Regulatory T Cells Induce Metastasis by Increasing Tgf-β and Enhancing the Epithelial–Mesenchymal Transition


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Mice
2.3. Isolation, Purification, and Differentiation of Regulatory T Cells (Tregs) from Draining Lymph Nodes
2.4. Migration and Invasion Assays
2.5. Quantification of Transforming Growth Factor-β (TGF-β) Expression
2.6. Western Blot Analysis
2.7. B16-BL6 Spontaneous Lung Metastasis Model
2.8. Immunohistochemical Analysis
2.9. Fluorescence-Activated Cell-Sorting Analysis
2.10. Statistical Analysis
3. Results
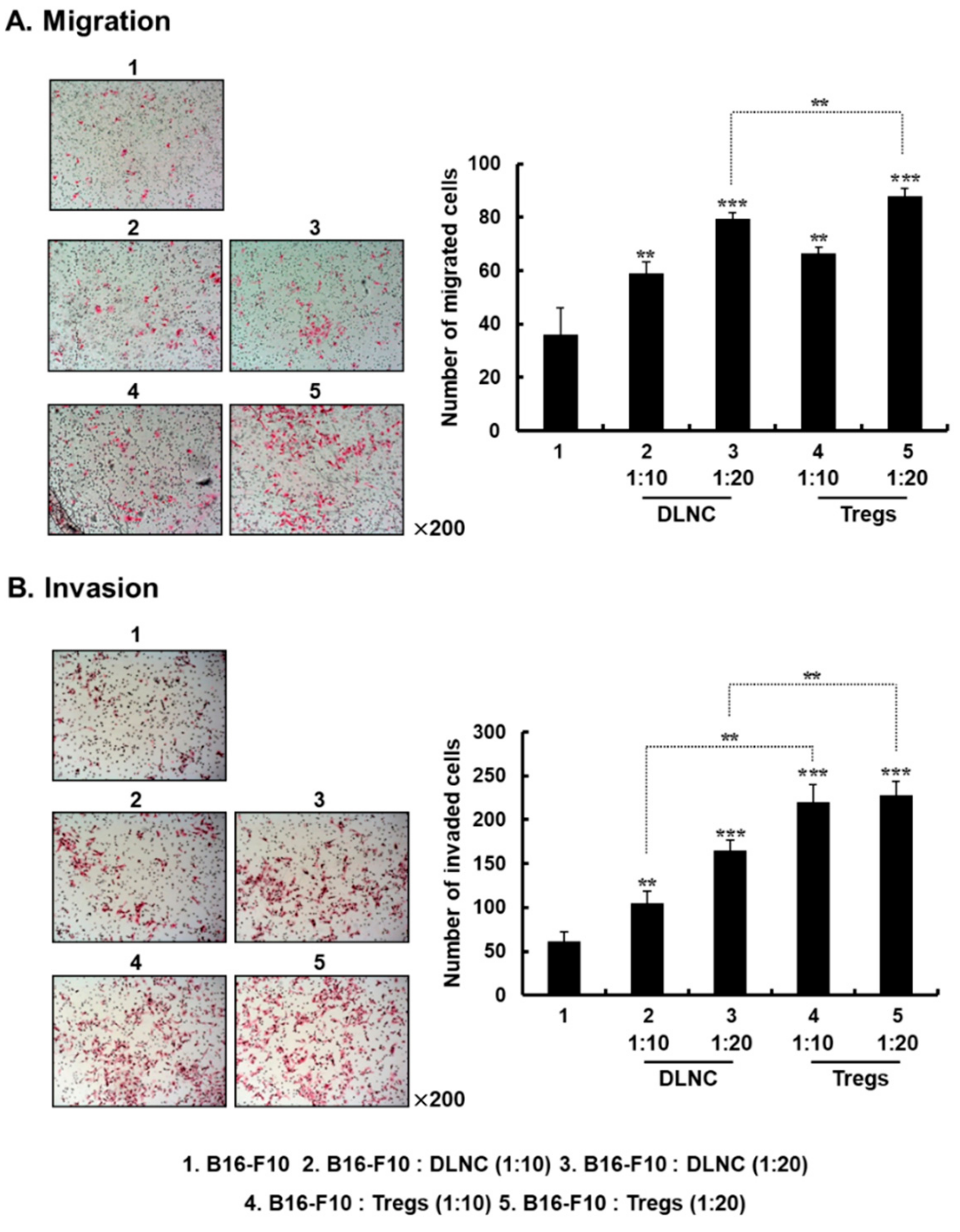
3.1. Effect of Tregs on Melanoma Migration and Invasion In Vitro
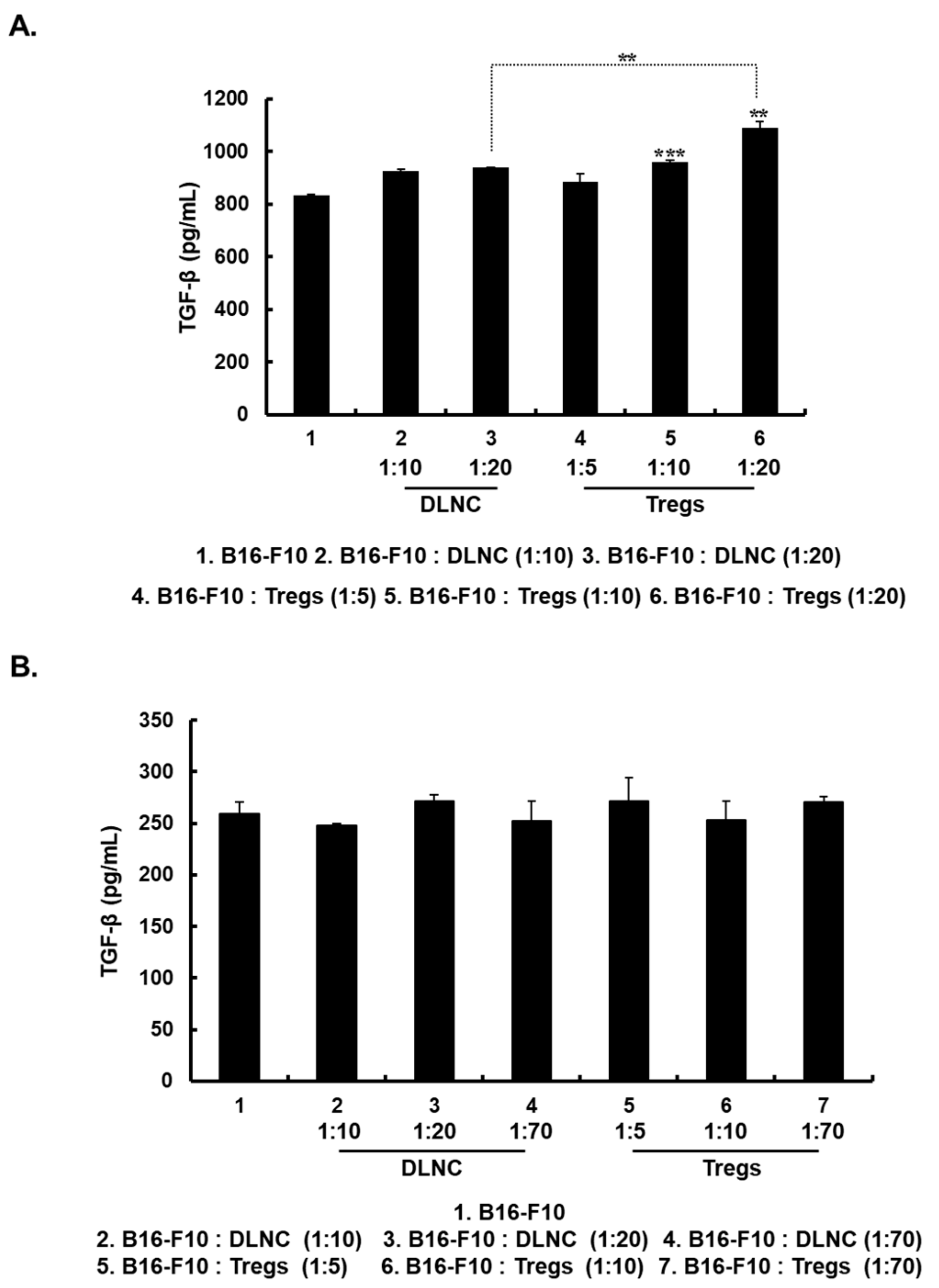
3.2. Increased Expression of TGF-β in Melanoma Cells Following Co-Culture with Tregs
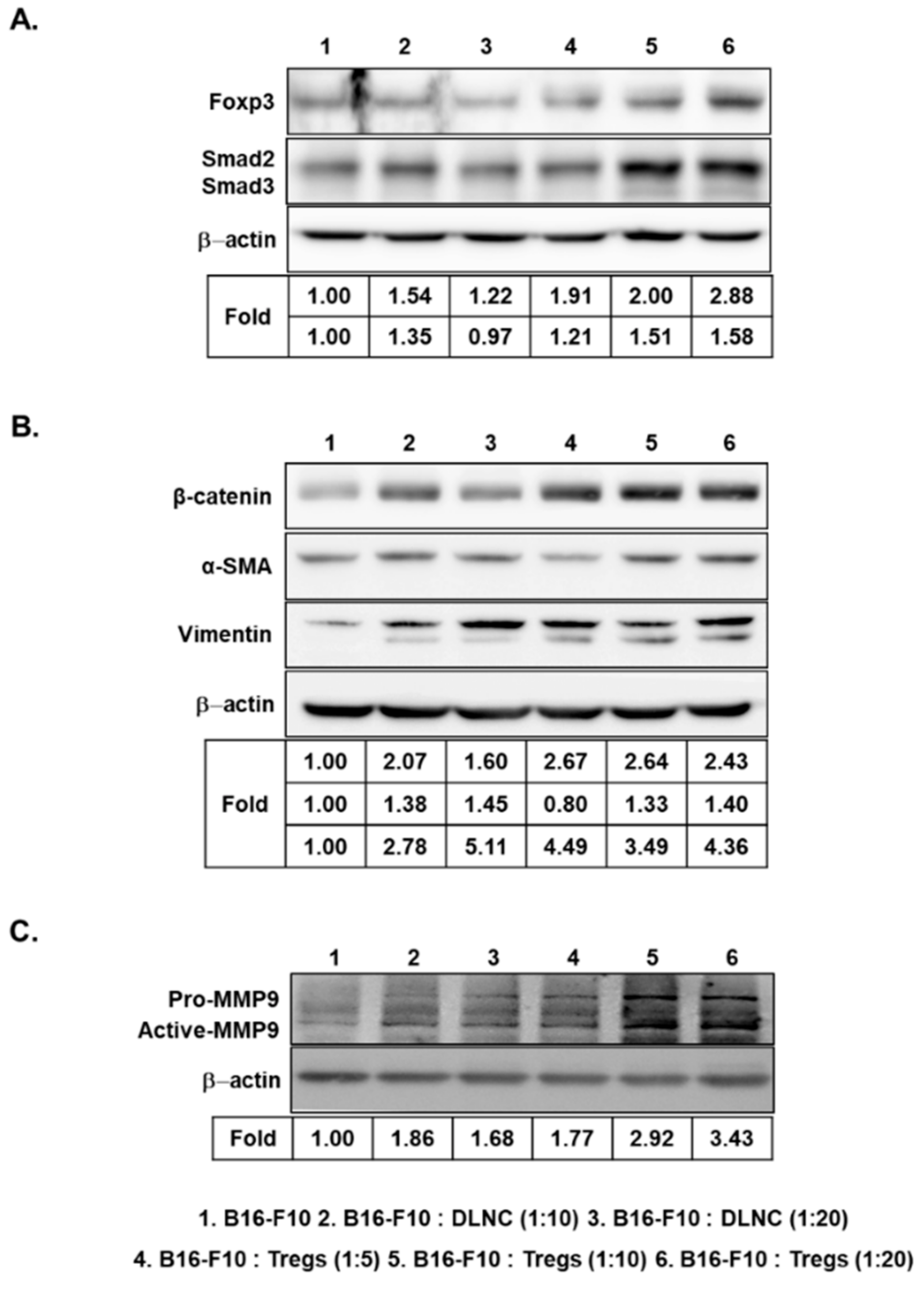
3.3. Increased Expression of Foxp3, Smad2/3, EMT-Related Markers, and MMP9 in Melanoma Cells Following Co-Culture with Tregs
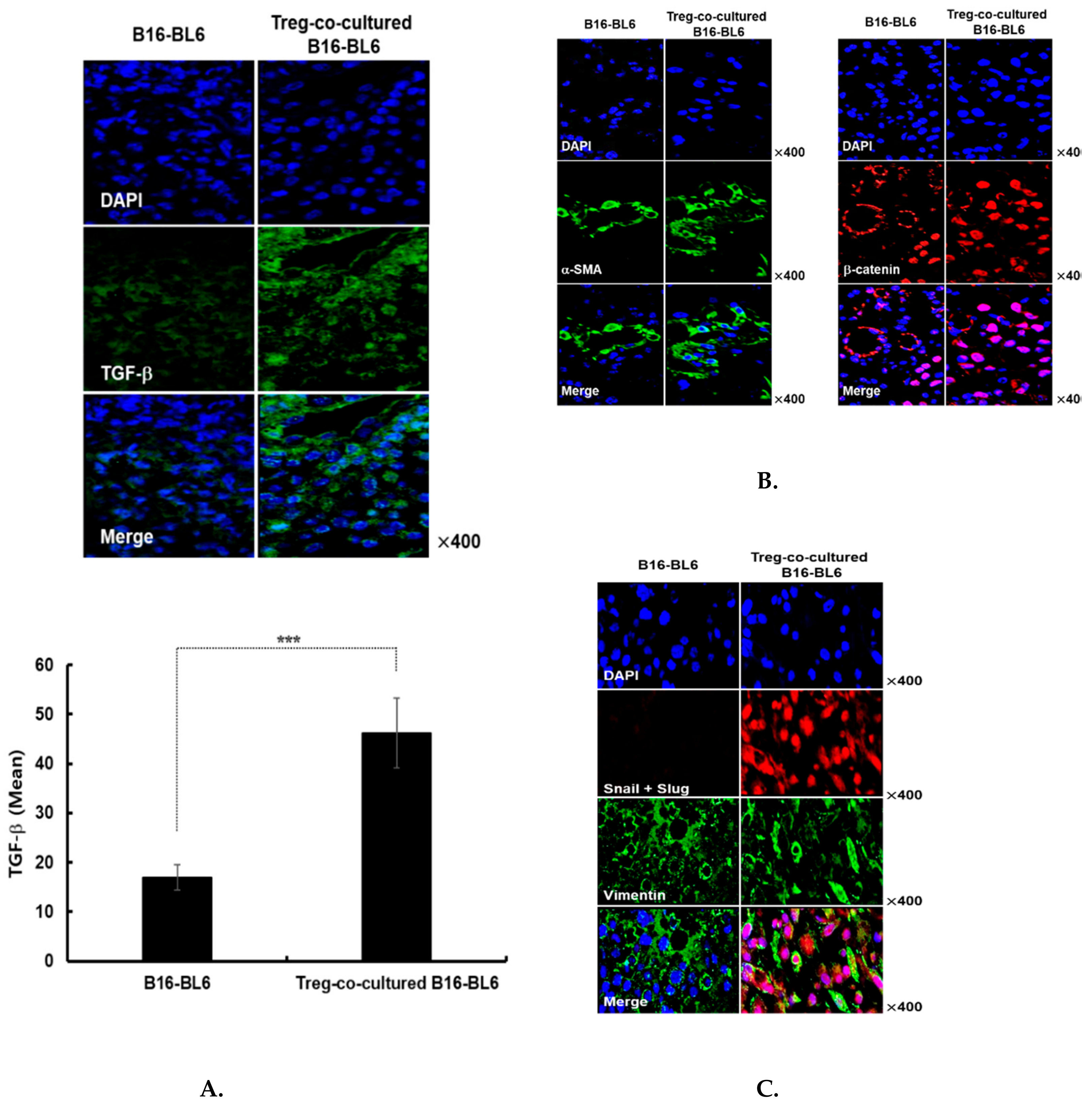
3.4. Immunohistochemical Assessment of Treg Co-Cultured B16-Bl6 Derived Tumor Tissue
3.5. Increased Migration of Cancer Cells from Either Established Treg Co-Cultured B16-Bl6 Derived Tumors or Treg-Injected Tumors
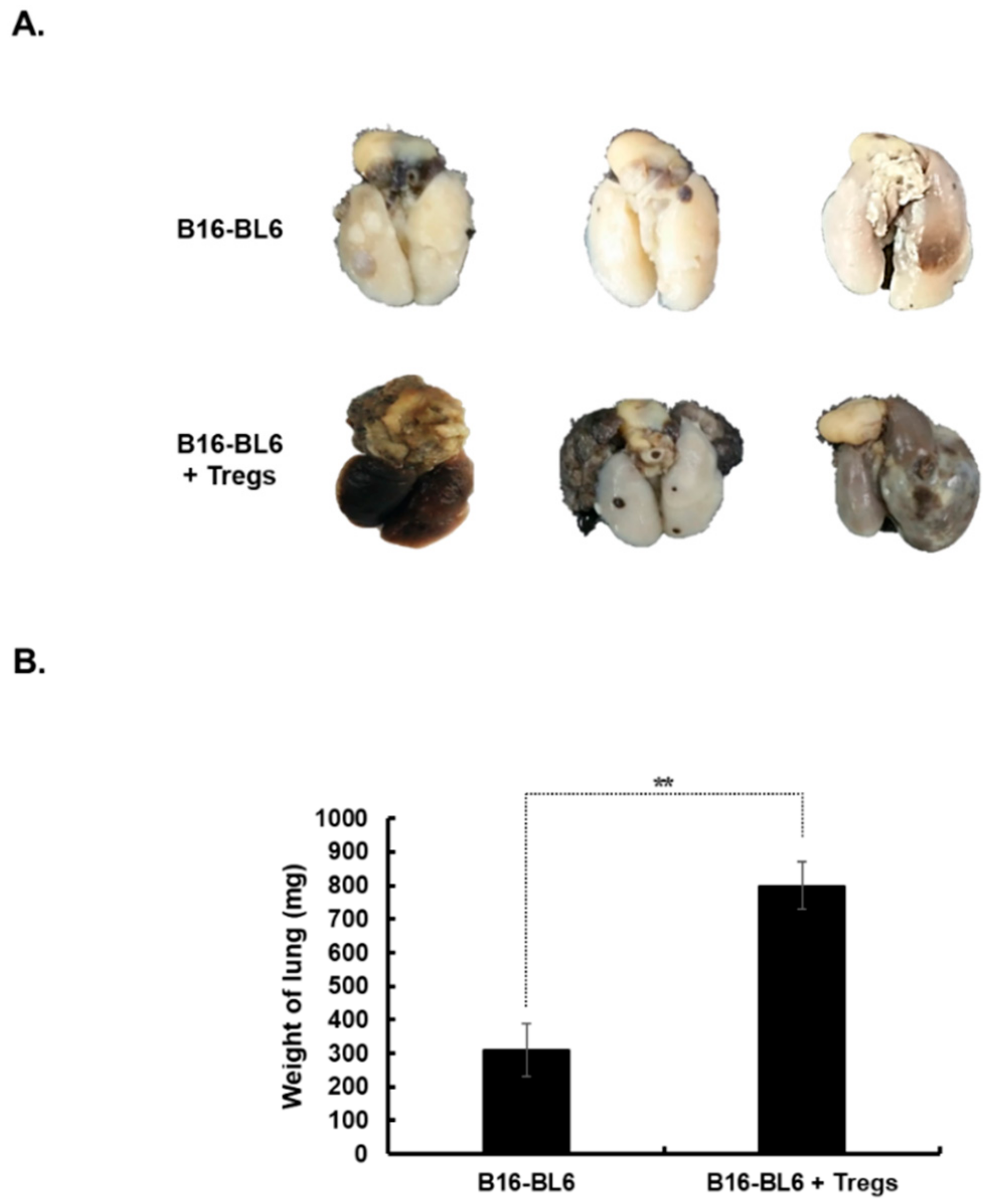
3.6. Promotion of Tumor Metastasis by Treg Injection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Singh, S.; Zafar, A.; Khan, S.; Naseem, I. Towards therapeutic advances in melanoma management: An overview. Life Sci. 2017, 174, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Buzaid, A.C.; Soong, S.-J.; Atkins, M.B.; Cascinelli, N.; Coit, D.G.; Fleming, I.D.; Gershenwald, J.E.; Houghton, A.; Kirkwood, J.M.; et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J. Clin. Oncol. 2001, 19, 3635–3648. [Google Scholar] [CrossRef] [PubMed]
- Apalla, Z.; Lallas, A.; Sotiriou, E.; Lazaridou, E.; Ioannides, D. Epidemiological trends in skin cancer. Dermatol. Pr. Concept. 2017, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Erickson, C.; Driscoll, M.S. Melanoma epidemic: Facts and controversies. Clin. Dermatol. 2010, 28, 281–286. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.-J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef]
