hiPSCs Derived Cardiac Cells for Drug and Toxicity Screening and Disease Modeling: What Micro- Electrode-Array Analyses Can Tell Us

Abstract
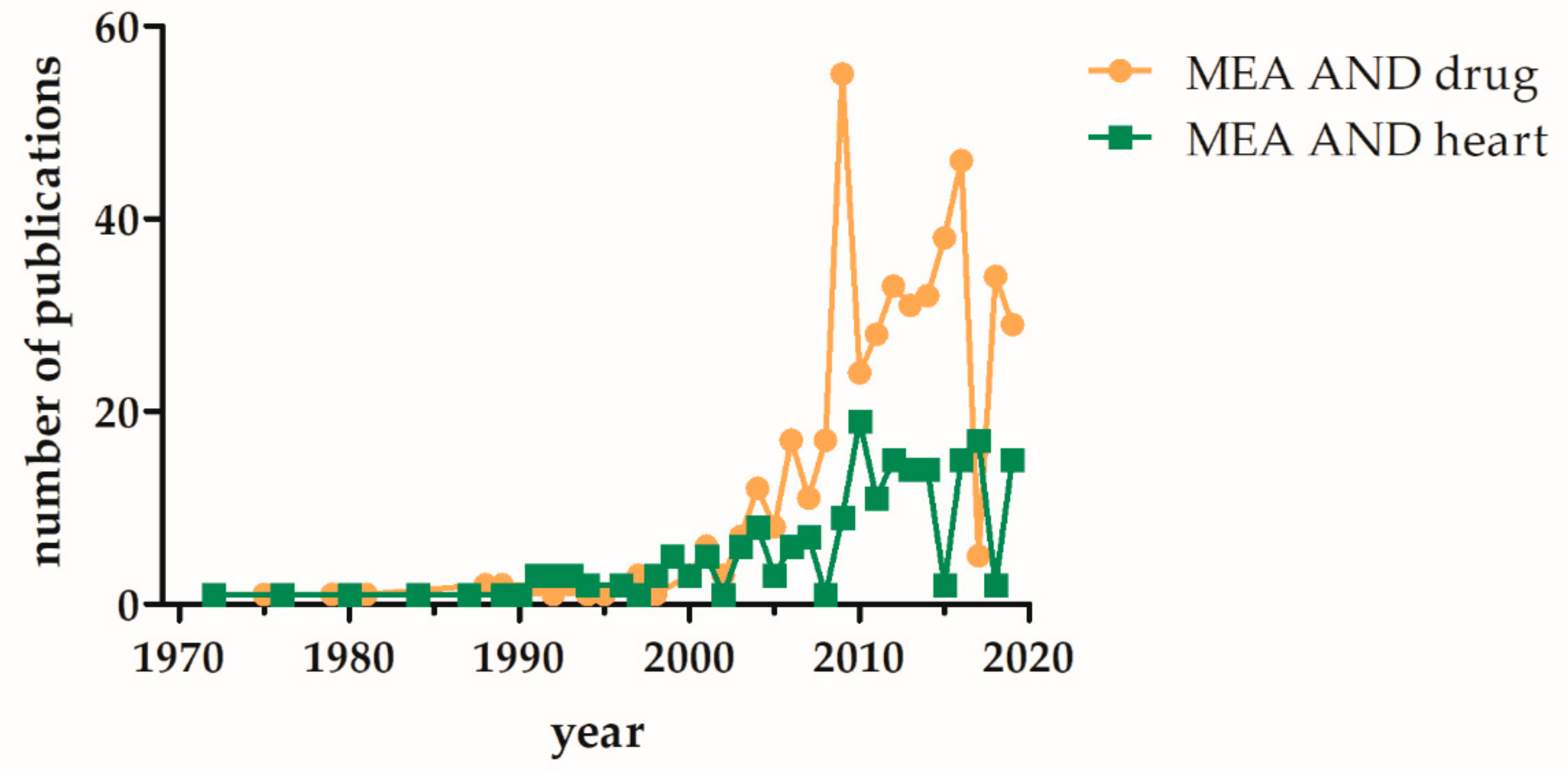
1. Introduction
2. Methods for Electrophysiological Characterization of iPSC-CMs
2.1. Patch Clamping
2.2. Optical Recordings of the Membrane Potential
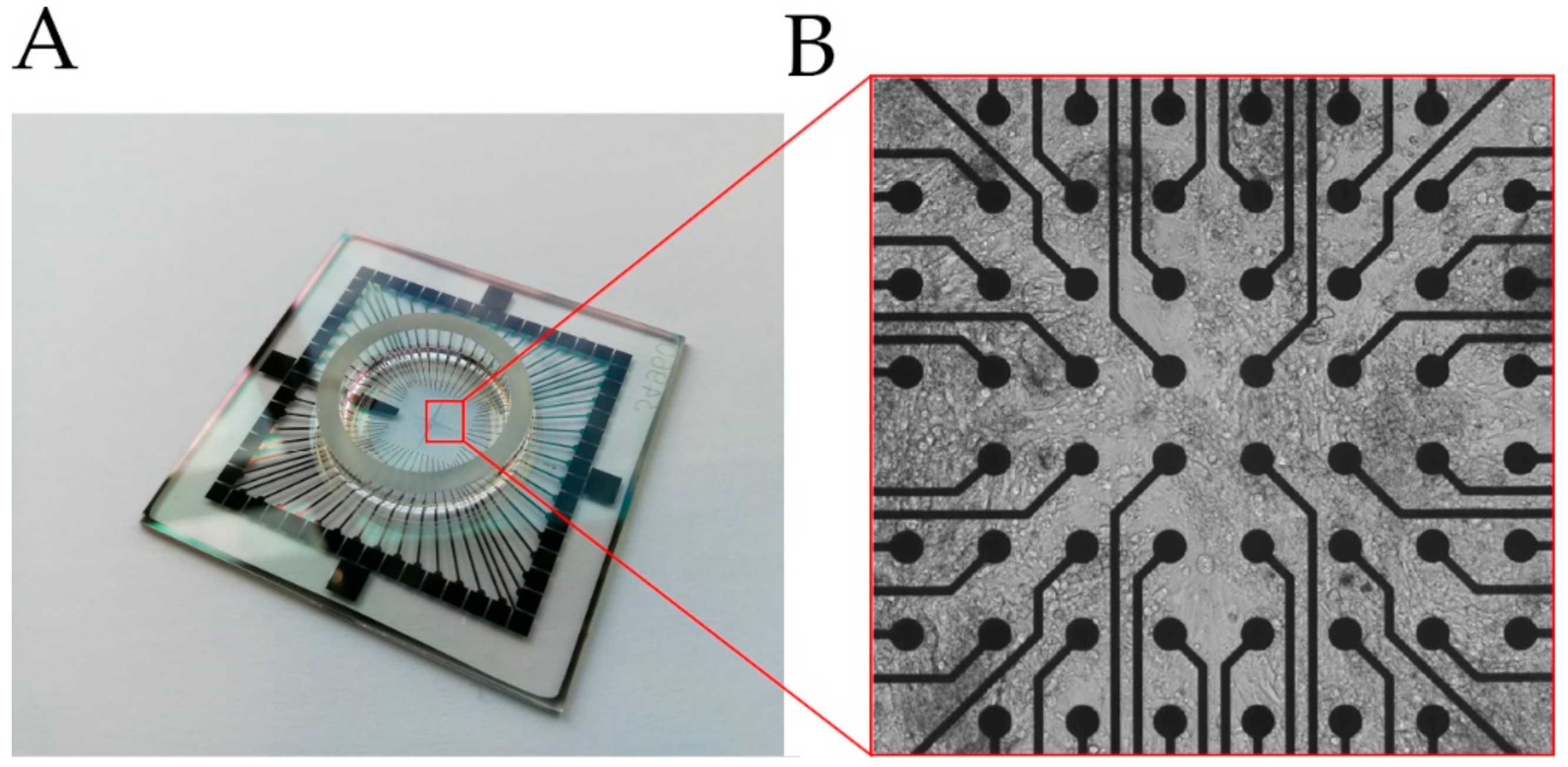
2.3. MEA-Based Analysis of Cell Behavior
3. Action Potential vs. Field Potential
3.1. Action Potential in Native Cardiac Cells and iPSC Derived CMs
3.2. Field Potential
4. Application of MEAs for Cardiotoxic Risk Assessment

{kind=link}
{kind=link}
{kind=link}
| Substance | (Site of) Action | Effect | Min. Effective Conc. | Cell Type/ Subtype | Differentiation Protocol | Age/ Maturation State | Platform | Reference |
|---|---|---|---|---|---|---|---|---|
| Alfuzosin | Treatment of benign prostatic enlargement, a hERG-channel blocker | Clinical QT prolongation | 30 nM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87,88] |
| Astemizole | Antihistaminergic drug, H1 receptor antagonist, multi-channel block | Repolarization prolongation/arrhythmogenic effects, hERG channel blockade | 3–10 nM | hiPSC-CM iCell™/iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation (+15–26 days) n/a | MEA | [87,88,93] |
| BaCl2 | Digitalis like activity, stimulation tonic contraction in muscle, used as contrast agent | Chronotropic effect K+ and Ca2+ modulation | - | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87] |
| Blebbistatin | Myosin II ATPase inhibitor | Increase in beating frequency, beating arrest (30 µM) | 1–30 µM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | Min. 32 days of differentiation | MEA | [88] |
| Carbachol | Parasympathomimetic drug cholinergic agonist KAch- channel, glaucoma treatment | Negative chronotropic effects, FPDc prolongation, decrease in beating frequency | 10 µM | Double reporter cell line, subtypes: ventricular, atrial, nodal, TBX5 Nkx2.5/hiPSC-CM (iCell™ mixture of ventricular, atrial, nodal cells) | 2D n/a | 35-40 day of differentiation 32 days of differentiation | Patch clamp, MEA | [21,88] |
| Chlorpromazine | Anti-psychotic drug, multi-channel block | Early afterdepolarization, beating arrest | 10 µM | hiPSC-CM iCell™ mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation | MEA | [88] |
