Alternative Splicing of RAD6B and Not RAD6A Is Selectively Increased in Melanoma: Identification and Functional Characterization



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Patient Samples
2.2. RT-PCR, Subcloning and Sequence Analysis
2.3. Expression Analysis
2.4. Western Blot Analysis
2.5. Homology/Template Modeling of Rad6B Splice Variant Structures
2.6. TCGA Analysis
2.7. Whole Exome Sequence (WES) Analysis
3. Results
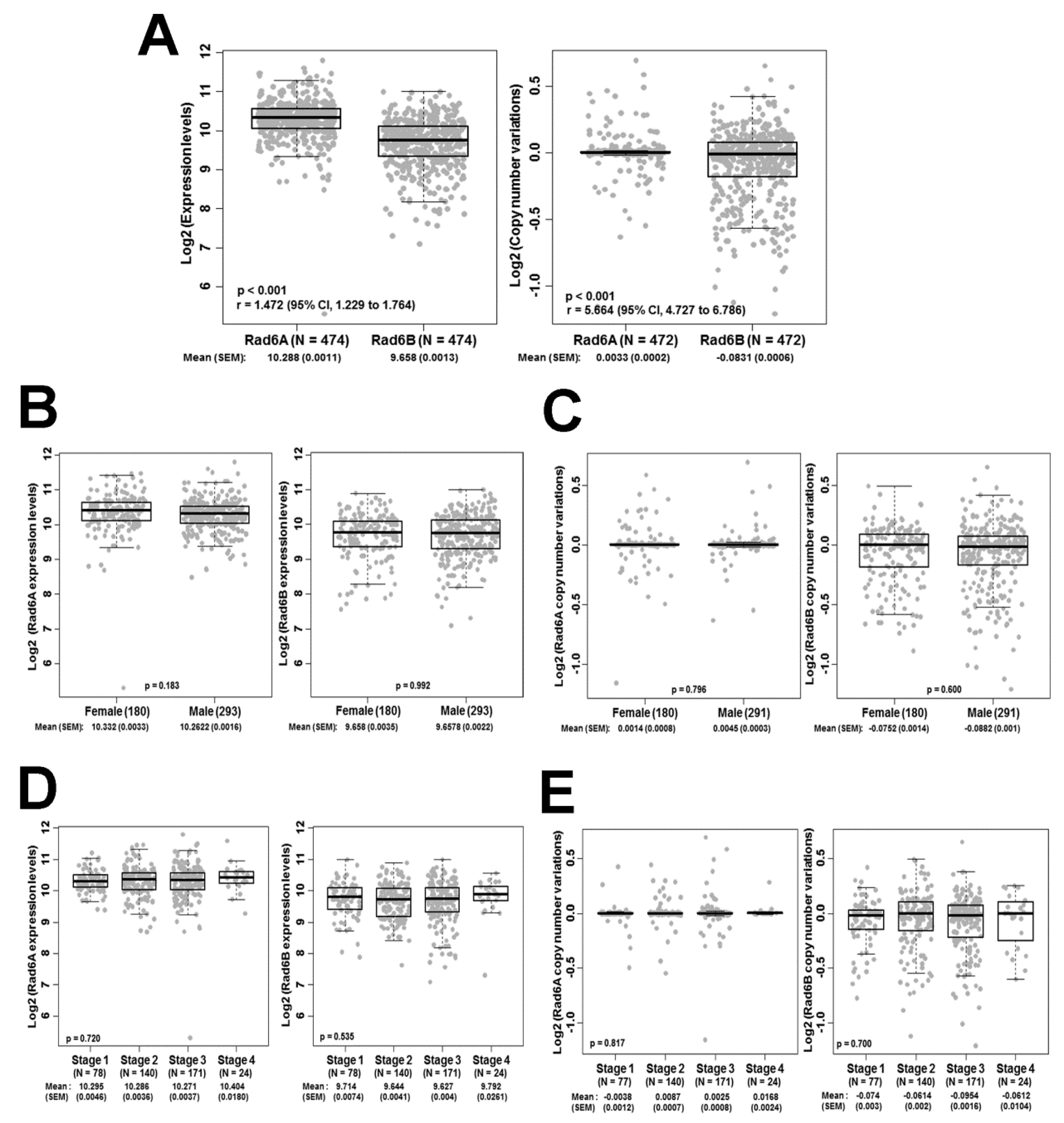
3.1. The RAD6B Paralog Displays Greater Heterogeneity in Expression Levels and Copy Number Compared to RAD6A in Melanoma Patients
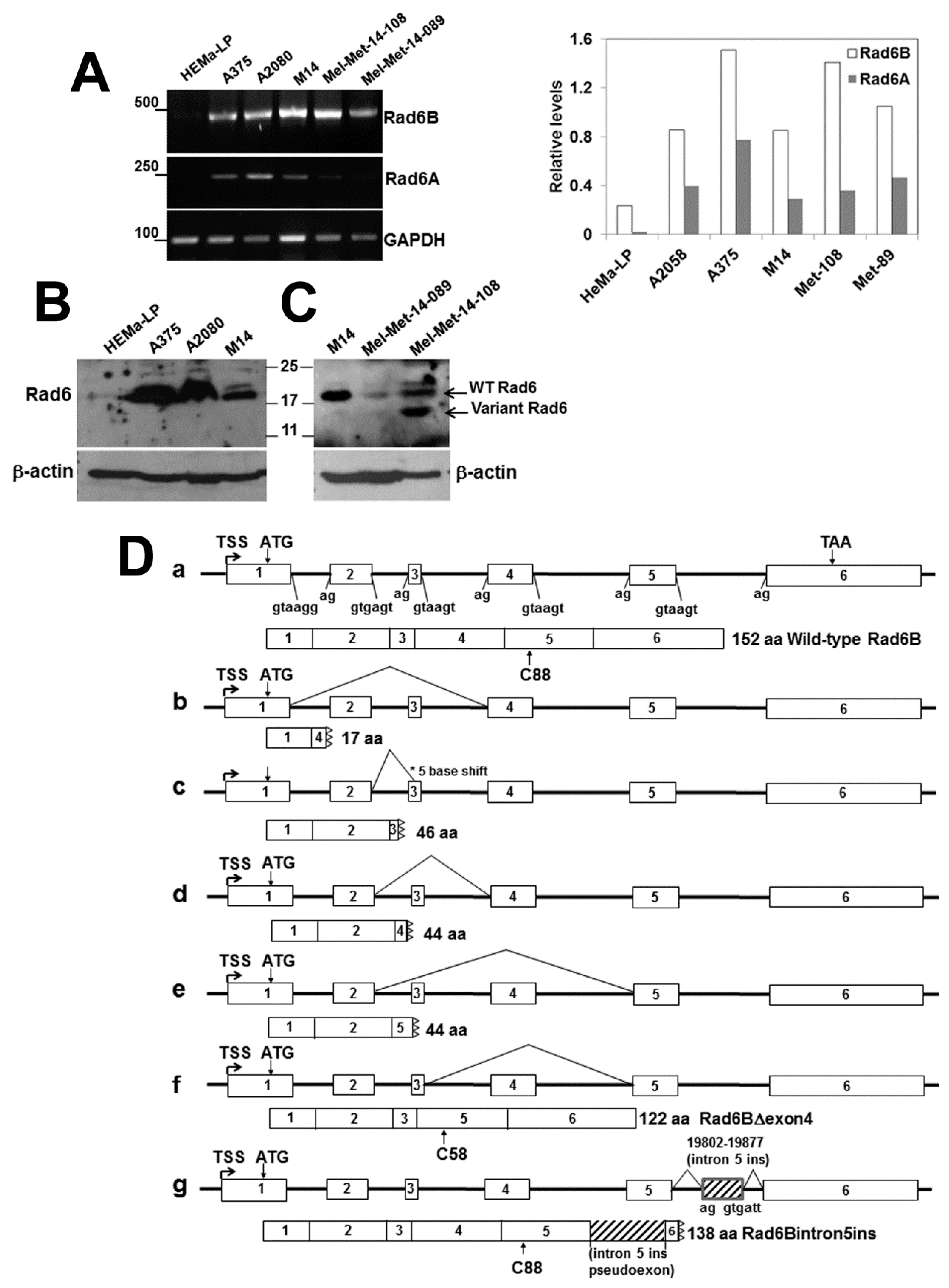
3.2. Melanoma Cell Lines and Patient-Derived Metastatic Melanomas show RAD6B mRNA Alterations Resulting from Alternative Splicing
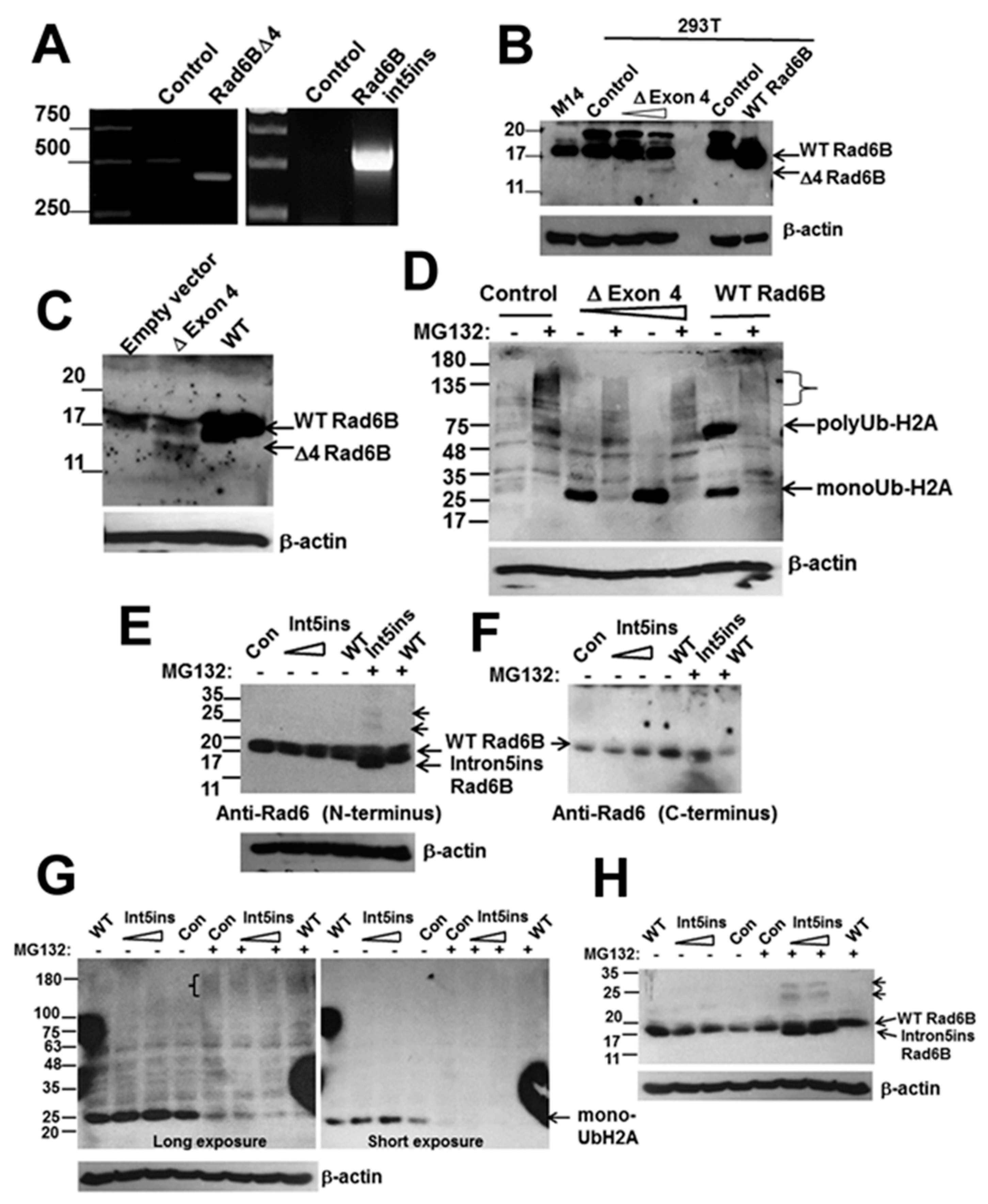
3.3. RAD6BΔExon4 and RAD6Bintron5ins Splice Variants are Functional
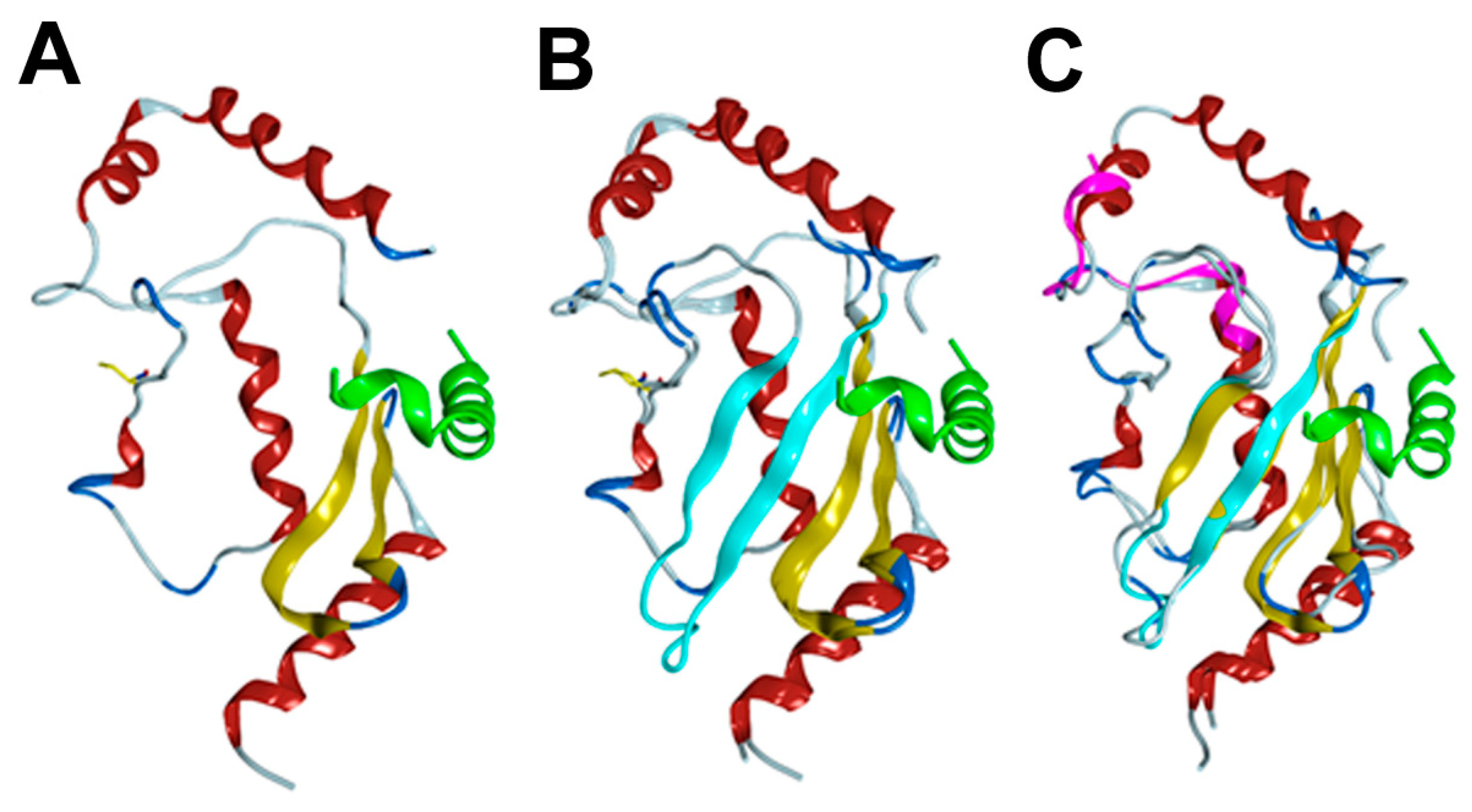
3.4. Homology/Template-Based Modeling of RAD6BΔexon4 and RAD6Bintron5ins 3D Structures
3.5. RAD6B Variant Expression is a Hallmark of Clinical Melanomas
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Candido, S.; Rapisarda, V.; Marconi, A.; Malaponte, G.; Bevelacqua, V.; Gangemi, P.; Scalisi, A.; McCubrey, J.A.; Maestro, R.; Spandidos, D.A.; et al. Analysis of the B-RafV600E mutation in cutaneous melanoma patients with occupational sun exposure. Oncol. Rep. 2014, 31, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Brocker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Gilchrest, B.A.; Eller, M.S.; Geller, A.C.; Yaar, M. The pathogenesis of melanoma induced by ultraviolet radiation. N. Engl. J. Med. 1999, 340, 1341–1348. [Google Scholar] [CrossRef]
- Pennello, G.; Devesa, S.; Gail, M. Association of surface ultraviolet B radiation levels with melanoma and nonmelanoma skin cancer in United States blacks. Cancer Epidemiol. Biomarkers Prev. 2000, 9, 291–297. [Google Scholar] [PubMed]
- Hedglin, M.; Benkovic, S.J. Regulation of Rad6/Rad18 activity during DNA damage tolerance. Annu. Rev. Biophys. 2015, 44, 207–228. [Google Scholar] [CrossRef]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Lange, S.S.; Takata, K.; Wood, R.D. DNA polymerases and cancer. Nat. Rev. Cancer 2011, 11, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R. Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett. 2005, 579, 873–876. [Google Scholar] [CrossRef]
- Prakash, S.; Johnson, R.E.; Prakash, L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 2005, 74, 317–353. [Google Scholar] [CrossRef]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Koken, M.H.; Reynolds, P.; Jaspers-Dekker, I.; Prakash, L.; Prakash, S.; Bootsma, D.; Hoeijmakers, J.H. Structural and functional conservation of two human homologs of the yeast DNA repair gene RAD6. Proc. Natl. Acad. Sci. USA 1991, 88, 8865–8869. [Google Scholar] [CrossRef] [PubMed]