- Winder, M.; Virós, A. Mechanisms of Drug Resistance in Melanoma. In Calcitonin Gene-Related Peptide (CGRP) Mechanisms; Springer: Cham, Switzerland, 2017; Volume 3, pp. 91–108. [Google Scholar]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: from mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef]
- Kwong, L.N.; Davies, M.A. Targeted therapy for melanoma: rational combinatorial approaches. Oncogene 2014, 33, 1–9. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Christofori, G. New signals from the invasive front. Nature 2006, 441, 444–450. [Google Scholar] [CrossRef]
- Suman, S.; Kurisetty, V.; Das, T.P.; Vadodkar, A.; Ramos, G.; Lakshmanaswamy, R.; Damodaran, C. Activation of AKT signaling promotes epithelial-mesenchymal transition and tumor growth in colorectal cancer cells. Mol. Carcinog. 2014, 5, E151–E160. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.; Ackland, M.L.; Blick, T.; Lawrence, M.G.; Clements, J.A.; Williams, E.D.; Thompson, E.W. Epithelial—Mesenchymal and mesenchymal—Epithelial transitions in carcinoma progression. J. Cell. Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C. Epithelial-mesenchymal transition. J. Cell Sci. 2005, 118, 4325–4326. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Ibaragi, S.; Hu, G.-F. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res. 2009, 69, 7135–7139. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Suzuki, M.; Nishino, Y.; Funaba, M. Regulatory expression of genes related to metastasis by TGF-beta and activin A in B16 murine melanoma cells. Mol. Biol. Rep. 2010, 37, 1279–1286. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.-H. Signaling networks guiding epithelial?mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef]
- Busse, A.; Keilholz, U. Role of TGF-beta in melanoma. Curr. Pharm. Biotechnol. 2011, 12, 2165–2175. [Google Scholar] [CrossRef]
