TRPC6-Mediated ERK1/2 Activation Increases Dentate Granule Cell Resistance to Status Epilepticus via Regulating Lon Protease-1 Expression and Mitochondrial Dynamics

Abstract

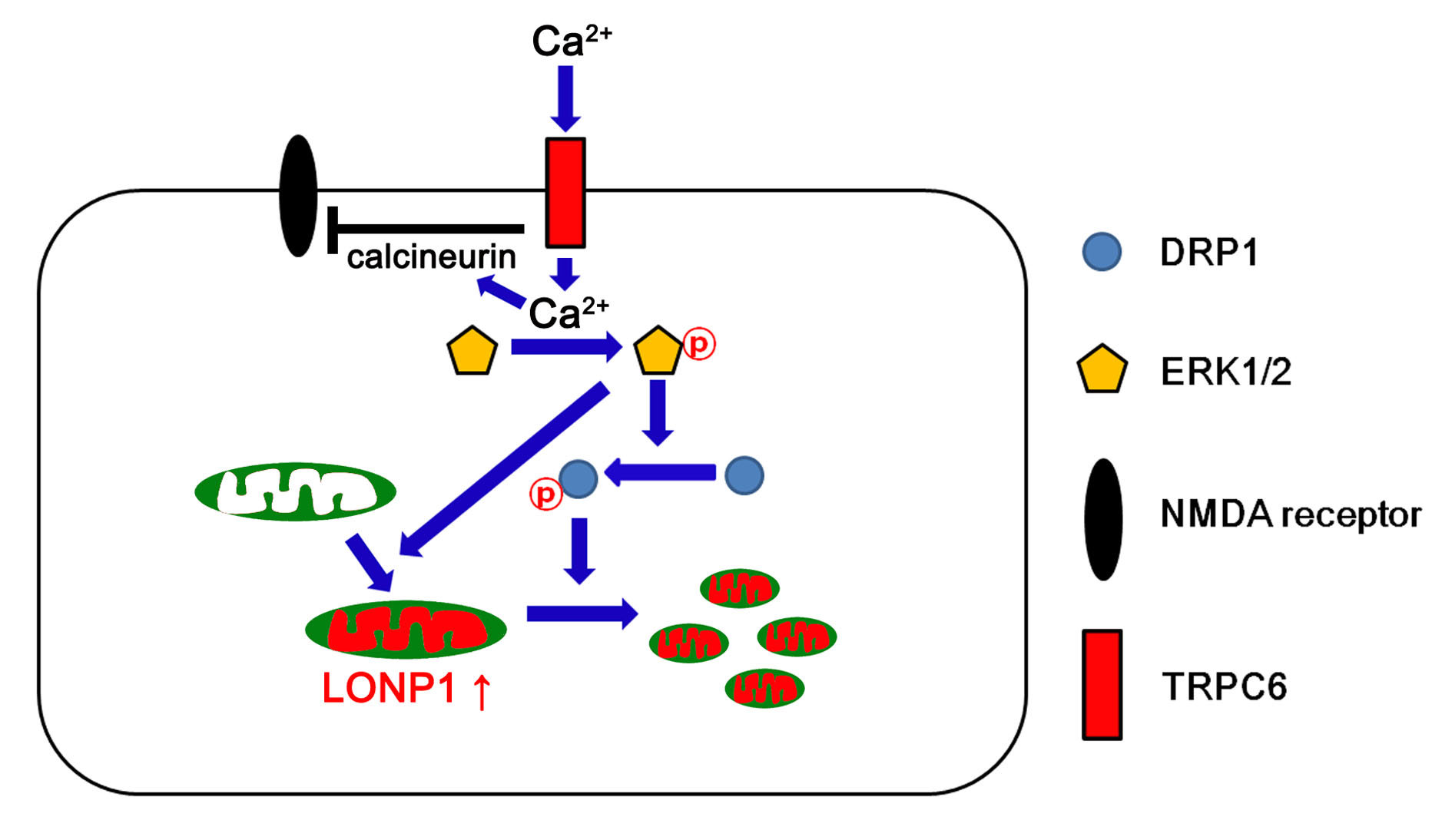
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Chemicals
2.2. siRNA and Drug Infusion
2.3. SE Induction and EEG Analysis
2.4. Tissue Processing
2.5. Western Blot
2.6. Immunohistochemistry and Fluoro-Jade B Staining
2.7. Cell Count and Measurement of Mitochondrial Length
2.8. Quantification of Data and Statistical Analysis
3. Results
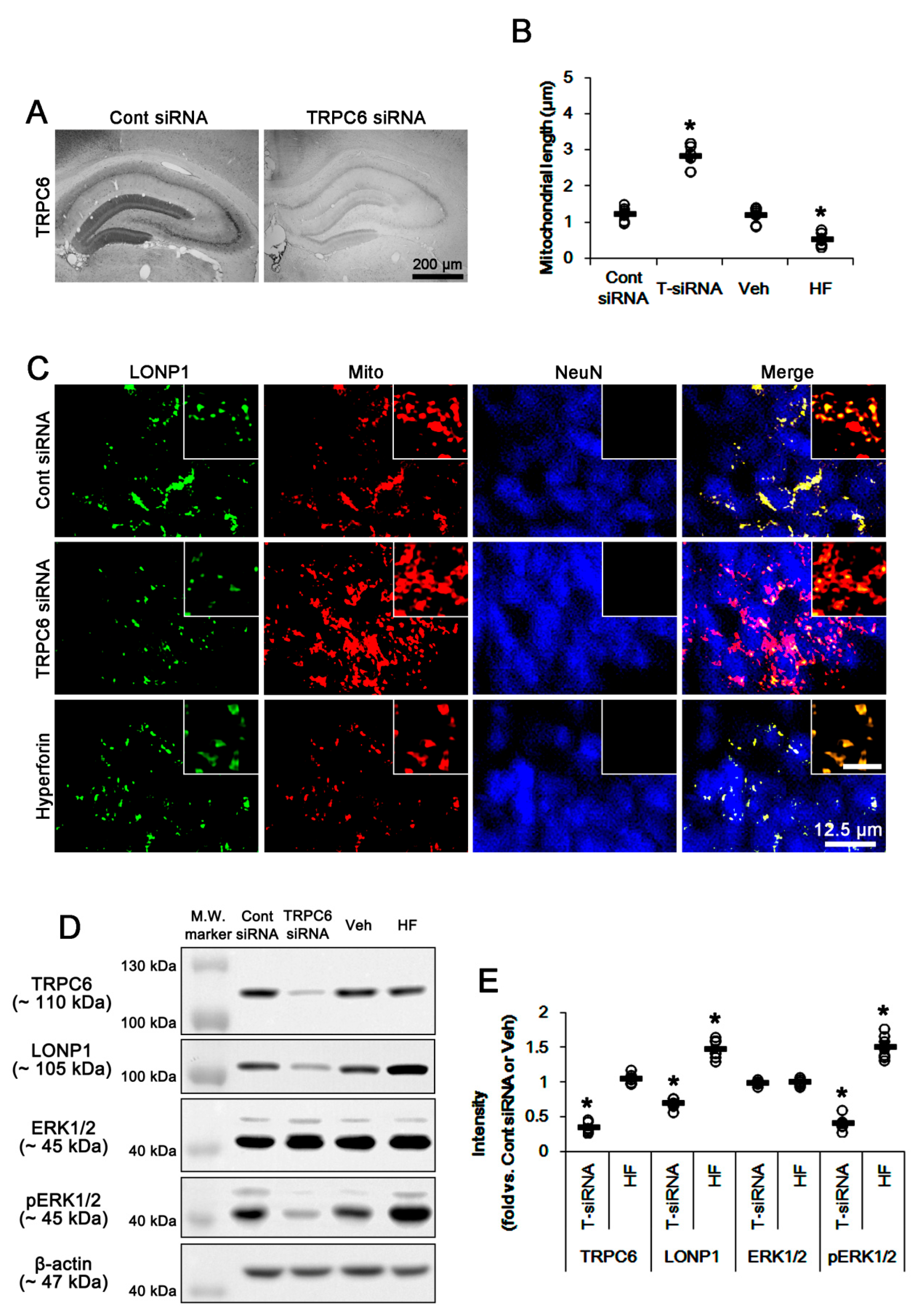
3.1. TRPC6 Knockdown and Hyperforin Reversely Regulate LONP1 Expression, ERK1/2 Phosphorylation, and Mitochondrial Length in DGC under Physiological Conditions
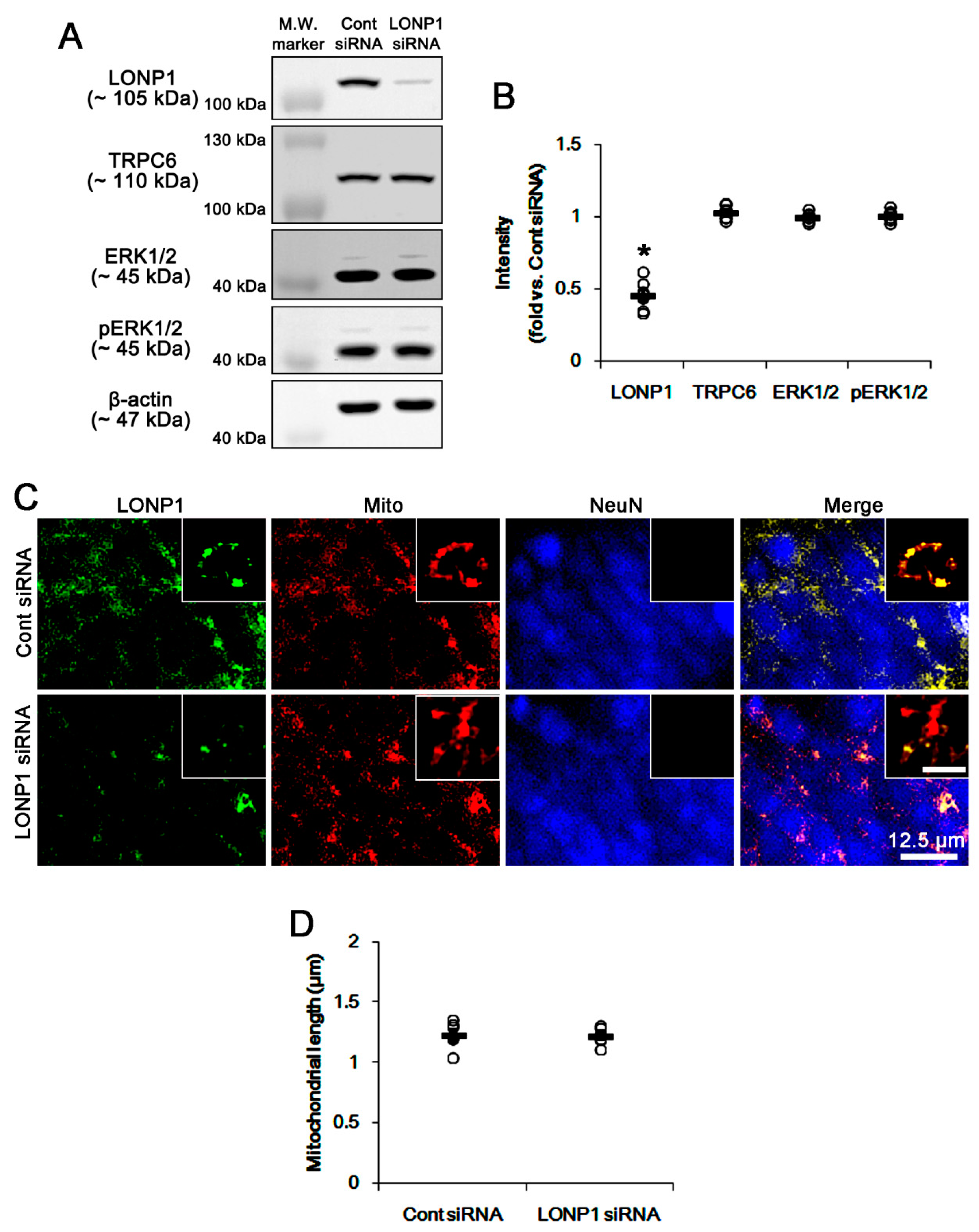
3.2. LONP1 siRNA Does Not Influence TRPC6 Expression, ERK1/2 Phosphorylation and Mitochondrial Length under Physiological Condition
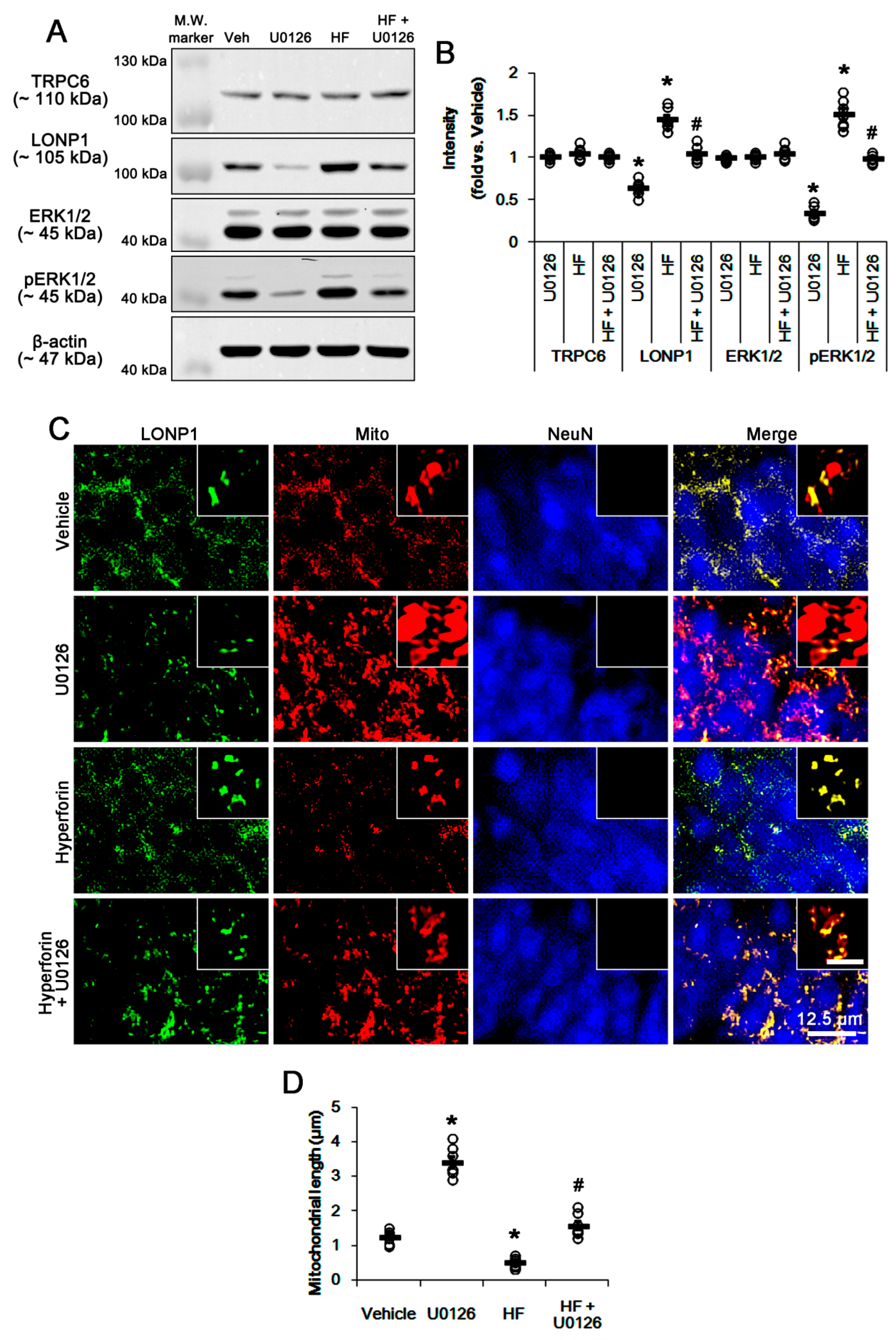
3.3. U0126 Abrogates Mitochondrial LONP1 Expression under Physiological Condition and after Hyperforin Treatment
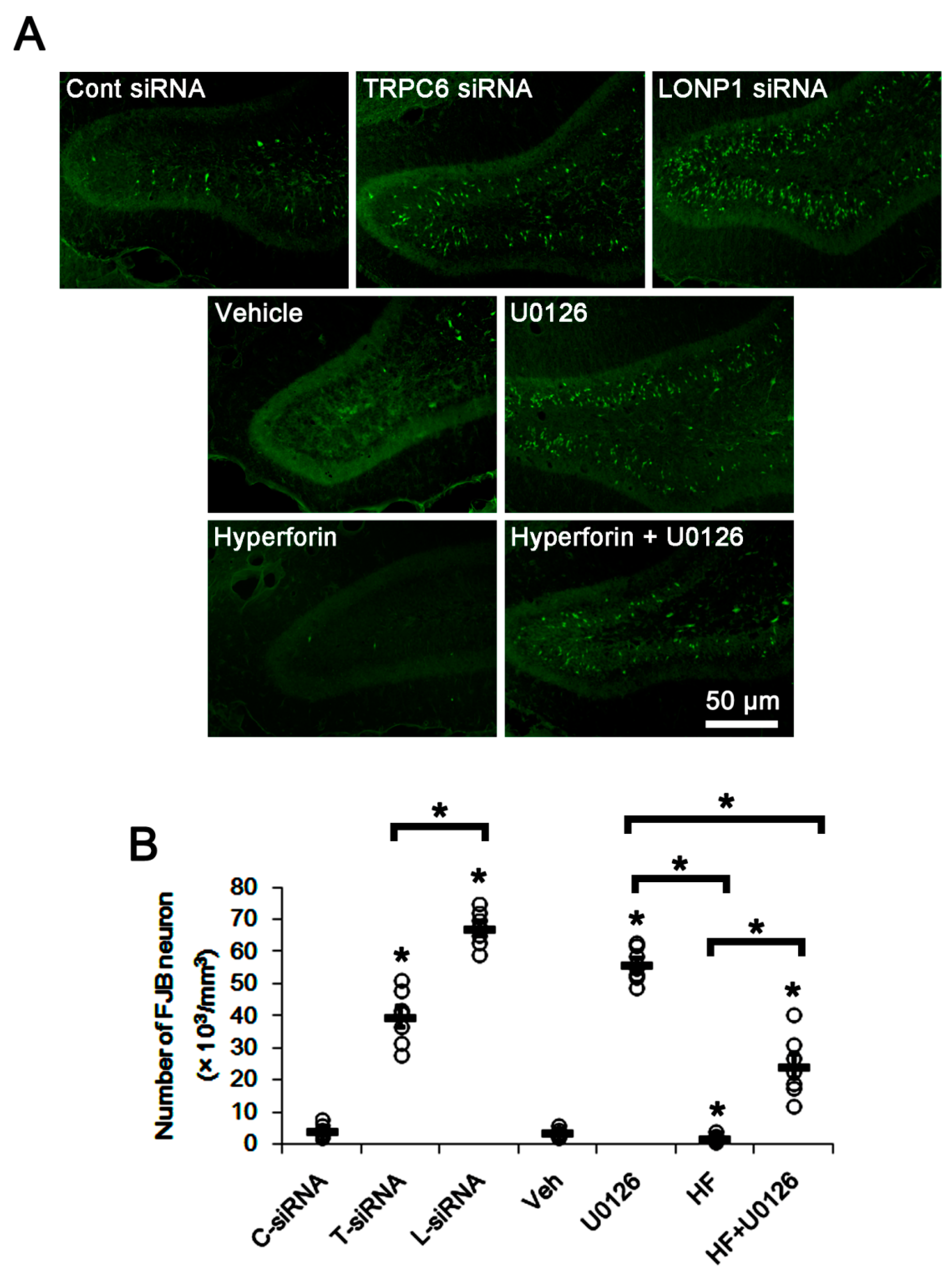
3.4. The TRPC6-ERK1/2-LONP1 Signaling Pathway Inhibits SE-Induced DGC Degeneration, Independent of Seizure Severity
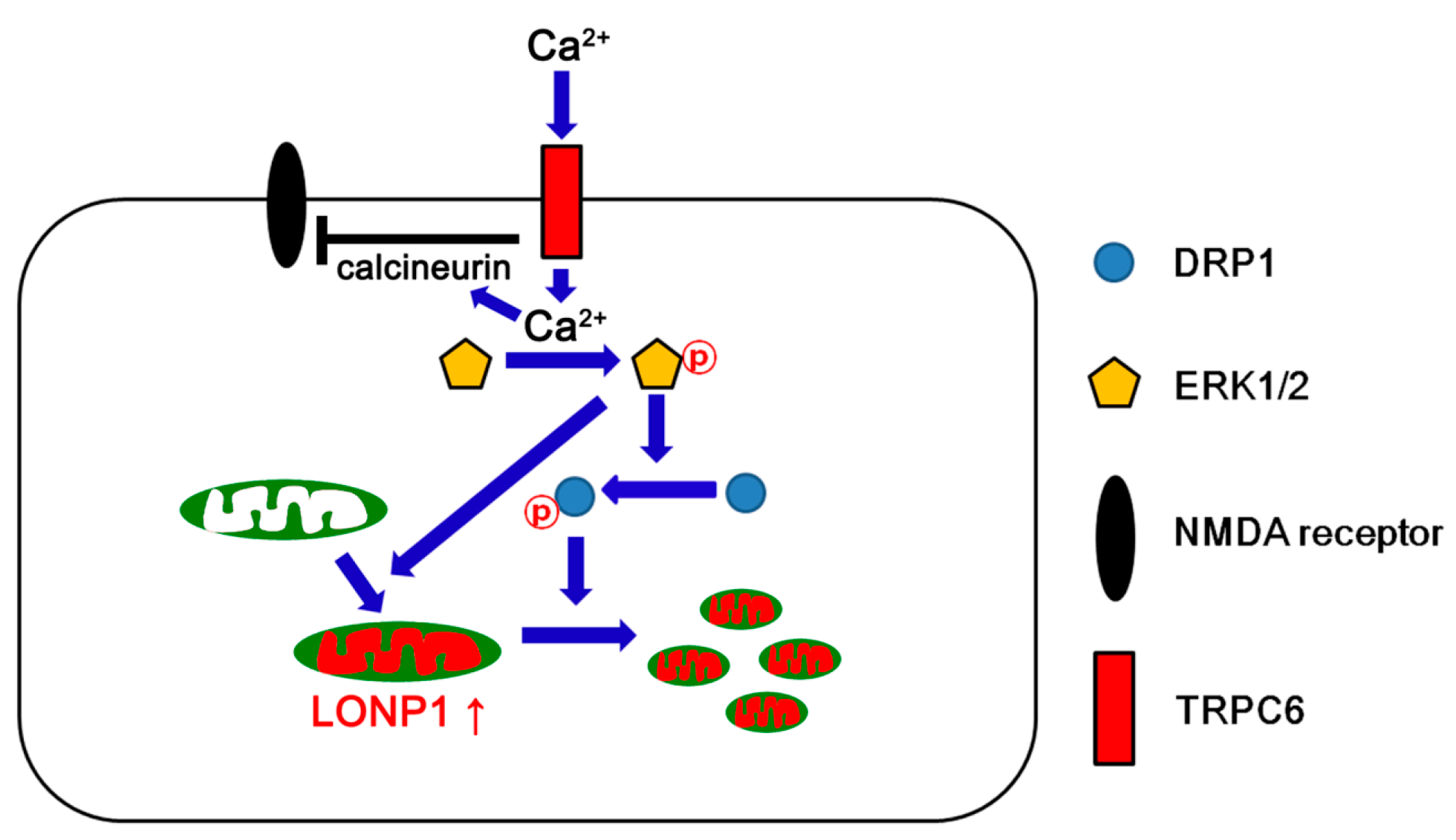
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Pinti, M.; Boraldi, F.; Giorgio, V.; Bernardi, P.; Bartolomeo, R.; Nasi, M.; De Biasi, S.; Missiroli, S.; Carnevale, G.; et al. Silencing of mitochondrial Lon protease deeply impairs mitochondrial proteome and function in colon cancer cells. FASEB J. 2014, 28, 5122–5135. [Google Scholar] [CrossRef] [PubMed]