- Koken, M.H.; Smit, E.M.; Jaspers-Dekker, I.; Oostra, B.A.; Hagemeijer, A.; Bootsma, D.; Hoeijmakers, J.H. Localization of two human homologs, HHR6A and HHR6B, of the yeast DNA repair gene RAD6 to chromosomes Xq24-q25 and 5q23-q31. Genomics 1992, 12, 447–453. [Google Scholar] [CrossRef]
- Jentsch, S.; McGrath, J.P.; Varshavsky, A. The yeast DNA repair gene RAD6 encodes a ubiquitin-conjugating enzyme. Nature 1987, 329, 131–134. [Google Scholar] [CrossRef]
- Rosner, K.; Adsule, S.; Haynes, B.; Kirou, E.; Kato, I.; Mehregan, D.R.; Shekhar, M.P. Rad6 is a Potential Early Marker of Melanoma Development. Transl. Oncol. 2014, 7, 384–392. [Google Scholar] [CrossRef]
- Rosner, K.; Mehregan, D.R.; Kirou, E.; Abrams, J.; Kim, S.; Campbell, M.; Frieder, J.; Lawrence, K.; Haynes, B.; Shekhar, M.P. Melanoma development and progression are associated with Rad6 upregulation and beta-catenin relocation to the cell membrane. J. Skin Cancer 2014, 2014, 439205. [Google Scholar] [CrossRef]
- Sarma, A.P.S.; Liu, F.; Nangia-Makker, P.; Mao, G.; Shekhar, M. Targeting Rad6B suppresses melanomagenesis. Cancer Res. 2017, 77. [Google Scholar] [CrossRef]
- Shekhar, M.P.; Tait, L.; Gerard, B. Essential role of T-cell factor/beta-catenin in regulation of Rad6B: a potential mechanism for Rad6B overexpression in breast cancer cells. Mol. Cancer Res. 2006, 4, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, M.P.; Gerard, B.; Pauley, R.J.; Williams, B.O.; Tait, L. Rad6B is a positive regulator of beta-catenin stabilization. Cancer Res. 2008, 68, 1741–1750. [Google Scholar] [CrossRef]