- Javelaud, D.; Alexaki, V.-I.; Mauviel, A. Transforming growth factor-β in cutaneous melanoma. Pigment. Cell Melanoma Res. 2008, 21, 123–132. [Google Scholar] [CrossRef]
- Javelaud, D.; Mohammad, K.S.; McKenna, C.R.; Fournier, P.; Luciani, F.; Niewolna, M.; Andre, J.; Delmas, V.; LaRue, L.; Guise, T.A.; et al. Stable Overexpression of Smad7 in Human Melanoma Cells Impairs Bone Metastasis. Cancer Res. 2007, 67, 2317–2324. [Google Scholar] [CrossRef]
- Javelaud, D.; Delmas, V.; Moller, M.; Sextius, P.; André, J.; Menashi, S.; LaRue, L.; Mauviel, A. Stable overexpression of Smad7 in human melanoma cells inhibits their tumorigenicity in vitro and in vivo. Oncogene 2005, 24, 7624–7629. [Google Scholar] [CrossRef]
- Ebert, L.M.; Tan, B.S.; Browning, J.; Svobodova, S.; Russell, S.E.; Kirkpatrick, N.; Gedye, C.; Moss, D.; Ng, S.P.; MacGregor, D.; et al. The Regulatory T Cell-Associated Transcription Factor FoxP3 Is Expressed by Tumor Cells. Cancer Res. 2008, 68, 3001–3009. [Google Scholar] [CrossRef] [PubMed]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer Metastasis Is Accelerated through Immunosuppression during Snail-Induced EMT of Cancer Cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Ishii, G.; Hiraoka, N.; Hirayama, S.; Yamauchi, C.; Aokage, K.; Hishida, T.; Yoshida, J.; Nagai, K.; Ochiai, A. Forkhead box P3 regulatory T cells coexisting with cancer associated fibroblasts are correlated with a poor outcome in lung adenocarcinoma. Cancer Sci. 2013, 104, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Li, Q.-J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-β-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Chopra, M.; Riedel, S.S.; Biehl, M.; Krieger, S.; Von Krosigk, V.; Bäuerlein, C.A.; Brede, C.; Garrote, A.-L.J.; Kraus, S.; Schäfer, V.; et al. Tumor necrosis factor receptor 2-dependent homeostasis of regulatory T cells as a player in TNF-induced experimental metastasis. Carcinogenesis 2013, 34, 1296–1303. [Google Scholar] [CrossRef]