- Marcillat, O.; Zhang, Y.; Lin, S.W.; Davies, K.J. Mitochondria contain a proteolytic system which can recognize and degrade oxidatively-denatured proteins. Biochem. J. 1988, 254, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Gur, E.; Sauer, R.T. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev. 2008, 22, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Lundberg, K.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc. Natl. Acad. Sci. USA 2005, 102, 5987–5991. [Google Scholar] [CrossRef]
- Hori, O.; Ichinoda, F.; Tamatani, T.; Yamaguchi, A.; Sato, N.; Ozawa, K.; Kitao, Y.; Miyazaki, M.; Harding, H.P.; Ron, D.; et al. Transmission of cell stress from endoplasmic reticulum to mitochondria: Enhanced expression of Lon protease. J. Cell Biol. 2002, 157, 1151–1160. [Google Scholar] [CrossRef]
- Cheng, C.W.; Kuo, C.Y.; Fan, C.C.; Fang, W.C.; Jiang, S.S.; Lo, Y.K.; Wang, T.Y.; Kao, M.C.; Lee, A.Y. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013, 4, e681. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, M.; Lin, H.; Huang, C.; Shi, X.; Luo, J. Epidermal growth factor up-regulates the transcription of mouse lon homology ATP-dependent protease through extracellular signal-regulated protein kinase- and phosphatidylinositol-3-kinase-dependent pathways. Exp. Cell Res. 2002, 280, 97–106. [Google Scholar] [CrossRef]
- Du, W.; Huang, J.; Yao, H.; Zhou, K.; Duan, B.; Wang, Y. Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats. J. Clin. Investig. 2010, 120, 3480–3492. [Google Scholar] [CrossRef]
- Li, H.; Huang, J.; Du, W.; Jia, C.; Yao, H.; Wang, Y. TRPC6 inhibited NMDA receptor activities and protected neurons from ischemic excitotoxicity. J. Neurochem. 2012, 123, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Ryu, H.J.; Kim, J.E.; Kang, T.C. The reverse roles of transient receptor potential canonical channel-3 and -6 in neuronal death following pilocarpine-induced status epilepticus. Cell Mol. Neurobiol. 