| Chromanol 293B | IKv7.1 channel Blocker | Prolong FPD in control cells, | LQTS cells and control (patient- derived cells) n/a | 3D | 30-60 days of differentiation +50 days | MEA | [97] | |
| Cisapride | Prokinetic gastrointestinal drug, multi-channel block | Prolongation of FPD, Repolarization delays/arrhythmogenic effects Prolongation of QT from patients with long QT syndrome | 100 nM | hiPSC-CM Cor4U® and iCell™ mixture of ventricular, atrial, nodal cells Wt iPSC, and from patient with LQTS (n/a) | n/a 3D | 10 days after differentiation, min. 32 days of differentiation n/a | MEA, automated patch clamp | [26,87,88,93,98,99] |
| Clarithromycin | Antibiotic drug | Repolarization prolongation, arrhythmogenic effects | - | hiPSC-CM iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation n/a | MEA/VSO | [93] |
| Clozapine | Anti-psychotic drug, multi-channel block | Shortening of FPDc, increase in beat frequency | 0.3–1 µM | hiPSC-CM iCell™ mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation | MEA | [88] |
| Domperidone | Dopamine-antagonist, anti-nausea drug hERG- channel blocker | Repolarization prolongation, arrhythmogenic effects | 10 nM | hiPSC-CM iCell™/iCell2™/Cor4U® mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation n/a | MEA/VSO | [88,93] |
| Doxorubicin | anthracycline chemotherapy agent | Decrease in FPD, beat frequency and spike amplitude | 1 µM | hiPSC-CMs iCell™ 50% ventricular, 10% atrial cells patient derived cells (n/a) | n/a 2D | 32 days of differentiation 20–30 days of differentiation | MEA | [100,101] |
| Droperidol | Neuroleptic drug | Repolarization prolongation, arrhythmogenic effects | - | hiPSC-CM iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation n/a | MEA/VSO | [93] |
| Fluoxetine | Anti-depressant drug | Clinical QT prolongation | - | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87] |
| Isoproterenol | Bronchodilator | Chronotropic effect, K+ and Ca2+ Modulation, FPDc shortening, increasing beating frequency | 3–100 nM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87,88] |
| Loratadine | Anti- histaminergic drug, H1 receptor block, multi-channel block | Increase in beating frequency | 0.1–3 µM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation | MEA | [88] |
| Moxifloxacin | Anti-biotic drug, multi-channel block | Repolarization delay | 10 µM | hiPSC-CM iCell™/Cor4U® (mixture of ventricular, atrial, nodal cells) GE Healthcare (Cytiva™), Stanford Cardiac Institute | n/a | 32 days of differentiation +14–24 days n/a | MEA | [85] |
