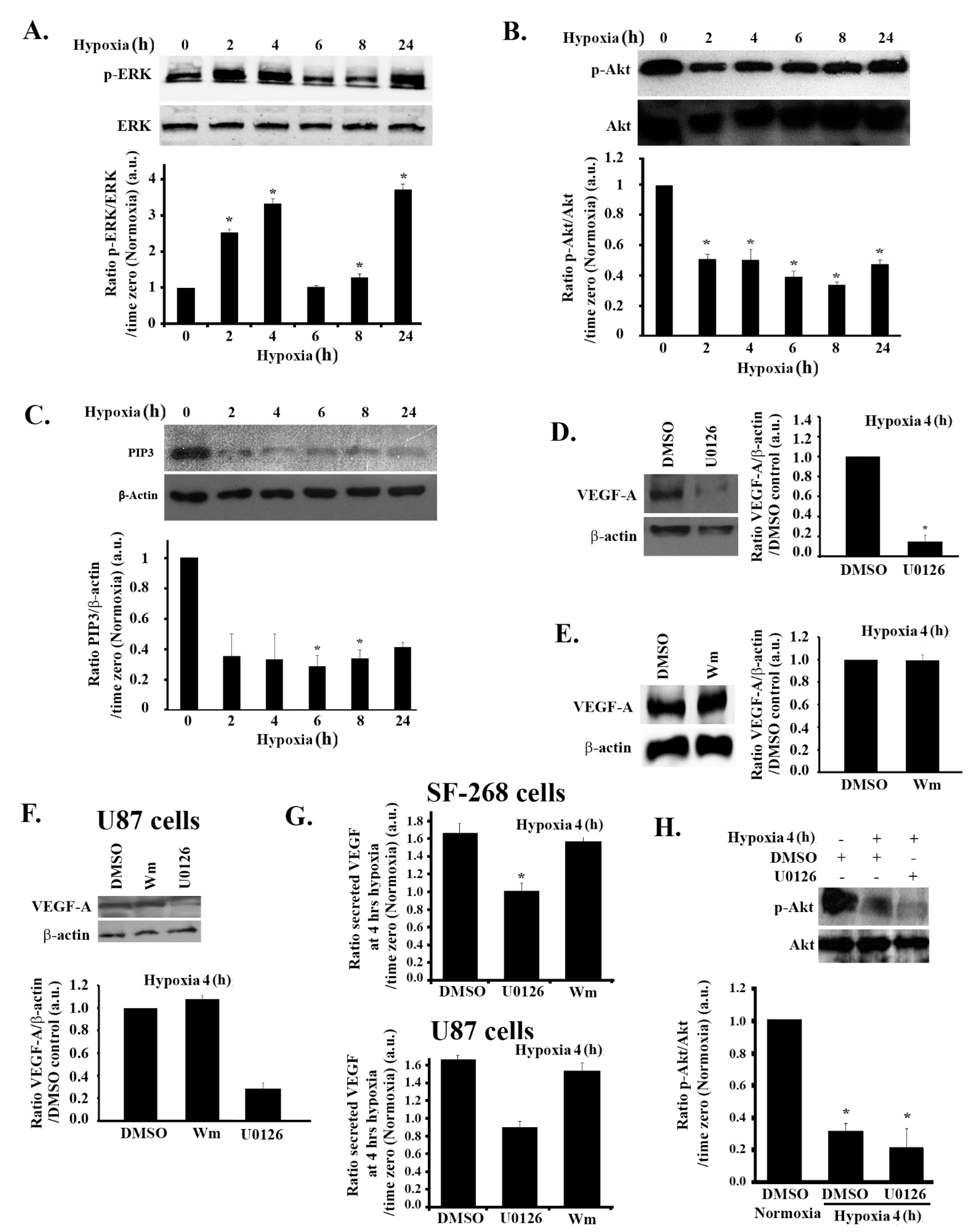
3.1. Hypoxia Mimicking Increases the Expression and Secretion of Vascular Endothelial Growth Factor (VEGF) in GBM Cells
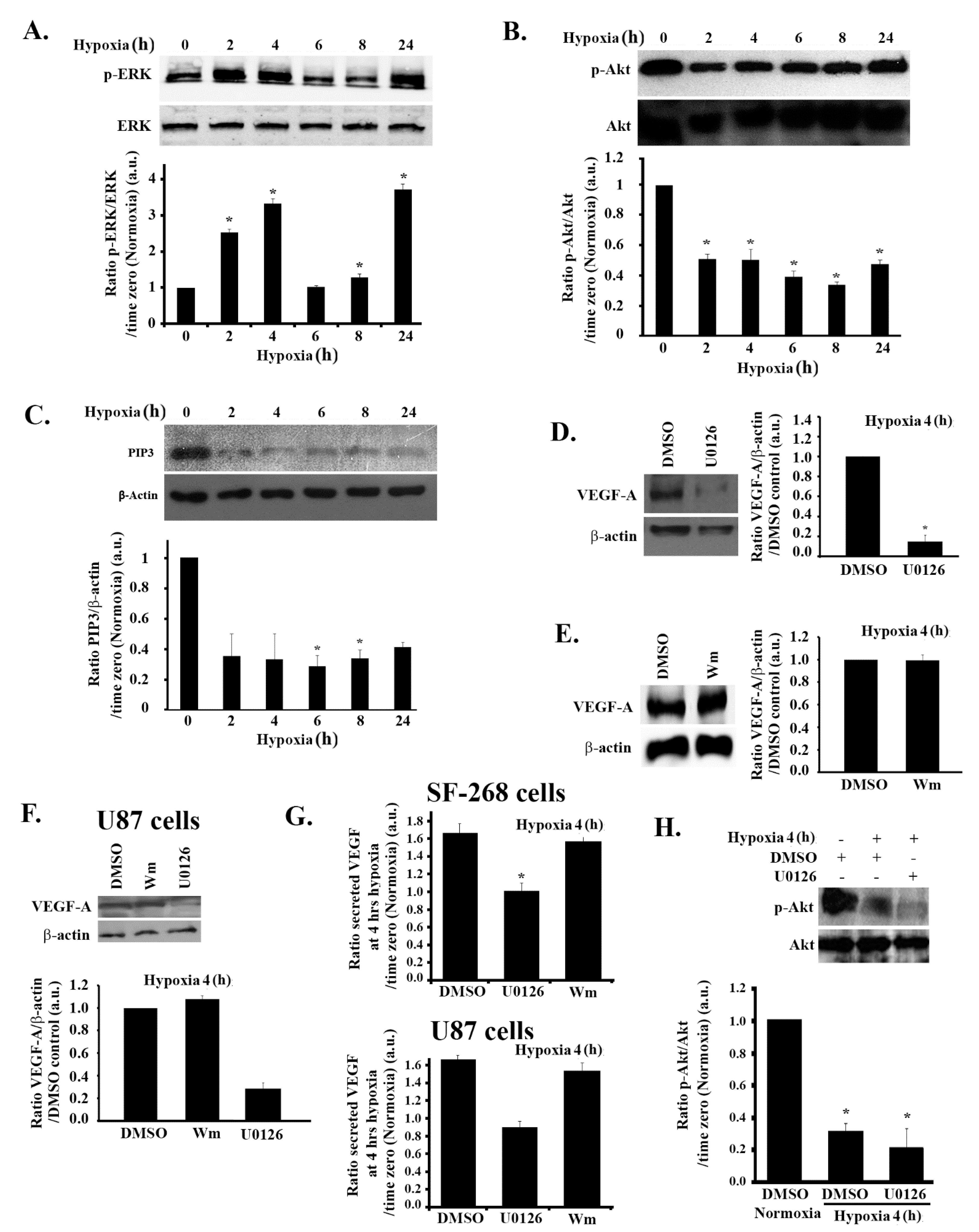
3.2. Hypoxia-Induced Increase in VEGF Expression and Secretion Is ERK-Dependent and PI3K-Independent in GBM Cells
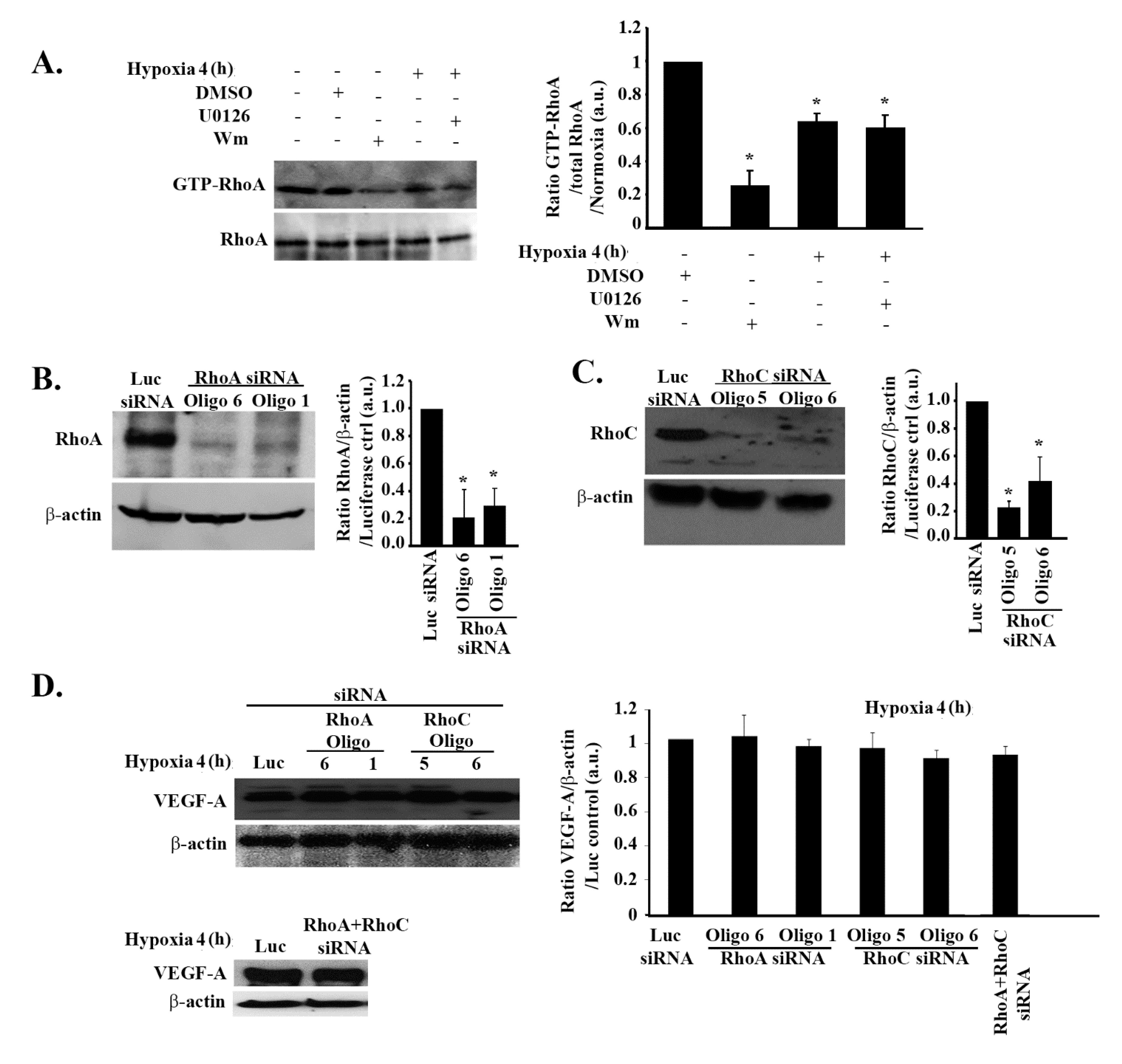
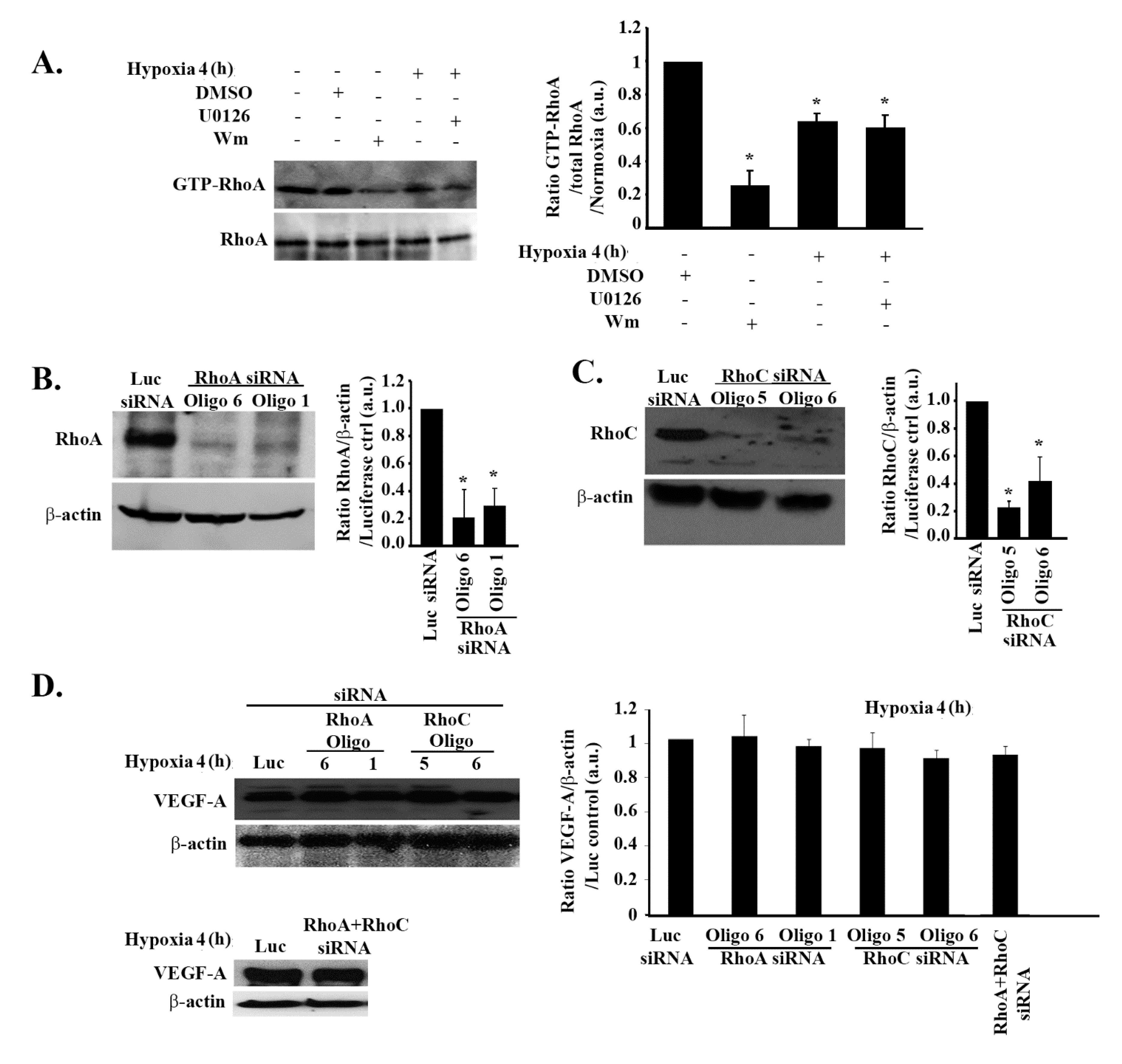
3.3. The Increase in VEGF Expression in Response to Hypoxia in GBM Cells Is Independent of Rho-GTPases
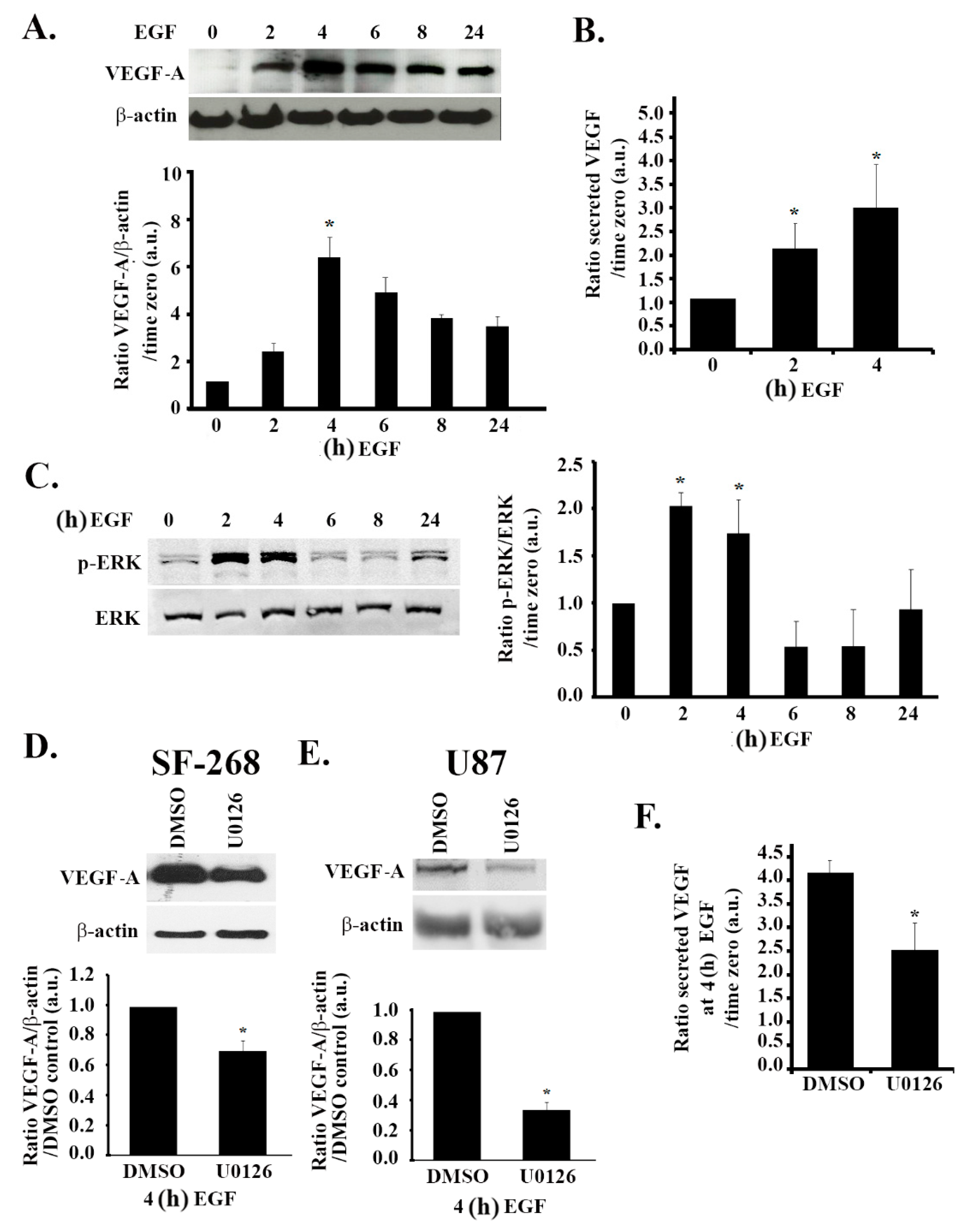
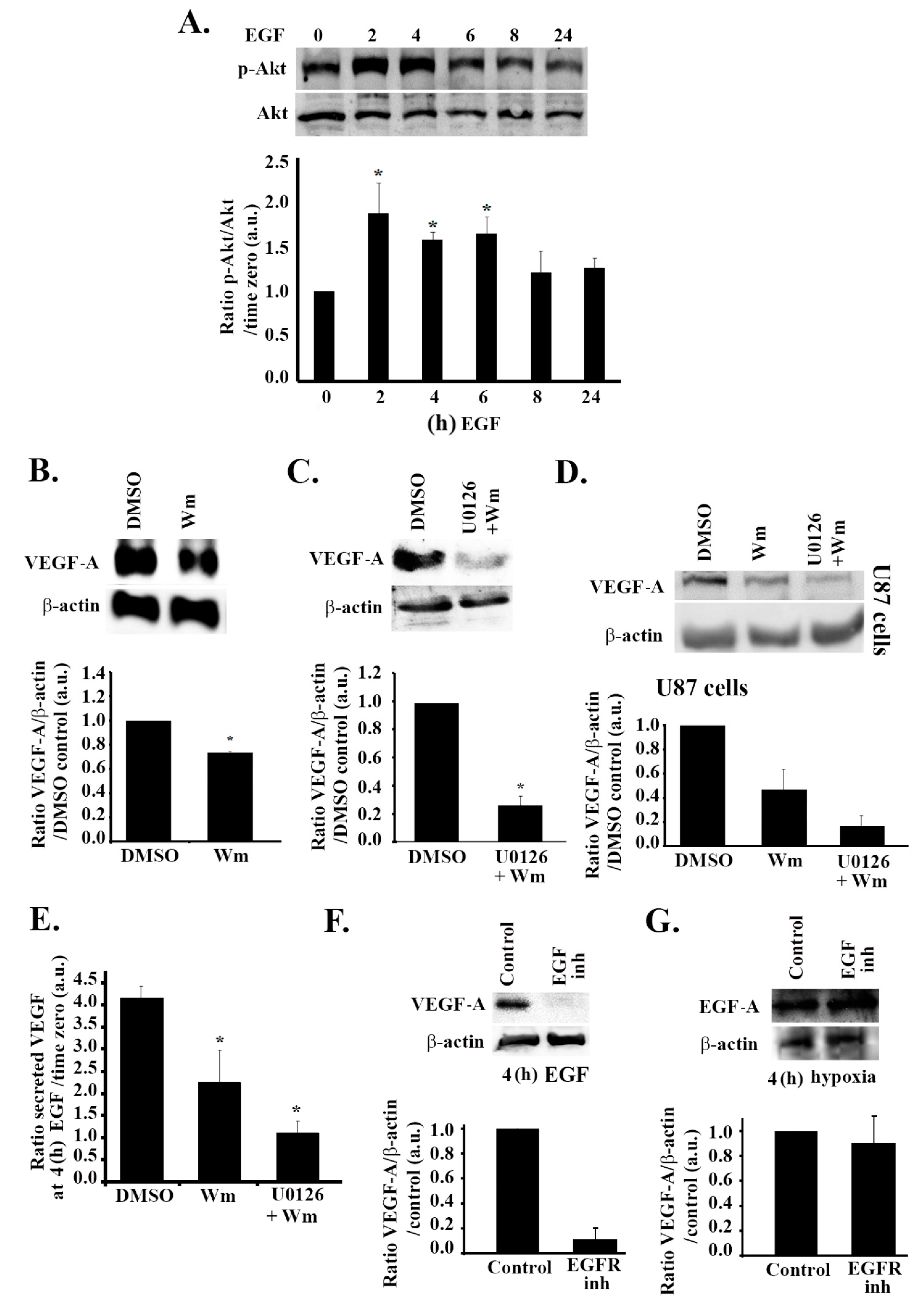
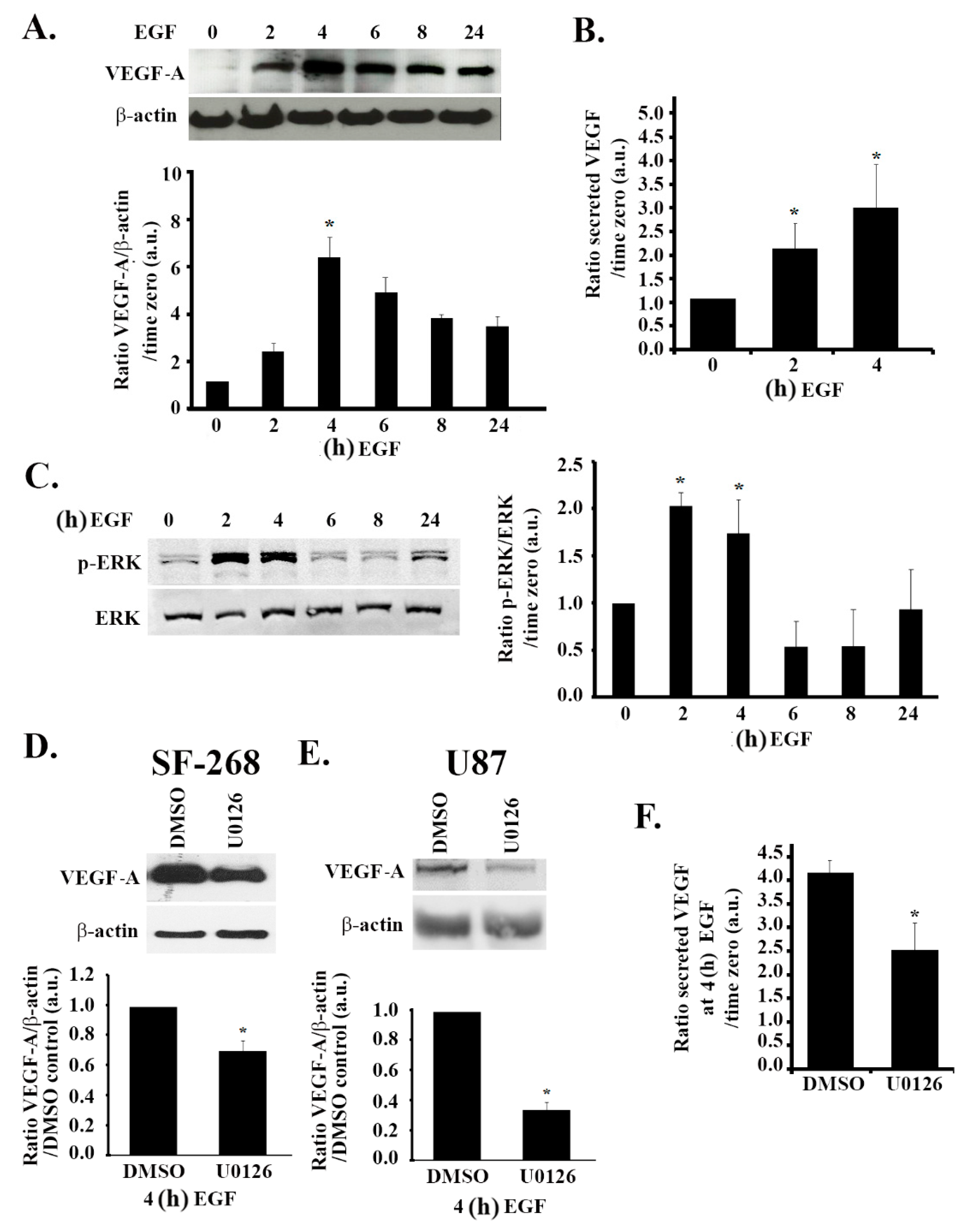
3.4. The MAPK Pathway Positively Regulates VEGF Expression and Secretion in Response to EGF Stimulation
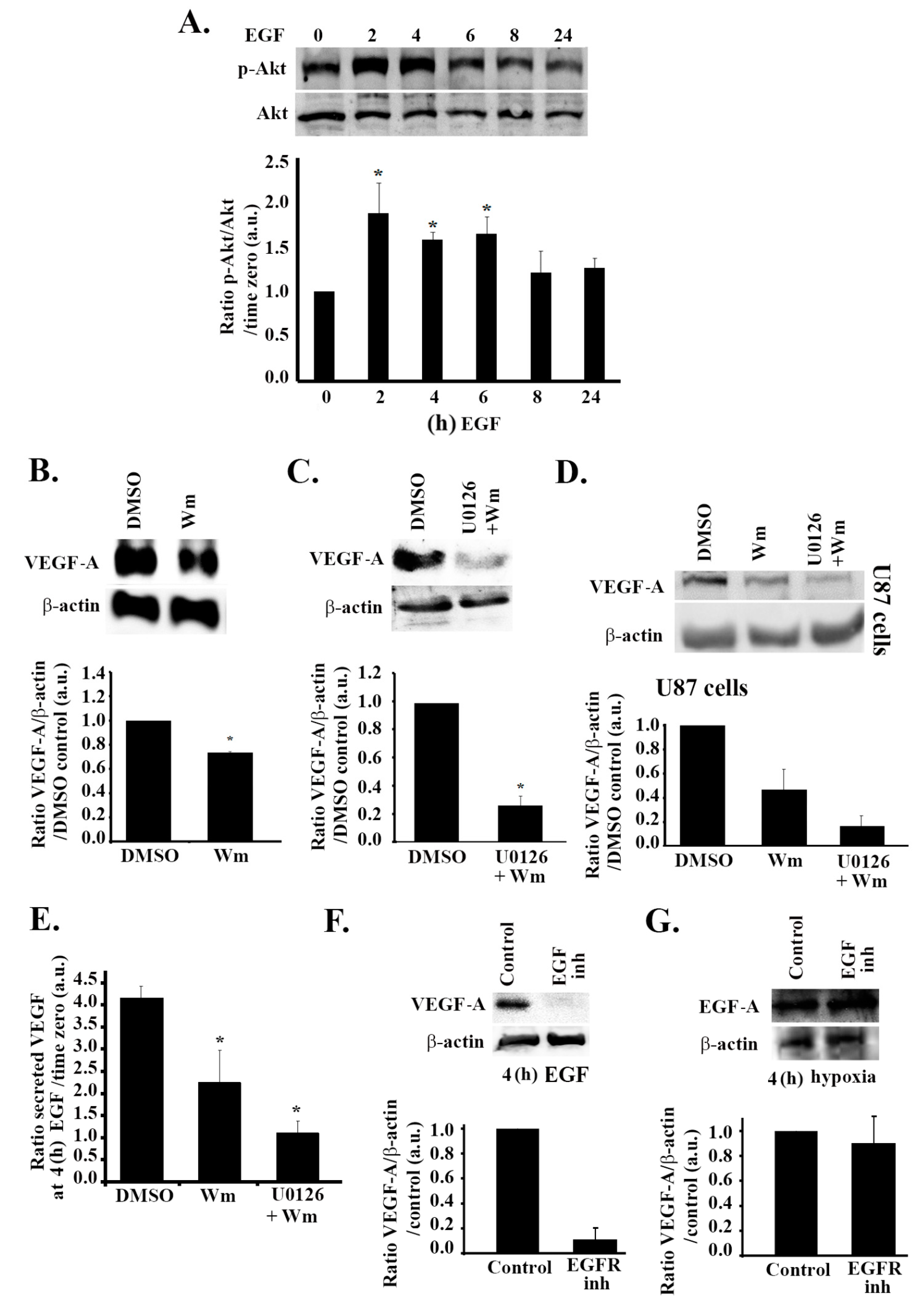
3.5. The PI3K Pathway Is also Involved in EGF Stimulation-Mediated VEGF Expression and Secretion
3.6. Hypoxia and the EGF Receptor Signal in Parallel Leading to VEGF Expression in Glioblastoma Cells
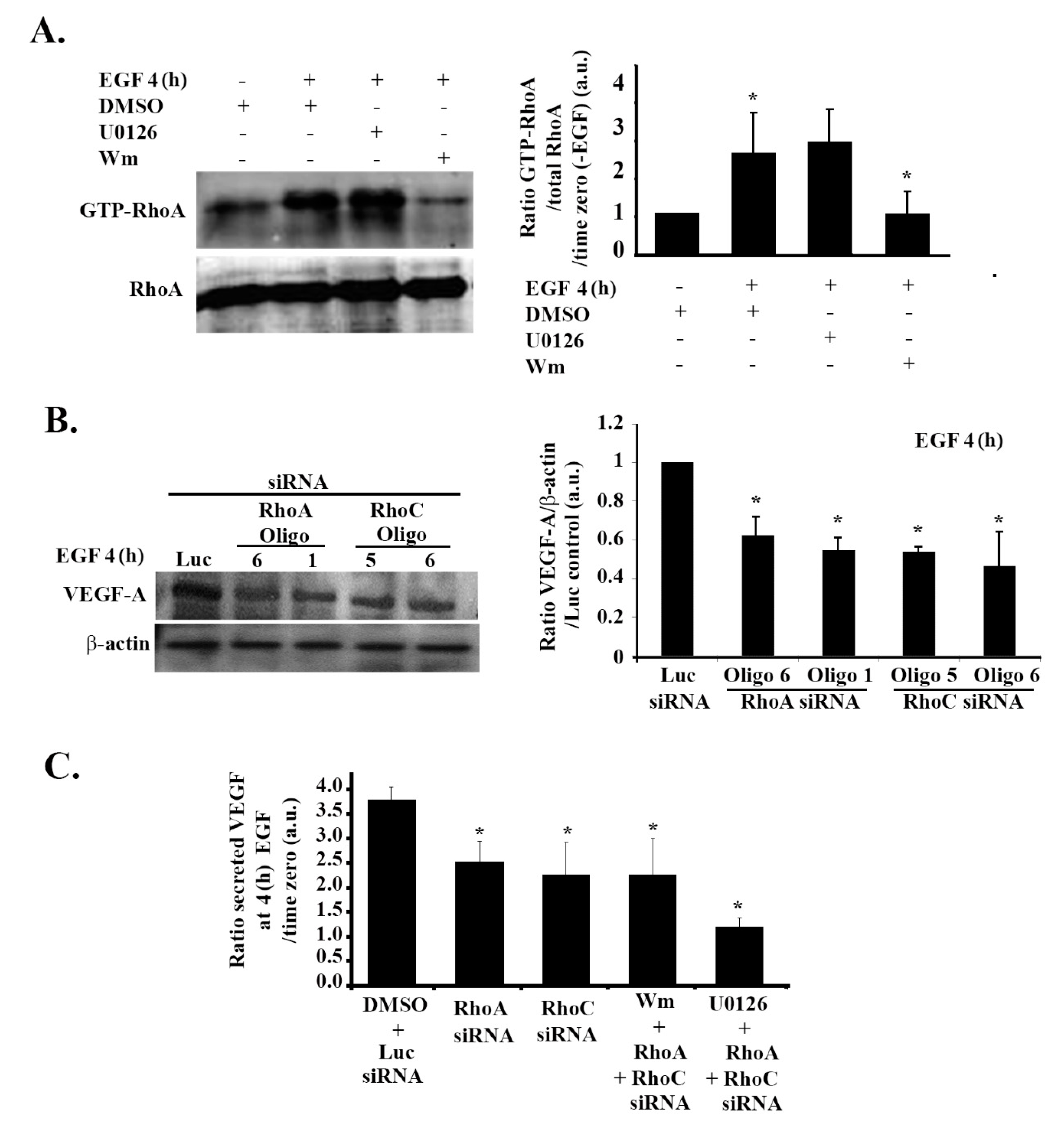
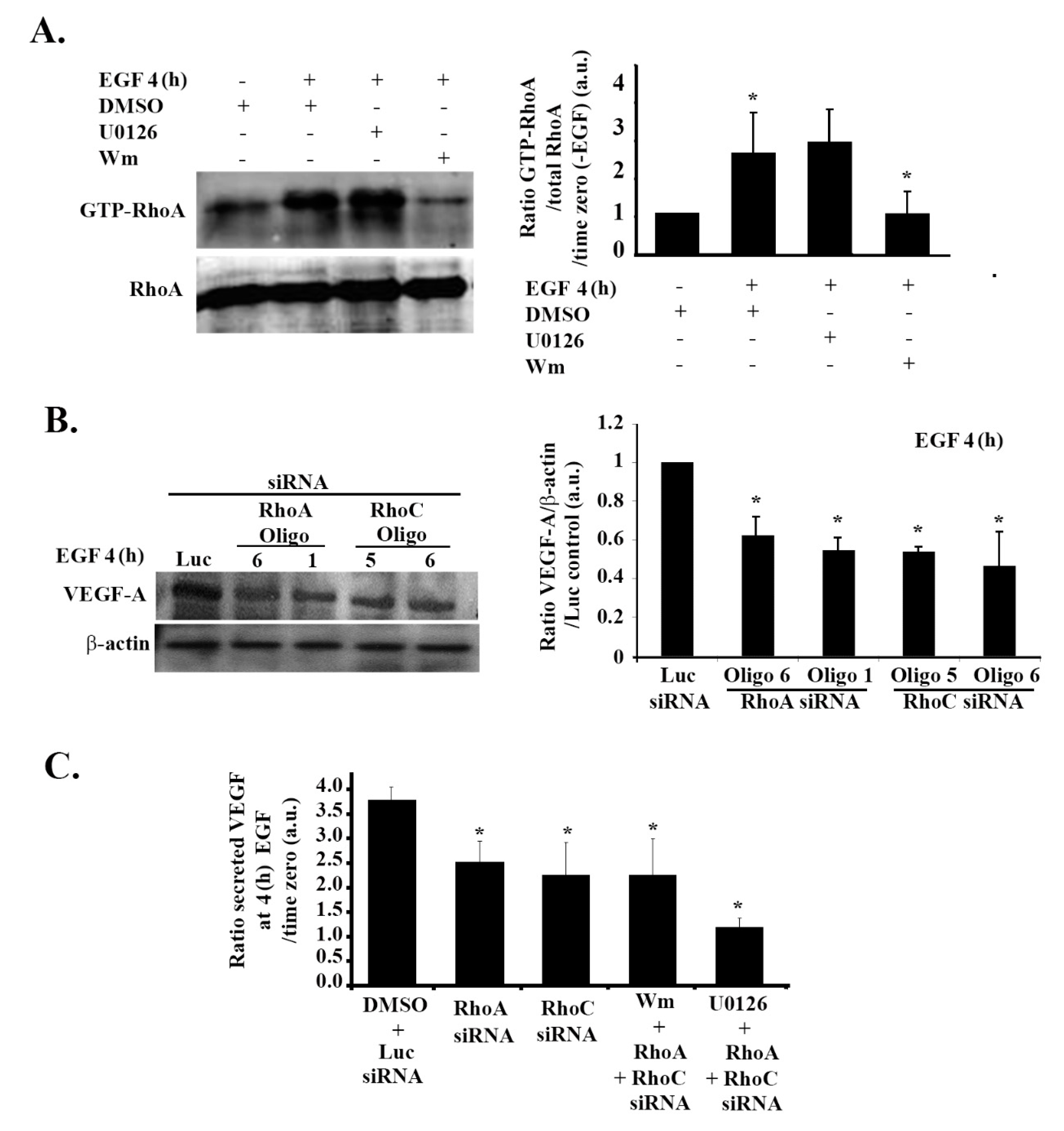
3.7. RhoA and RhoC Mediate PI3K-Regulated Increase in VEGF Expression and Secretion in Response to EGF Stimulation in Glioblastoma Cells
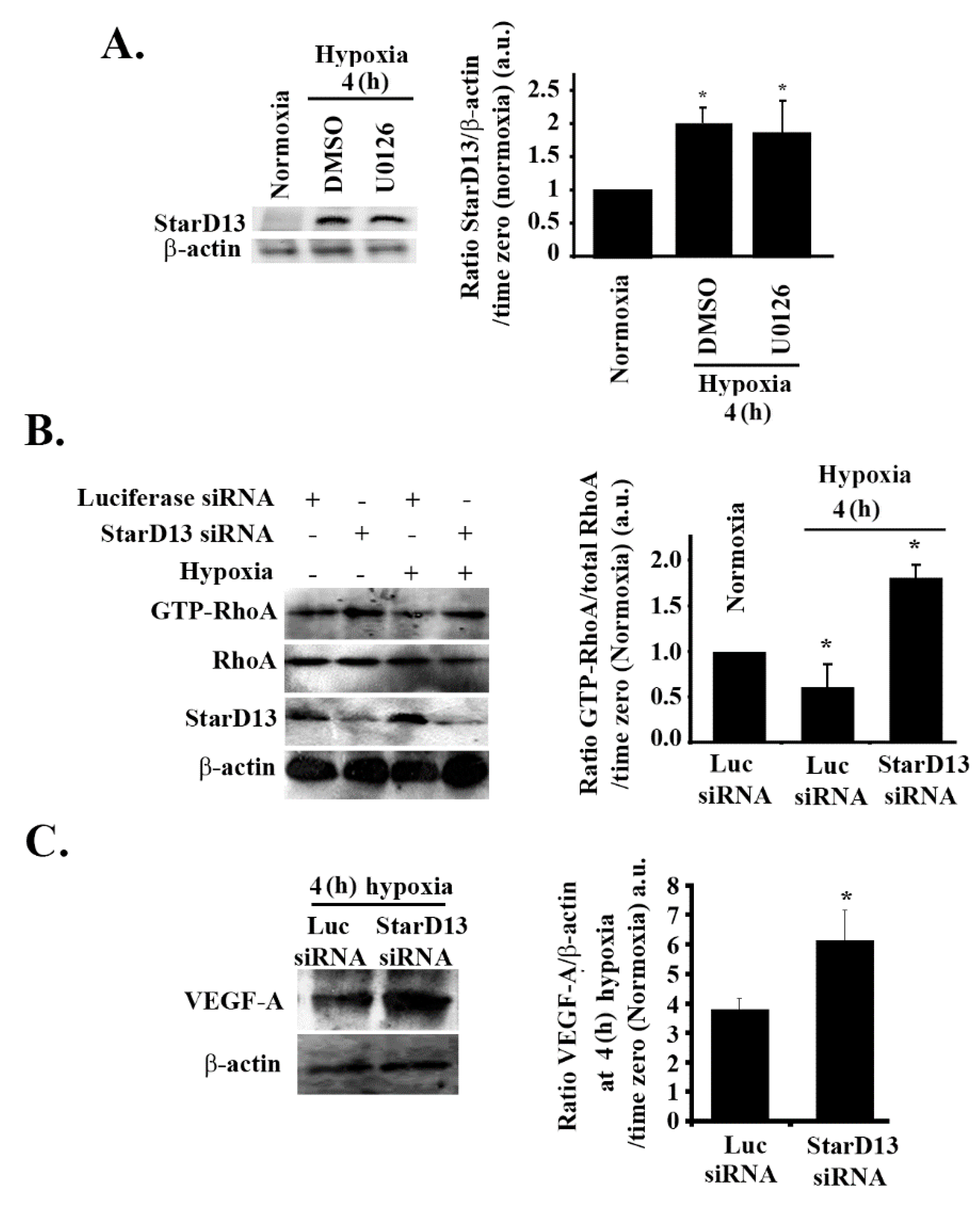
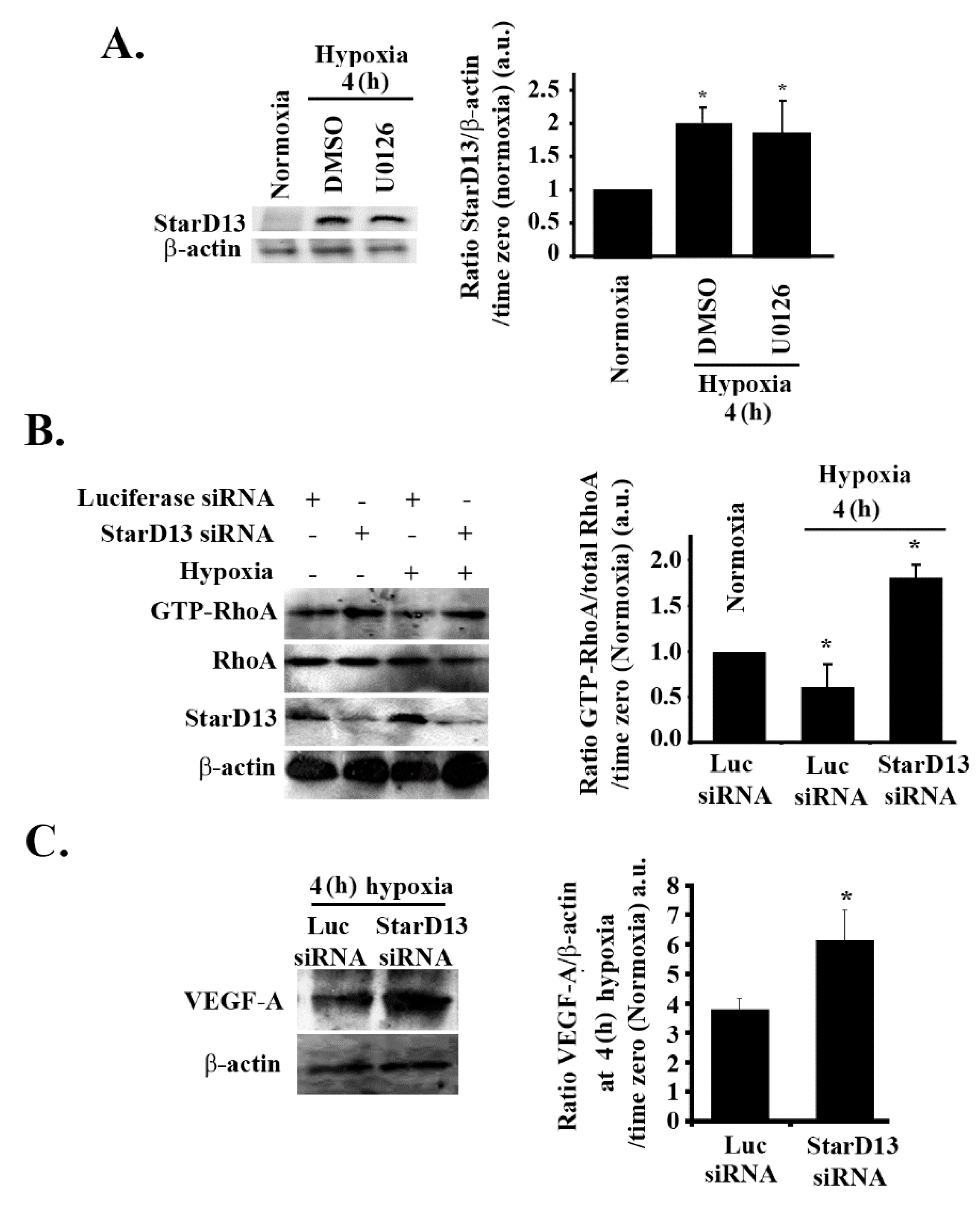
3.8. Hypoxia Suppresses RhoA/C through StarD13
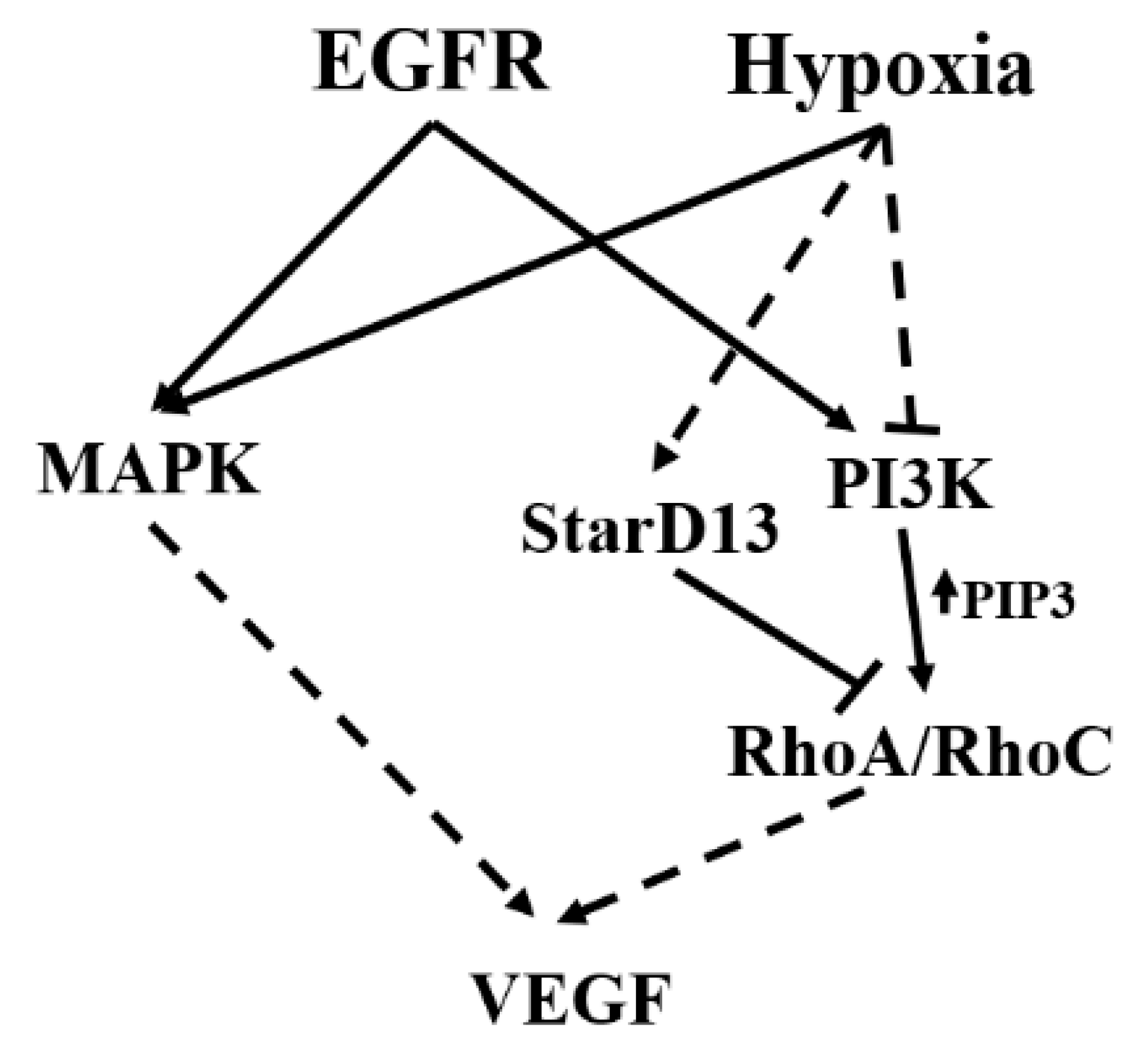
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abou-Antoun, T.J.; Hale, J.S.; Lathia, J.D.; Dombrowski, S.M. Brain Cancer Stem Cells in Adults and Children: Cell Biology and Therapeutic Implications. Neurotherapeutics 2017, 14, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Thews, O.; Kelleher, D.K.; Hoeckel, M. Current status of knowledge and critical issues in tumor oxygenation. Results from 25 years research in tumor pathophysiology. Adv. Exp. Med. Biol. 1998, 454, 591–602. [Google Scholar] [PubMed]
- Musah-Eroje, A.; Watson, S. Adaptive Changes of Glioblastoma Cells Following Exposure to Hypoxic (1% Oxygen) Tumour Microenvironment. Int. J. Mol. Sci. 2019, 20, 2091. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Gerber, H.P.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J. Biol. Chem. 1998, 273, 13313–13316. [Google Scholar] [CrossRef]
- Ikeda, E.; Achen, M.G.; Breier, G.; Risau, W. Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J. Biol. Chem. 1995, 270, 19761–19766. [Google Scholar] [CrossRef]