2013, 33, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.A.; Botond, G.; Borhegyi, Z.; Plummer, N.W.; Freund, T.F.; Hájos, N. DAG-sensitive and Ca2+ permeable TRPC6 channels are expressed in dentate granule cells and interneurons in the hippocampal formation. Hippocampus 2013, 23, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kang, T.C. The role of TRPC6 in seizure susceptibility and seizure-related neuronal damage in the rat dentate gyrus. Neuroscience 2015, 307, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.R.; Kang, T.C. TRPC6-mediated ERK1/2 phosphorylation prevents dentate granule cell degeneration via inhibiting mitochondrial elongation. Neuropharmacology 2017, 121, 120–129. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, J.Y.; Kang, T.C. TRPC6-mediated ERK1/2 activation regulates neuronal excitability via subcellular Kv4.3 localization in the rat hippocampus. Front. Cell. Neurosci. 2017, 11, 413. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. CDDO-Me selectively attenuates CA1 neuronal death induced by status epilepticus via facilitating mitochondrial fission independent of LONP1. Cells 2019, 8, 833. [Google Scholar] [CrossRef]
- Lee, S.K.; Kim, J.E.; Kim, Y.J.; Kim, M.J.; Kang, T.C. Hyperforin attenuates microglia activation and inhibits p65-Ser276 NFκB phosphorylation in the rat piriform cortex following status epilepticus. Neurosci. Res. 2014, 85, 39–50. [Google Scholar] [CrossRef]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Heiser, J.H.; Schuwald, A.M.; Sillani, G.; Ye, L.; Müller, W.E.; Leuner, K. TRPC6 channel-mediated neurite outgrowth in PC12 cells and hippocampal neurons involves activation of RAS/MEK/ERK, PI3K, and CAMKIV signaling. J. Neurochem. 2013, 127, 303–313. [Google Scholar] [CrossRef]
- Do Val-da Silva, R.A.; Peixoto-Santos, J.E.; Kandratavicius, L.; De Ross, J.B.; Esteves, I.; De Martinis, B.S.; Alves, M.N.; Scandiuzzi, R.C.; Hallak, J.E.; Zuardi, A.W.; et al. Protective effects of cannabidiol against seizures and neuronal death in a rat model of mesial temporal lobe epilepsy. Front. Pharmacol. 