| Ondansetron | Antiemetic drug, serotonin-receptor block | Repolarization prolongation, arrhythmogenic effects | 30 nM | hiPSC-CM iCell2 ™/Cor4U® (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation n/a | MEA/VSO | [93] |
| Pimozide | Anti-psychotic drug, multi-channel block | Repolarization prolongation/arrhythmogenic effects | 3–10 nM | hiPSC-CM iCell™/iCell2 ™/Cor4U® (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days n/a | MEA | [87,93] |
| Risperidon | Anti-psychotic drug, serotonin-receptor block | Repolarization prolongation | 3–30 nM | hiPSC-CM iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation n/a | MEA | [93] |
| Sunitinib | Anti-cancer drug, tyrosine kinase inhibitor | FPDc prolongation, early afterdepolarization | 0.3–10 µM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation | MEA | [88] |
| Terfenadine | Anti-histaminergic drug, H1 receptor block | FPDc prolongation, decrease in spike amplitude, repolarization prolongation | 100–1000 nM | hiPSC-CM iCell™/iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation n/a | MEA | [93,98] |
| Tetrodotoxin (TTX) | Neurotoxic drug, (voltage sensitive) Nav (1.1, 1.7, 1.5)- channel block | Decrease in slope, depolarization potential and action potential duration | 10 µM | hiPSC-CM Cor4U® mixture of ventricular, atrial, nodal cells | n/a | 10 days after differentiation | Automated patch clamp | [26] |
| Thioridazine | Sedative, anti- psychotic drug, multi-channel block | Repolarization delays/arrhythmogenic effects | 100 nM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87,88] |
| Tolterodine | Treatment of urinary incontinence, muscarinic receptor antagonist | clinical QT prolongation, early afterdepolarization | 100–300 nM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87] |
| Vanoxerine | Serotonin-dopamine reuptake inhibitor | Clinical QT prolongation, multiple ion-channel effects, early afterdepolarizations | 100 nM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87] |
| Vandetanib | Anti-cancer drug for thyroid gland, kinase inhibitor | Repolarization prolongation, arrhythmia like events | 0.1–1 µM | hiPSC-CM iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation n/a | MEA/VSO | [93] |
| Substance | (Side of) Action | Effect | Min. Effective Conc. | Cell Type | Differentiation Protocol | Age/Maturation State | Platform | Reference |