- Maity, A.; Pore, N.; Lee, J.; Solomon, D.; O’Rourke, D.M. Epidermal growth factor receptor transcriptionally up-regulates vascular endothelial growth factor expression in human glioblastoma cells via a pathway involving phosphatidylinositol 3′-kinase and distinct from that induced by hypoxia. Cancer Res. 2000, 60, 5879–5886. [Google Scholar]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef]
- Chan, A.S.; Leung, S.Y.; Wong, M.P.; Yuen, S.T.; Cheung, N.; Fan, Y.W.; Chung, L.P. Expression of vascular endothelial growth factor and its receptors in the anaplastic progression of astrocytoma, oligodendroglioma, and ependymoma. Am. J. Surg. Pathol. 1998, 22, 816–826. [Google Scholar] [CrossRef]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Weindel, K.; Moringlane, J.R.; Marme, D.; Weich, H.A. Detection and quantification of vascular endothelial growth factor/vascular permeability factor in brain tumor tissue and cyst fluid: The key to angiogenesis? Neurosurgery 1994, 35, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Berkman, R.A.; Merrill, M.J.; Reinhold, W.C.; Monacci, W.T.; Saxena, A.; Clark, W.C.; Robertson, J.T.; Ali, I.U.; Oldfield, E.H. Expression of the vascular permeability factor/vascular endothelial growth factor gene in central nervous system neoplasms. J. Clin. Investig. 1993, 91, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Veikkola, T.; Alitalo, K. VEGFs, receptors and angiogenesis. Semin. Cancer Biol. 1999, 9, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Goldman, C.K.; Kim, J.; Wong, W.L.; King, V.; Brock, T.; Gillespie, G.Y. Epidermal growth factor stimulates vascular endothelial growth factor production by human malignant glioma cells: A model of glioblastoma multiforme pathophysiology. Mol. Biol. Cell 1993, 4, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Chen, Y.; Dellian, M.; Safabakhsh, N.; Ferrara, N.; Jain, R.K. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc. Natl. Acad. Sci. USA 1996, 93, 14765–14770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machein, M.R.; Risau, W.; Plate, K.H. Antiangiogenic gene therapy in a rat glioma model using a dominant-negative vascular endothelial growth factor receptor 2. Hum. Gene Ther. 1999, 10, 1117–1128. [Google Scholar] [CrossRef]
- Weathers, S.P.; de Groot, J. VEGF Manipulation in Glioblastoma. Oncology (Williston Park N.Y.) 2015, 29, 720–727. [Google Scholar]