- Hari, L.; Miescher, I.; Shakhova, O.; Suter, U.; Chin, L.; Taketo, M.; Richardson, W.D.; Kessaris, N.; Sommer, L. Temporal control of neural crest lineage generation by Wnt/beta-catenin signaling. Development 2012, 139, 2107–2117. [Google Scholar] [CrossRef]
- Bonomi, S.; Gallo, S.; Catillo, M.; Pignataro, D.; Biamonti, G.; Ghigna, C. Oncogenic alternative splicing switches: role in cancer progression and prospects for therapy. Int. J. Cell Biol 2013, 2013, 962038. [Google Scholar] [CrossRef]
- Hagen, R.M.; Ladomery, M.R. Role of splice variants in the metastatic progression of prostate cancer. Biochem. Soc. Trans. 2012, 40, 870–874. [Google Scholar] [CrossRef]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef]
- Shekhar, M.P.; Lyakhovich, A.; Visscher, D.W.; Heng, H.; Kondrat, N. Rad6 overexpression induces multinucleation, centrosome amplification, abnormal mitosis, aneuploidy, and transformation. Cancer Res. 2002, 62, 2115–2124. [Google Scholar] [PubMed]
- Sanders, M.A.; Haynes, B.; Nangia-Makker, P.; Polin, L.A.; Shekhar, M.P. Pharmacological targeting of RAD6 enzyme-mediated translesion synthesis overcomes resistance to platinum-based drugs. J. Biol. Chem. 2017, 292, 10347–10363. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Kim, J.; Guermah, M.; McGinty, R.K.; Lee, J.S.; Tang, Z.; Milne, T.A.; Shilatifard, A.; Muir, T.W.; Roeder, R.G. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell 2009, 137, 459–471. [Google Scholar] [CrossRef]