2017, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Min, S.J.; Hyun, H.W.; Kang, T.C. Leptomycin B attenuates neuronal death via PKA- and PP2B-mediated ERK1/2 activation in the rat hippocampus following status epilepticus. Brain Res. 2017, 1670, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, S. Proteases and their targets in Escherichia coli. Ann. Rev. Genet. 1996, 30, 465–506. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.K.; Pomatto, L.C.; Davies, K.J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Davies, K.J.A. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 2002, 4, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Luciakova, K.; Sokolikova, B.; Chloupkova, M.; Nelson, B.D. Enhanced mitochondrial biogenesis is associated with increased expression of the mitochondrial ATP-dependent Lon protease. FEBS Lett. 1999, 444, 186–188. [Google Scholar] [CrossRef]
- Bota, D.A.; Ngo, J.K.; Davies, K.J. Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radic. Biol. Med. 2005, 38, 665–677. [Google Scholar] [CrossRef]
- Graham, S.; Ding, M.; Sours-Brothers, S.; Yorio, T.; Ma, J.X.; Ma, R. Downregulation of TRPC6 protein expression by high glucose, a possible mechanism for the impaired Ca2+ signaling in glomerular mesangial cells in diabetes. Am. J. Physiol. Renal Physiol. 2007, 293, F1381–F1390. [Google Scholar] [CrossRef]
- Zhou, J.; Du, W.; Zhou, K.; Tai, Y.; Yao, H.; Jia, Y.; Ding, Y.; Wang, Y. Critical role of TRPC6 channels in the formation of excitatory synapses. Nat. Neurosci. 2008, 11, 741–743. [Google Scholar] [CrossRef]
- Chiluiza, D.; Krishna, S.; Schumacher, V.A.; Schlöndorff, J. Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular signal-regulated kinases 1/2 (ERK1/2). J. Biol. Chem. 2013, 288, 18407–18420. [Google Scholar] [CrossRef]