- Robles Irizarry, L.; Hambardzumyan, D.; Nakano, I.; Gladson, C.L.; Ahluwalia, M.S. Therapeutic targeting of VEGF in the treatment of glioblastoma. Expert Opin. Ther. Targets 2012, 16, 973–984. [Google Scholar] [CrossRef]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985, 313, 144–147. [Google Scholar] [CrossRef]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.; Smith, K.; Gullick, W.J.; Harris, A.L. Mutant epidermal growth factor receptor enhances induction of vascular endothelial growth factor by hypoxia and insulin-like growth factor-1 via a PI3 kinase dependent pathway. Br. J. Cancer 2001, 84, 1322–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, R.L. Growth factor-mediated angiogenesis in the malignant progression of glial tumors: A review. Surg. Neurol. 1998, 49, 189–195. [Google Scholar] [CrossRef]
- Liang, X.; Ding, Y.; Lin, F.; Zhang, Y.; Zhou, X.; Meng, Q.; Lu, X.; Jiang, G.; Zhu, H.; Chen, Y.; et al. Overexpression of ERBB4 rejuvenates aged mesenchymal stem cell and enhances angiogenesis via PI3K/AKT and MAPK/ERK pathway. FASEB J. 2018, 33, 4559–4570. [Google Scholar] [CrossRef] [PubMed]
- Petit, A.M.; Rak, J.; Hung, M.C.; Rockwell, P.; Goldstein, N.; Fendly, B.; Kerbel, R.S. Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: Angiogenic implications for signal transduction therapy of solid tumors. Am. J. Pathol. 1997, 151, 1523–1530. [Google Scholar] [PubMed]
- Nagane, M.; Coufal, F.; Lin, H.; Bogler, O.; Cavenee, W.K.; Huang, H.J. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996, 56, 5079–5086. [Google Scholar]
- Swinson, D.E.; O’Byrne, K.J. Interactions between hypoxia and epidermal growth factor receptor in non-small-cell lung cancer. Clin. Lung. Cancer 2006, 7, 250–256. [Google Scholar] [CrossRef]
- Wang, Y.; Roche, O.; Xu, C.; Moriyama, E.H.; Heir, P.; Chung, J.; Roos, F.C.; Chen, Y.; Finak, G.; Milosevic, M.; et al. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor-mediated upregulation of caveolin-1. Proc. Natl. Acad. Sci. USA 2012, 109, 4892–4897. [Google Scholar] [CrossRef]
- Munk, M.; Memon, A.A.; Goetze, J.P.; Nielsen, L.B.; Nexo, E.; Sorensen, B.S. Hypoxia changes the expression of the epidermal growth factor (EGF) system in human hearts and cultured cardiomyocytes. PLoS ONE 2012, 7, e40243. [Google Scholar] [CrossRef]
- Wu, C.J.; Qian, X.; O’Rourke, D.M. Sustained mitogen-activated protein kinase activation is induced by transforming erbB receptor complexes. DNA Cell. Biol. 1999, 18, 731–741. [Google Scholar] [CrossRef]