|---|---|---|---|---|---|---|---|---|
| Amiodarone | Class III anti-arrhythmic drug, multi-channel block | Clinical QT prolongation | 0.1–1 µM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87,88] |
| Azimilide | Class III anti-arrhythmic drug | FPDc prolongation, decrease in beating frequency, early after depolarization | 0.3–1 µM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation | MEA | [88] |
| Bay K 8644 | Agonist of voltage sensitive dihydropyridine (DHP; L-Typ) Calcium channel | FPDc prolongation, decrease in beat frequency, positive inotropic | 0.3–3 nM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87,88] |
| Bepridil | Class IV anti-arrhythmic drug, multi-channel block | Repolarization delays/arrhythmogenic effects | 0.1–1 µM | hiPSC-CM iCell™/iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days n/a | MEA | [87,88,93] |
| Dofetilide | Class III anti-arrhythmic drug, multi-channel block | Increase in FPD, TdP arrhythmias | 3–100 nM | hiPSC-CM iCell™/iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) Pluricytes™ | n/a | 32 days of differentiation n/a n/a | MEA | [88,93,102] |
| E-4031 | Class III anti-arrhythmic drug, hERG- channel block | prolonged FPD, severe arrhythmia in LQTS iPSC-CM | 30–100 nM | hiPSC-CM iCell™/Cor4U® (mixture of ventricular, atrial, nodal cells) GE Healthcare (Cytiva™), Stanford Cardiac Institute LQTS cells and control | n/a3D | 32 days of differentiation +14–24 days n/a 30–60 days of differentiation +50 days | MEA | [85,97,98] |
| Flecainide | Class Ic anti-arrhythmic drug, multi-channel block | Decrease in spike amplitude, FPDc prolongation | 1 µM | hiPSC-CM iCell™/Cor4U® (mixture of ventricular, atrial, nodal cells) GE Healthcare (Cytiva™), Stanford Cardiac InstituteCPVT cells and control | n/a 2D/3D | 32 days of differentiation +14–24 days n/a 20–30 days of beating | MEA Patch clamp | [85,98,103] |
| Ibutilide | Class III Anti-arrhythmic drug, multi-channel block | Arrhythmia like events, early after depolarizations | 1–100 nM | hiPSC-CM iCell™/iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation n/a | MEA/VSO | [88,93] |
| Ivabradin | Treatment of stable angina pectoris, If-channel inhibitor, heart rate reducing drug | Prolongation in APD, decrease in beating frequency | 1 µM | Double reporter cell line, subtypes: ventricular, atrial, nodal, TBX5 Nkx2.5/hiPSC-CM | 2D | 35–40 days of differentiation | Patch clamp | [21] |