- Agell, N.; Bachs, O.; Rocamora, N.; Villalonga, P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca2+, and calmodulin. Cell Signal 2002, 14, 649–654. [Google Scholar] [CrossRef]
- Cheung, E.C.; Slack, R.S. Emerging role for ERK as a key regulator of neuronal apoptosis. Sci. STKE. 2004, 2004, PE45. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Ifuku, M.; Take, S.; Kawamura, J.; Miake, K.; Katafuchi, T. Plasmalogens rescue neuronal cell death through an activation of AKT and ERK survival signaling. PLoS ONE 2013, 8, e83508. [Google Scholar] [CrossRef] [PubMed]
- Ortuño-Sahagún, D.; González, R.M.; Verdaguer, E.; Huerta, V.C.; Torres-Mendoza, B.M.; Lemus, L.; Rivera-Cervantes, M.C.; Camins, A.; Zárate, C.B. Glutamate excitotoxicity activates the MAPK/ERK signaling pathway and induces the survival of rat hippocampal neurons in vivo. J. Mol. Neurosci. 2014, 52, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-García, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef]
- Leuner, K.; Li, W.; Amaral, M.D.; Rudolph, S.; Calfa, G.; Schuwald, A.M.; Harteneck, C.; Inoue, T.; Pozzo-Miller, L. Hyperforin modulates dendritic spine morphology in hippocampal pyramidal neurons by activating Ca2+ -permeable TRPC6 channels. Hippocampus 2013, 23, 40–52. [Google Scholar] [CrossRef]
- Wong-Riley, M.T. Cytochrome oxidase: An endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989, 12, 94–101. [Google Scholar] [CrossRef]
- Wong-Riley, M.T. Bigenomic regulation of cytochrome c oxidase in neurons and the tight coupling between neuronal activity and energy metabolism. Adv. Exp. Med. Biol. 2012, 748, 283–304. [Google Scholar]
- Sepuri, N.B.V.; Angireddy, R.; Srinivasan, S.; Guha, M.; Spear, J.; Lu, B.; Anandatheerthavarada, H.K.; Suzuki, C.K.; Avadhani, N.G. Mitochondrial LON protease-dependent degradation of cytochrome c oxidase subunits under hypoxia and myocardial ischemia. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 519–528. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.A.; Durisic, N.; Mativetsky, J.M.; Costantino, S.; Hancock, M.A.; Grutter, P.; Shoubridge, E.A. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol. Biol. Cell. 2007, 18, 3225–3236. [Google Scholar] [CrossRef] [PubMed]