- Wu, C.J.; Chen, Z.; Ullrich, A.; Greene, M.I.; O’Rourke, D.M. Inhibition of EGFR-mediated phosphoinositide-3-OH kinase (PI3-K) signaling and glioblastoma phenotype by signal-regulatory proteins (SIRPs). Oncogene 2000, 19, 3999–4010. [Google Scholar] [CrossRef] [PubMed]
- Dise, R.S.; Frey, M.R.; Whitehead, R.H.; Polk, D.B. Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G276–G285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbach, J.P.; Klumpp, A.; Wolburg, H.; Weller, M. Inhibition of epidermal growth factor receptor signaling protects human malignant glioma cells from hypoxia-induced cell death. Cancer Res. 2004, 64, 1575–1578. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Bosse, T.; Ehinger, J.; Czuchra, A.; Benesch, S.; Steffen, A.; Wu, X.; Schloen, K.; Niemann, H.H.; Scita, G.; Stradal, T.E.; et al. Cdc42 and phosphoinositide 3-kinase drive Rac-mediated actin polymerization downstream of c-Met in distinct and common pathways. Mol. Cell. Biol. 2007, 27, 6615–6628. [Google Scholar] [CrossRef]
- Davies, E.M.; Gurung, R.; Le, K.Q.; Mitchell, C.A. Effective angiogenesis requires regulation of phosphoinositide signaling. Adv. Biol. Regul. 2018, 71, 69–78. [Google Scholar] [CrossRef]
- Yip, S.C.; El-Sibai, M.; Coniglio, S.J.; Mouneimne, G.; Eddy, R.J.; Drees, B.E.; Neilsen, P.O.; Goswami, S.; Symons, M.; Condeelis, J.S.; et al. The distinct roles of Ras and Rac in PI 3-kinase-dependent protrusion during EGF-stimulated cell migration. J. Cell. Sci. 2007, 120, 3138–3146. [Google Scholar] [CrossRef] [Green Version]
- Mizukami, Y.; Fujiki, K.; Duerr, E.M.; Gala, M.; Jo, W.S.; Zhang, X.; Chung, D.C. Hypoxic regulation of vascular endothelial growth factor through the induction of phosphatidylinositol 3-kinase/Rho/ROCK and c-Myc. J. Biol. Chem. 2006, 281, 13957–13963. [Google Scholar] [CrossRef]
- Ping, Y.F.; Yao, X.H.; Jiang, J.Y.; Zhao, L.T.; Yu, S.C.; Jiang, T.; Lin, M.C.; Chen, J.H.; Wang, B.; Zhang, R.; et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef]
- Grugel, S.; Finkenzeller, G.; Weindel, K.; Barleon, B.; Marme, D. Both v-Ha-Ras and v-Raf stimulate expression of the vascular endothelial growth factor in NIH 3T3 cells. J. Biol. Chem. 1995, 270, 25915–25919. [Google Scholar] [CrossRef]