- Melikova, M.S.; Kondratov, K.A.; Kornilova, E.S. Two different stages of epidermal growth factor (EGF) receptor endocytosis are sensitive to free ubiquitin depletion produced by proteasome inhibitor MG132. Cell Biol. Int. 2006, 30, 31–43. [Google Scholar] [CrossRef]
- Bailly, V.; Prakash, S.; Prakash, L. Domains required for dimerization of yeast Rad6 ubiquitin-conjugating enzyme and Rad18 DNA binding protein. Mol. Cell Biol. 1997, 17, 4536–4543. [Google Scholar] [CrossRef]
- Brogna, S.; Wen, J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Biol. 2009, 16, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Yatsenko, A.N.; Georgiadis, A.P.; Murthy, L.J.; Lamb, D.J.; Matzuk, M.M. UBE2B mRNA alterations are associated with severe oligozoospermia in infertile men. Mol. Hum. Reprod. 2013, 19, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Kalnina, Z.; Zayakin, P.; Silina, K.; Line, A. Alterations of pre-mRNA splicing in cancer. Genes Chromosomes Cancer 2005, 42, 342–357. [Google Scholar] [CrossRef]
- Orban, T.I.; Olah, E. Emerging roles of BRCA1 alternative splicing. Mol. Pathol 2003, 56, 191–197. [Google Scholar] [CrossRef]
- Mackay, C.R.; Terpe, H.J.; Stauder, R.; Marston, W.L.; Stark, H.; Gunthert, U. Expression and modulation of CD44 variant isoforms in humans. J. Cell Biol. 1994, 124, 71–82. [Google Scholar] [CrossRef]
- Rosso, M.; Okoro, D.E.; Bargonetti, J. Splice variants of MDM2 in oncogenesis. Subcell. Biochem. 2014, 85, 247–261. [Google Scholar]