- Olkhanud, P.B.; Baatar, D.; Bodogai, M.; Hakim, F.; Gress, R.; Anderson, R.L.; Deng, J.; Xu, M.; Briest, S.; Biragyn, A. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009, 69, 5996–6004. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Pasetto, A.; Jia, L.; Deniger, D.C.; Stevanovic, S.; Robbins, P.F.; Rosenberg, S.A. Tumor-infiltrating human CD4(+) regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci. Immunol. 2019, 4, eaao4310. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, J.-H.; Kim, J.; Huang, J.-H.; Zhang, S.N.; Kang, Y.-A.; Kim, H.; Yun, C.-O. Short hairpin RNA-expressing oncolytic adenovirus-mediated inhibition of IL-8: effects on antiangiogenesis and tumor growth inhibition. Gene Ther. 2008, 15, 635–651. [Google Scholar] [CrossRef]
- Lee, J.-S.; Hur, M.-W.; Lee, S.K.; Choi, W.-I.; Kwon, Y.-G.; Yun, C.-O. A Novel sLRP6E1E2 Inhibits Canonical Wnt Signaling, Epithelial-to-Mesenchymal Transition, and Induces Mitochondria-Dependent Apoptosis in Lung Cancer. PLoS ONE 2012, 7, e36520. [Google Scholar] [CrossRef]
- Choi, I.-K.; Li, Y.; Oh, E.; Kim, J.; Yun, C.-O. Oncolytic Adenovirus Expressing IL-23 and p35 Elicits IFN-γ- and TNF-α-Co-Producing T Cell-Mediated Antitumor Immunity. PLoS ONE 2013, 8, e67512. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, J.-H.; Kwon, Y.-G.; Kim, E.-C.; Kim, N.K.; Choi, H.J.; Yun, C.-O. VEGF-specific Short Hairpin RNA–expressing Oncolytic Adenovirus Elicits Potent Inhibition of Angiogenesis and Tumor Growth. Mol. Ther. 2007, 15, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Zhang, S.N.; Choi, K.J.; Choi, I.K.; Kim, J.H.; Lee, M.G.; Kim, H.; Yun, C.O. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Molecules 2010, 18, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Yoon, A.-R.; Kim, J.-H.; Lee, Y.-S.; Huang, J.-H.; Yun, C.-O. Relaxin Expression From Tumor-Targeting Adenoviruses and Its Intratumoral Spread, Apoptosis Induction, and Efficacy. J. Natl. Cancer Inst. 2006, 98, 1482–1493. [Google Scholar]