- Rak, J.; Mitsuhashi, Y.; Sheehan, C.; Tamir, A.; Viloria-Petit, A.; Filmus, J.; Mansour, S.J.; Ahn, N.G.; Kerbel, R.S. Oncogenes and tumor angiogenesis: Differential modes of vascular endothelial growth factor up-regulation in ras-transformed epithelial cells and fibroblasts. Cancer Res. 2000, 60, 490–498. [Google Scholar] [PubMed]
- Namkoong, S.; Kim, C.K.; Cho, Y.L.; Kim, J.H.; Lee, H.; Ha, K.S.; Choe, J.; Kim, P.H.; Won, M.H.; Kwon, Y.G.; et al. Forskolin increases angiogenesis through the coordinated cross-talk of PKA-dependent VEGF expression and Epac-mediated PI3K/Akt/eNOS signaling. Cell. Signal. 2009, 21, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Bryan, B.A.; D’Amore, P.A. What tangled webs they weave: Rho-GTPase control of angiogenesis. Cell. Mol. Life Sci. 2007, 64, 2053–2065. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell. Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, rac and cdc42 GTPases: Regulators of actin structures, cell adhesion and motility. Biochem. Soc. Trans. 1995, 23, 456–459. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; Sharma, V.P.; Roh-Johnson, M.; Chen, X.; Eddy, R.; Condeelis, J.; Hodgson, L. Spatial regulation of RhoC activity defines protrusion formation in migrating cells. J. Cell Sci. 2013, 126, 3356–3369. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Hall, A. Guanine nucleotide exchange factors for Rho GTPases: Turning on the switch. Genes Dev. 2002, 16, 1587–1609. [Google Scholar] [CrossRef]
- Hall, A.; Nobes, C.D. Rho GTPases: Molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos. Trans. R Soc. Lond. B Biol. Sci. 2000, 355, 965–970. [Google Scholar] [CrossRef]
- Hotchin, N.A.; Hall, A. Regulation of the actin cytoskeleton, integrins and cell growth by the Rho family of small GTPases. Cancer Surv. 1996, 27, 311–322. [Google Scholar]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, F.; Fang, F.; Tao, Y.; Yang, L. RhoC is essential for angiogenesis induced by hepatocellular carcinoma cells via regulation of endothelial cell organization. Cancer Sci. 2008, 99, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Holinstat, M.; Knezevic, N.; Broman, M.; Samarel, A.M.; Malik, A.B.; Mehta, D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: Role in regulation of endothelial permeability. J. Biol. Chem. 2006, 281, 2296–2305. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Monaghan-Benson, E.; Burridge, K. The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J. Biol. Chem. 2009, 284, 25602–25611. [Google Scholar] [CrossRef] [PubMed]