- Roest, H.P.; van Klaveren, J.; de Wit, J.; van Gurp, C.G.; Koken, M.H.; Vermey, M.; van Roijen, J.H.; Hoogerbrugge, J.W.; Vreeburg, J.T.; Baarends, W.M.; et al. Inactivation of the HR6B ubiquitin-conjugating DNA repair enzyme in mice causes male sterility associated with chromatin modification. Cell 1996, 86, 799–810. [Google Scholar] [CrossRef]
- Mulugeta Achame, E.; Wassenaar, E.; Hoogerbrugge, J.W.; Sleddens-Linkels, E.; Ooms, M.; Sun, Z.W.; van, I.W.F.; Grootegoed, J.A.; Baarends, W.M. The ubiquitin-conjugating enzyme HR6B is required for maintenance of X chromosome silencing in mouse spermatocytes and spermatids. BMC Genomics 2010, 11, 36. [Google Scholar] [CrossRef]
- Singh, R.K.; Cooper, T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012, 18, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Thakran, P. Intron specificity in pre-mRNA splicing. Curr. Genet. 2018, 64, 777–784. [Google Scholar] [CrossRef]
- Kong, Y.; Krauthammer, M.; Halaban, R. Rare SF3B1 R625 mutations in cutaneous melanoma. Melanoma Res. 2014, 24, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Hintzsche, J.D.; Gorden, N.T.; Amato, C.M.; Kim, J.; Wuensch, K.E.; Robinson, S.E.; Applegate, A.J.; Couts, K.L.; Medina, T.M.; Wells, K.R.; et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017, 27, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Aslanzadeh, V.; Huang, Y.; Sanguinetti, G.; Beggs, J.D. Transcription rate strongly affects splicing fidelity and cotranscriptionality in budding yeast. Genome Res. 2018, 28, 203–213. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gajan, A.; Martin, C.E.; Kim, S.; Joshi, M.; Michelhaugh, S.K.; Sloma, I.; Mittal, S.; Firestine, S.; Shekhar, M.P.V. Alternative Splicing of RAD6B and Not RAD6A Is Selectively Increased in Melanoma: Identification and Functional Characterization. Cells 2019, 8, 1375. https://doi.org/10.3390/cells8111375
Gajan A, Martin CE, Kim S, Joshi M, Michelhaugh SK, Sloma I, Mittal S, Firestine S, Shekhar MPV. Alternative Splicing of RAD6B and Not RAD6A Is Selectively Increased in Melanoma: Identification and Functional Characterization. Cells. 2019; 8(11):1375. https://doi.org/10.3390/cells8111375
Chicago/Turabian StyleGajan, Ambikai, Carly E. Martin, Seongho Kim, Milap Joshi, Sharon K. Michelhaugh, Ido Sloma, Sandeep Mittal, Steven Firestine, and Malathy P. V. Shekhar. 2019. "Alternative Splicing of RAD6B and Not RAD6A Is Selectively Increased in Melanoma: Identification and Functional Characterization" Cells 8, no. 11: 1375. https://doi.org/10.3390/cells8111375
APA StyleGajan, A., Martin, C. E., Kim, S., Joshi, M., Michelhaugh, S. K., Sloma, I., Mittal, S., Firestine, S., & Shekhar, M. P. V. (2019). Alternative Splicing of RAD6B and Not RAD6A Is Selectively Increased in Melanoma: Identification and Functional Characterization. Cells, 8(11), 1375. https://doi.org/10.3390/cells8111375