- Oh, E.; Choi, I.-K.; Hong, J.; Yun, C.-O. Oncolytic adenovirus coexpressing interleukin-12 and decorin overcomes Treg-mediated immunosuppression inducing potent antitumor effects in a weakly immunogenic tumor model. Oncotarget 2017, 8, 4730–4746. [Google Scholar] [CrossRef]
- Gupta, G.P.; Massagué, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Leivonen, S.-K.; Kähäri, V.-M.; Leivonen, S.; Kähäri, V. Transforming growth factor-β signaling in cancer invasion and metastasis. Int. J. Cancer 2007, 121, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Crampton, S.P.; Hughes, C.C. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity 2007, 26, 227–239. [Google Scholar] [CrossRef]
- Bjørnland, K.; Flatmark, K.; Pettersen, S.; Aaasen, A.O.; Fodstad, Ø.; Mælandsmo, G.M. Matrix Metalloproteinases Participate in Osteosarcoma Invasion. J. Surg. Res. 2005, 127, 151–156. [Google Scholar]
- Handsley, M.M.; Edwards, D.R. Metalloproteinases and their inhibitors in tumor angiogenesis. Int. J. Cancer 2005, 115, 849–860. [Google Scholar] [CrossRef]
- Turpeenniemi-Hujanen, T. Gelatinases (MMP-2 and -9) and their natural inhibitors as prognostic indicators in solid cancers. Biochimie 2005, 87, 287–297. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.C.; A Bonner, J.; Kline, R.W. Impact of beam energy and field margin on penumbra at lung tumor-lung parenchyma interfaces. Int. J. Radiat. Oncol. 1998, 41, 707–713. [Google Scholar] [CrossRef]
- Angelucci, C.; Maulucci, G.; Lama, G.; Proietti, G.; Colabianchi, A.; Papi, M.; Maiorana, A.; De Spirito, M.; Micera, A.; Balzamino, O.B.; et al. Epithelial-Stromal Interactions in Human Breast Cancer: Effects on Adhesion, Plasma Membrane Fluidity and Migration Speed and Directness. PLoS ONE 2012, 7, e50804. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Bain, G.; Wang, X.; Papkoff, J. Regulation of epithelial cell migration and tumor formation by beta-catenin signaling. Exp. Cell Res. 2002, 280, 119–133. [Google Scholar] [CrossRef]
- Kim, K.; Lu, Z.; Hay, E.D. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Boil. Int. 2002, 26, 463–476. [Google Scholar] [CrossRef]