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell. 2013, 49, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, S.H.; Venkatesh, S.; Li, M.; Lee, J.; Lu, B.; Hilchey, S.; Morse, K.M.; Metcalfe, H.M.; Andreeff, M.; Brookes, P.S.; et al. The mitochondrial ATP-dependent Lon protease: A novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 2012, 119, 3321–3329. [Google Scholar] [CrossRef]
- Wang, H.M.; Cheng, K.C.; Lin, C.J.; Hsu, S.W.; Fang, W.C.; Hsu, T.F.; Chiu, C.C.; Chang, H.W.; Hsu, C.H.; Lee, Y.L. Obtusilactone A and (−)-sesamin induce apoptosis in human lung cancer cells by inhibiting mitochondrial Lon protease and activating DNA damage checkpoints. Cancer Sci. 2010, 101, 2612–2620. [Google Scholar] [CrossRef]
- Flippo, K.H.; Strack, S. Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 2017, 130, 671–681. [Google Scholar] [CrossRef]
- Campello, S.; Scorrano, L. Mitochondrial shape changes: Orchestrating cell pathophysiology. EMBO Rep. 2010, 11, 678–684. [Google Scholar] [CrossRef]
- Kim, J.E.; Kang, T.C. p47Phox/CDK5/DRP1-mediated mitochondrial fission evokes PV cell degeneration in the rat dentate gyrus following status epilepticus. Front. Cell. Neurosci. 2017, 11, 267. [Google Scholar] [CrossRef]
- Kim, J.E.; Kang, T.C. Differential roles of mitochondrial translocation of active caspase-3 and HMGB1 in neuronal death induced by status epilepticus. Front. Cell. Neurosci. 2018, 12, 301. [Google Scholar] [CrossRef]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012, 197, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Ryu, H.J.; Kim, M.J.; Kang, T.C. LIM kinase-2 induces programmed necrotic neuronal death via dysfunction of DRP1-mediated mitochondrial fission. Cell Death Differ. 