| JNJ303 | IKv7.1- channel inhibitor | Small prolongation of FPDc | 300 nM | hiPSC-CM iCell™/Cor4U® (mixture of ventricular, atrial, nodal cells) GE Healthcare (Cytiva™), Stanford Cardiac Institute | n/a | 32 days of differentiation n/a | MEA | [85] |
| Levocromakalim | Vasodilating drug, KATP opener | Membrane hyperpolarization, decrease in FPDc and beating frequency | 1–3 µM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87,88] |
| Metoprolol | Anti- arrhythmic, anti- hypertonic drug, ß1-adreno receptor block | Induced arrhythmias, hERG block at higher concentrations | 100 µM | hIPSC CM iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) CPVT cells and control | n/a 2D/3D | 32 days of differentiation n/a 20–30 days of beating | MEA/VSO Patch clamp | [93,103] |
| Mexiletine | Class Ib anti-arrhythmic drug, Inhibiting Nav1.5- also hERG block | Reduce spike amplitude, cessation of spontaneous beating (100 µM) | 1–10 µM, | hIPSC-CM iCell™/iCell2™ Cor4U® (mixture of ventricular, atrial, nodal cells) GE Healthcare (Cytiva™), Stanford Cardiac Institute | n/a | 32 days of differentiation +14–24 days n/a | MEA | [85,88,93,98] |
| Mibefradil | Treatment of angina pectoris and hypertension, multi-channel block | Shortening in FPDc, increase in beat frequency | 0.3–1 µM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87,88] |
| Nifedipin | Vasodilating drug, ICaL block | Shortening of FPDc, increase in beating rate | 0.3–1 µM | hiPSC-CM iCell™/Cor4U® (mixture of ventricular, atrial, nodal cells) GE Healthcare (Cytiva™), Stanford Cardiac Institute | n/a | 32 days of differentiation +10 days n/a | MEA, automated patch clamp | [26,85,98] |
| NS- 1643 | hERG-channel activator | Repolarization effect, decrease in FPDc, increase in beating frequency | 3 µM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87,88] |
| Ouabain | Cardiac glycoside, Na+-K+- ATPase inhibitor | Repolarization effects, decrease in FPDc | 10–100 nM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87,88] |
| Propranolol | Class II anti-arrhythmic drug, beta- receptor block | Early afterdepolarization, decrease in beating frequency | 10 µM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation | MEA | [88] |