- Mariadason, J.M.; Bordonaro, M.; Aslam, F.; Shi, L.; Kuraguchi, M.; Velcich, A.; Augenlicht, L.H. Down-regulation of beta-catenin TCF signaling is linked to colonic epithelial cell differentiation. Cancer Res. 2001, 61, 3465–3471. [Google Scholar]
- Naishiro, Y.; Yamada, T.; Takaoka, A.S.; Hayashi, R.; Hasegawa, F.; Imai, K.; Hirohashi, S. Restoration of epithelial cell polarity in a colorectal cancer cell line by suppression of beta-catenin/T-cell factor 4-mediated gene transactivation. Cancer Res. 2001, 61, 2751–2758. [Google Scholar]
- Ivaska, J. Vimentin: Central hub in EMT induction? Small Gtpases 2011, 2, 51–53. [Google Scholar] [CrossRef]
- Xu, X.; Sun, P.-L.; Li, J.-Z.; Jheon, S.; Lee, C.-T.; Chung, J.-H. Aberrant Wnt1/β-Catenin Expression is an Independent Poor Prognostic Marker of Non-small Cell Lung Cancer After Surgery. J. Thorac. Oncol. 2011, 6, 716–724. [Google Scholar] [CrossRef]
- Prudkin, L.; Liu, D.D.; Ozburn, N.C.; Sun, M.; Behrens, C.; Tang, X.; Brown, K.C.; Bekele, B.N.; Moran, C.; Wistuba, I.I. Epithelial-to-mesenchymal transition in the development and progression of adenocarcinoma and squamous cell carcinoma of the lung. Mod. Pathol. 2009, 22, 668. [Google Scholar] [CrossRef]
- Soltermann, A.; Tischler, V.; Arbogast, S.; Braun, J.; Probst-Hensch, N.; Weder, W.; Moch, H.; Kristiansen, G. Prognostic Significance of Epithelial-Mesenchymal and Mesenchymal-Epithelial Transition Protein Expression in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 7430–7437. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Merkel, S.; Goehl, J.; Hohenberger, W. Surgical therapy for distant metastases of malignant melanoma. Cancer 2000, 89, 1983–1991. [Google Scholar] [CrossRef]
- Sharpless, S.M.; Das Gupta, T.K. Surgery for metastatic melanoma. Semin. Surg. Oncol. 1998, 14, 311–318. [Google Scholar] [CrossRef]
- Barth, A.; A Wanek, L.; Morton, D.L. Prognostic factors in 1,521 melanoma patients with distant metastases. J. Am. Coll. Surg. 1995, 181, 193–201. [Google Scholar]