2014, 21, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Polo, M.; Alegre, F.; Moragrega, A.B.; Gibellini, L.; Marti-Rodrigo, A.; Blas-Garcia, A.; Esplugues, J.V.; Apostolova, N. Lon protease: A novel mitochondrial matrix protein in the interconnection between drug-induced mitochondrial dysfunction and endoplasmic reticulum stress. Br. J. Pharmacol. 2017, 174, 4409–4429. [Google Scholar] [CrossRef] [PubMed]
- Staff, N.P.; Spruston, N. Intracellular correlate of EPSP-spike potentiation in CA1 pyramidal neurons is controlled by GABAergic modulation. Hippocampus 2003, 13, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Langosch, J.M.; Zhou, X.Y.; Heinen, M.; Kupferschmid, S.; Chatterjee, S.S.; Nöldner, M.; Walden, J. St John’s wort (Hypericum perforatum) modulates evoked potentials in guinea pig hippocampal slices via AMPA and GABA receptors. Eur. Neuropsychopharmacol. 2002, 12, 209–216. [Google Scholar] [CrossRef]
- Sell, T.S.; Belkacemi, T.; Flockerzi, V.; Beck, A. Protonophore properties of hyperforin are essential for its pharmacological activity. Sci. Rep. 2014, 4, 7500. [Google Scholar] [CrossRef]
- Xiong, Z.Q.; Stringer, J.L. Extracellular pH responses in CA1 and the dentate gyrus during electrical stimulation, seizure discharges, and spreading depression. J. Neurophysiol. 2000, 83, 3519–3524. [Google Scholar] [CrossRef]
- Chatterjee, S.S.; Biber, A.; Weibezahn, C. Stimulation of glutamate, aspartate and gamma-aminobutyric acid release from synaptosomes by hyperforin. Pharmacopsychiatry 2001, 34, S11–S19. [Google Scholar] [CrossRef]
- Vance, K.M.; Ribnicky, D.M.; Hermann, G.E.; Rogers, R.C. St. John’s Wort enhances the synaptic activity of the nucleus of the solitary tract. Nutrition 2014, 30, S37–S42. [Google Scholar] [CrossRef]