- Vader, P.; van der Meel, R.; Symons, M.H.; Fens, M.H.; Pieters, E.; Wilschut, K.J.; Storm, G.; Jarzabek, M.; Gallagher, W.M.; Schiffelers, R.M.; et al. Examining the role of Rac1 in tumor angiogenesis and growth: A clinically relevant RNAi-mediated approach. Angiogenesis 2011, 14, 457–466. [Google Scholar] [CrossRef] [PubMed]
- van der Meel, R.; Symons, M.H.; Kudernatsch, R.; Kok, R.J.; Schiffelers, R.M.; Storm, G.; Gallagher, W.M.; Byrne, A.T. The VEGF/Rho GTPase signalling pathway: A promising target for anti-angiogenic/anti-invasion therapy. Drug. Discov. Today 2011, 16, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Fryer, B.H.; Field, J. Rho, Rac, Pak and angiogenesis: Old roles and newly identified responsibilities in endothelial cells. Cancer Lett. 2005, 229, 13–23. [Google Scholar] [CrossRef]
- Zhao, L.; Xu, G.; Zhou, J.; Xing, H.; Wang, S.; Wu, M.; Lu, Y.P.; Ma, D. The effect of RhoA on human umbilical vein endothelial cell migration and angiogenesis in vitro. Oncol. Rep. 2006, 15, 1147–1152. [Google Scholar] [CrossRef]
- Bryan, B.A.; Dennstedt, E.; Mitchell, D.C.; Walshe, T.E.; Noma, K.; Loureiro, R.; Saint-Geniez, M.; Campaigniac, J.P.; Liao, J.K.; D’Amore, P.A. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 2010, 24, 3186–3195. [Google Scholar] [CrossRef]
- Ramis, G.; Thomas-Moya, E.; Fernandez de Mattos, S.; Rodriguez, J.; Villalonga, P. EGFR inhibition in glioma cells modulates Rho signaling to inhibit cell motility and invasion and cooperates with temozolomide to reduce cell growth. PLoS ONE 2012, 7, e38770. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.V.; Whelan, M.C.; Senger, D.R. Rho activity critically and selectively regulates endothelial cell organization during angiogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 1874–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rani, A.; Prasad, S. CoCl2-induced biochemical hypoxia down regulates activities and expression of super oxide dismutase and catalase in cerebral cortex of mice. Neurochem. Res. 2014, 39, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.P.; Mottet, D.; Raes, M.; Michiels, C. CoCl2, a chemical inducer of hypoxia-inducible factor-1, and hypoxia reduce apoptotic cell death in hepatoma cell line HepG2. Ann. N. Y. Acad. Sci. 2002, 973, 443–447. [Google Scholar]