- Eccles, S.; Welch, D.R. Metastasis: recent discoveries and novel treatment strategies. Lancet 2007, 369, 1742–1757. [Google Scholar] [CrossRef]
- Kopfstein, L.; Christofori, G. Metastasis: cell-autonomous mechanisms versus contributions by the tumor microenvironment. Cell. Mol. Life Sci. 2006, 63, 449–468. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Trusolino, L. Cancer: the matrix is now in control. Nat. Med. 2005, 11, 1156–1158. [Google Scholar] [CrossRef]
- Mueller, M.M.; Fusenig, N.E. Friends or foes — bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial–mesenchymal transitions. Nat. Rev. Mol. Cell Boil. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Yasuda, H.; Saigusa, S.; Tanaka, K.; Inoue, Y.; Goel, A.; Kusunoki, M. Increased expression of Slug and Vimentin as novel predictive biomarkers for lymph node metastasis and poor prognosis in colorectal cancer. Carcinogenesis 2013, 34, 2548–2557. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.; Pagerols-Raluy, L.; Oberg, H.-H.; Ammerpohl, O.; Grüssel, S.; Sipos, B.; Grützmann, R.; Pilarsky, C.; Ungefroren, H.; Saeger, H.-D.; et al. Foxp3 Expression in Pancreatic Carcinoma Cells as a Novel Mechanism of Immune Evasion in Cancer. Cancer Res. 2007, 67, 8344–8350. [Google Scholar] [CrossRef] [PubMed]
- Merlo, A.; Casalini, P.; Carcangiu, M.L.; Malventano, C.; Triulzi, T.; Ménard, S.; Tagliabue, E.; Balsari, A. FOXP3 Expression and Overall Survival in Breast Cancer. J. Clin. Oncol. 2009, 27, 1746–1752. [Google Scholar] [CrossRef]
- Li, Y.; Li, D.; Yang, W.; Fu, H.; Liu, Y.; Li, Y. Overexpression of the transcription factor FOXP3 in lung adenocarcinoma sustains malignant character by promoting G1/S transition gene CCND1. Tumour Biol. 2016, 37, 7395–7404. [Google Scholar] [CrossRef]
- Chu, R.; Liu, S.Y.; Vlantis, A.C.; Van Hasselt, C.A.; Ng, E.K.; Fan, M.D.; Ng, S.K.; Chan, A.B.; Du, J.; Wei, W.; et al. Inhibition of Foxp3 in cancer cells induces apoptosis of thyroid cancer cells. Mol. Cell. Endocrinol. 2015, 399, 228–234. [Google Scholar] [CrossRef]