- Singer, A.; Wonnemann, M.; Müller, W.E. Hyperforin, a major antidepressant constituent of St. John’s Wort, inhibits serotonin uptake by elevating free intracellular Na+1. J. Pharmacol. Exp. Ther. 1999, 290, 1363–1368. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| ERK1/2 | Rabbit | Biorbyt (Orb160960) | 1:2000 (WB) |
| LONP1 | Rabbit | Proteintech (15440-1-AP) | 1:100 (IF) 1:1000 (WB) |
| Mitochondrial marker (Mitochondrial complex IV subunit 1, MTCO1) | Mouse | Abcam (#ab14705) | 1:500 (IF) |
| pERK1/2 | Rabbit | Bioss (bs-3330R) | 1:1000 (WB) |
| TRPC6 | Rabbit | Millipore (AB5574) | 1:100 (IHC) 1:1000 (WB) |
| β-actin | Mouse | Sigma (A5316) | 1:5000 (WB) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-E.; Park, H.; Choi, S.-H.; Kong, M.-J.; Kang, T.-C. TRPC6-Mediated ERK1/2 Activation Increases Dentate Granule Cell Resistance to Status Epilepticus via Regulating Lon Protease-1 Expression and Mitochondrial Dynamics. Cells 2019, 8, 1376. https://doi.org/10.3390/cells8111376
Kim J-E, Park H, Choi S-H, Kong M-J, Kang T-C. TRPC6-Mediated ERK1/2 Activation Increases Dentate Granule Cell Resistance to Status Epilepticus via Regulating Lon Protease-1 Expression and Mitochondrial Dynamics. Cells. 2019; 8(11):1376. https://doi.org/10.3390/cells8111376
Chicago/Turabian StyleKim, Ji-Eun, Hana Park, Seo-Hyeon Choi, Min-Jeong Kong, and Tae-Cheon Kang. 2019. "TRPC6-Mediated ERK1/2 Activation Increases Dentate Granule Cell Resistance to Status Epilepticus via Regulating Lon Protease-1 Expression and Mitochondrial Dynamics" Cells 8, no. 11: 1376. https://doi.org/10.3390/cells8111376
APA StyleKim, J.-E., Park, H., Choi, S.-H., Kong, M.-J., & Kang, T.-C. (2019). TRPC6-Mediated ERK1/2 Activation Increases Dentate Granule Cell Resistance to Status Epilepticus via Regulating Lon Protease-1 Expression and Mitochondrial Dynamics. Cells, 8(11), 1376. https://doi.org/10.3390/cells8111376