- Al-Dimassi, S.; Salloum, G.; Saykali, B.; Khoury, O.; Liu, S.; Leppla, S.H.; Abi-Habib, R.; El-Sibai, M. Targeting the MAP kinase pathway in astrocytoma cells using a recombinant anthrax lethal toxin as a way to inhibit cell motility and invasion. Int. J. Oncol. 2016, 48, 1913–1920. [Google Scholar] [CrossRef] [Green Version]
- Khalil, B.D.; Hanna, S.; Saykali, B.A.; El-Sitt, S.; Nasrallah, A.; Marston, D.; El-Sabban, M.; Hahn, K.M.; Symons, M.; El-Sibai, M. The regulation of RhoA at focal adhesions by StarD13 is important for astrocytoma cell motility. Exp. Cell Res. 2014, 321, 109–122. [Google Scholar] [CrossRef]
- Hanna, S.; Khalil, B.; Nasrallah, A.; Saykali, B.A.; Sobh, R.; Nasser, S.; El-Sibai, M. StarD13 is a tumor suppressor in breast cancer that regulates cell motility and invasion. Int. J. Oncol. 2014, 44, 1499–1511. [Google Scholar] [CrossRef] [Green Version]
- El-Sitt, S.; Khalil, B.D.; Hanna, S.; El-Sabban, M.; Fakhreddine, N.; El-Sibai, M. DLC2/StarD13 plays a role of a tumor suppressor in astrocytoma. Oncol. Rep. 2012, 28, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Tahergorabi, Z.; Khazaei, M. A review on angiogenesis and its assays. Iran. J. Basic Med. Sci. 2012, 15, 1110–1126. [Google Scholar]
- El Atat, O.; Fakih, A.; El-Sibai, M. RHOG Activates RAC1 through CDC42 Leading to Tube Formation in Vascular Endothelial Cells. Cells 2019, 8, 171. [Google Scholar] [CrossRef]
- Bisht, M.; Dhasmana, D.C.; Bist, S.S. Angiogenesis: Future of pharmacological modulation. Indian J. Pharmacol. 2010, 42, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Yotnda, P. Induction and testing of hypoxia in cell culture. J. Vis. Exp. 2011. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Hilliard, G.; Ferguson, T.; Millhorn, D.E. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J. Biol. Chem. 2003, 278, 15911–15916. [Google Scholar] [CrossRef] [PubMed]
- Guille, A.; Chaffanet, M.; Birnbaum, D. Signaling pathway switch in breast cancer. Cancer Cell Int. 2013, 13, 66. [Google Scholar] [CrossRef]
- Triantafyllou, A.; Liakos, P.; Tsakalof, A.; Georgatsou, E.; Simos, G.; Bonanou, S. Cobalt induces hypoxia-inducible factor-1alpha (HIF-1alpha) in HeLa cells by an iron-independent, but ROS-, PI-3K- and MAPK-dependent mechanism. Free Radic. Res. 2006, 40, 847–856. [Google Scholar] [CrossRef]