- Luo, Q.; Zhang, S.; Wei, H.; Pang, X.; Zhang, H. Roles of Foxp3 in the occurrence and development of cervical cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8717–8730. [Google Scholar]
- Altenberger, C.; Heller, G.; Ziegler, B.; Tomasich, E.; Marhold, M.; Topakian, T.; Müllauer, L.; Heffeter, P.; Lang, G.; End-Pfützenreuter, A.; et al. SPAG6 and L1TD1 are transcriptionally regulated by DNA methylation in non-small cell lung cancers. Mol. Cancer 2017, 16, 1. [Google Scholar] [CrossRef]
- Vihinen, P.; Kähäri, V.-M. Matrix metalloproteinases in cancer: Prognostic markers and therapeutic targets. Int. J. Cancer 2002, 99, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Kida, Y.; Mai, M.; Endo, Y.; Sasaki, T.; Tanaka, J.; Seiki, M. Expression of genes encoding type IV collagen-degrading metalloproteinases and tissue inhibitors of metalloproteinases in various human tumor cells. Oncogene 1992, 7, 77–83. [Google Scholar] [PubMed]
- Kondo, M.; Cubillo, E.; Tobiume, K.; Shirakihara, T.; Fukuda, N.; Suzuki, H.; Shimizu, K.; Takehara, K.; Cano, A.; Saitoh, M.; et al. A role for Id in the regulation of TGF-β-induced epithelial–mesenchymal transdifferentiation. Cell Death Differ. 2004, 11, 1092–1101. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, E.; Hong, J.; Yun, C.-O. Regulatory T Cells Induce Metastasis by Increasing Tgf-β and Enhancing the Epithelial–Mesenchymal Transition. Cells 2019, 8, 1387. https://doi.org/10.3390/cells8111387
Oh E, Hong J, Yun C-O. Regulatory T Cells Induce Metastasis by Increasing Tgf-β and Enhancing the Epithelial–Mesenchymal Transition. Cells. 2019; 8(11):1387. https://doi.org/10.3390/cells8111387
Chicago/Turabian StyleOh, Eonju, JinWoo Hong, and Chae-Ok Yun. 2019. "Regulatory T Cells Induce Metastasis by Increasing Tgf-β and Enhancing the Epithelial–Mesenchymal Transition" Cells 8, no. 11: 1387. https://doi.org/10.3390/cells8111387
APA StyleOh, E., Hong, J., & Yun, C.-O. (2019). Regulatory T Cells Induce Metastasis by Increasing Tgf-β and Enhancing the Epithelial–Mesenchymal Transition. Cells, 8(11), 1387. https://doi.org/10.3390/cells8111387