- Alvarez-Tejado, M.; Alfranca, A.; Aragones, J.; Vara, A.; Landazuri, M.O.; del Peso, L. Lack of evidence for the involvement of the phosphoinositide 3-kinase/Akt pathway in the activation of hypoxia-inducible factors by low oxygen tension. J. Biol. Chem. 2002, 277, 13508–13517. [Google Scholar] [CrossRef]
- Cheng, C.I.; Lee, Y.H.; Chen, P.H.; Lin, Y.C.; Chou, M.H.; Kao, Y.H. Cobalt chloride induces RhoA/ROCK activation and remodeling effect in H9c2 cardiomyoblasts: Involvement of PI3K/Akt and MAPK pathways. Cell. Signal. 2017, 36, 25–33. [Google Scholar] [CrossRef]
- Steinbach, J.P.; Wolburg, H.; Klumpp, A.; Probst, H.; Weller, M. Hypoxia-induced cell death in human malignant glioma cells: Energy deprivation promotes decoupling of mitochondrial cytochrome c release from caspase processing and necrotic cell death. Cell Death Differ. 2003, 10, 823–832. [Google Scholar] [CrossRef]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR signaling networks in glioma. Sci. Signal. 2009, 2, re6. [Google Scholar] [CrossRef]
- Marcoux, N.; Vuori, K. EGF receptor mediates adhesion-dependent activation of the Rac GTPase: A role for phosphatidylinositol 3-kinase and Vav2. Oncogene 2003, 22, 6100–6106. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolas, S.; Abdellatef, S.; Haddad, M.A.; Fakhoury, I.; El-Sibai, M. Hypoxia and EGF Stimulation Regulate VEGF Expression in Human Glioblastoma Multiforme (GBM) Cells by Differential Regulation of the PI3K/Rho-GTPase and MAPK Pathways. Cells 2019, 8, 1397. https://doi.org/10.3390/cells8111397
Nicolas S, Abdellatef S, Haddad MA, Fakhoury I, El-Sibai M. Hypoxia and EGF Stimulation Regulate VEGF Expression in Human Glioblastoma Multiforme (GBM) Cells by Differential Regulation of the PI3K/Rho-GTPase and MAPK Pathways. Cells. 2019; 8(11):1397. https://doi.org/10.3390/cells8111397
Chicago/Turabian StyleNicolas, Samer, Sandra Abdellatef, Maria Al Haddad, Isabelle Fakhoury, and Mirvat El-Sibai. 2019. "Hypoxia and EGF Stimulation Regulate VEGF Expression in Human Glioblastoma Multiforme (GBM) Cells by Differential Regulation of the PI3K/Rho-GTPase and MAPK Pathways" Cells 8, no. 11: 1397. https://doi.org/10.3390/cells8111397
APA StyleNicolas, S., Abdellatef, S., Haddad, M. A., Fakhoury, I., & El-Sibai, M. (2019). Hypoxia and EGF Stimulation Regulate VEGF Expression in Human Glioblastoma Multiforme (GBM) Cells by Differential Regulation of the PI3K/Rho-GTPase and MAPK Pathways. Cells, 8(11), 1397. https://doi.org/10.3390/cells8111397