| Quinidine | Class Ia anti-arrhythmic drug, multi-channel block (Nav1.5, Cav1.2, hERG) | FPDc prolongation, reduced spike amplitude, repolarization delays/arrhythmogenic effects | 0.3–10 µM | hiPSC-CM iCell™/iCell2™ Cor4U® (mixture of ventricular, atrial, nodal cells) GE Healthcare (Cytiva™), Stanford Cardiac Institute Fibroblast-derived iPSC-CM (67% ventricular, 5% nodal, 28% atrial) | n/a 3D | 32 days of differentiation +15–26 days 65–95 days after differentiation induction | MEA, Low impedance MEA | [85,87,88,93,98,104] |
| Ranolazine | Angina pectoris treatment, multichannel (Na and hERG block) | FPDc prolongation, clinical QT prolongation, repolarization prolongation | 0.3 µM, clinical conc. <100 µM | hiPSC-CM iCell iCell2™ Cor4U® (mixture of ventricular, atrial, nodal cells) GE Healthcare (Cytiva™), Stanford Cardiac Institute | 32 days of differentiation +15–26 days (14–24 days) n/a | MEA | [85,87,88,93] | |
| Sotalol | Anti-arrhythmic drug, beta adreno receptor block | Repolarization prolongation, arrhythmogenic effects, hERG- channel block | 15 µM | hiPSC-CMs iCell2™/Cor4U® (mixture of ventricular, atrial, nodal cells) Fibroblast-derived iPSC-CM (67% ventricular,5% nodal, 28% atrial) | n/a 3D | 32 days of differentiation n/a 65–95 days after differentiation induction | Low impedance MEA | [93,104] |
| Verapamil | Class VI anti-arrhythmic drug, inhibits hERG, ICal-typ calcium channels, Multi-channel block | Shortening of FPDc, increase in spontaneous beat rate; shortening in APD20 and APD90 | 0.1–0.3 µM; 1 µM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) Pluricytes™ | n/a | 32 days of differentiation n/a | MEA; patch clamp | [98,102] |
| Vernakalant | Class III anti-arrhythmic drug, used for cardioversion of atrial fibrillation, atrial potassium- channel block | APD prolongation, partly arrhythmogenic effects, | - | Double reporter cell line, different subtypes (ventricular phenotype) | 2D | 20–30 days post induction of differentiation | Patch clamp | [21] |
| ZD 7288 | Selective hyperpolarization-activated cyclic nucleotide-gated channel blocker, If- current inhibitor | Negative chronotropic effect, FPDc prolongation | 3–30 nM | hiPSC-CM iCell™ (mixture of ventricular, atrial, nodal cells) | n/a | 32 days of differentiation +15–26 days | MEA | [87,88] |
5. Disease Modeling Using hiPSC-CM
6. Overview of Developed Disease Models
7. Conclusions and Outlook
Supplementary Materials
Funding
Conflicts of Interest
Abbreviations
| 2D | Two-dimensional |
| ADPKD | Autosomal dominant polycystic kidney disease |
| AFM | Atomic force microscopy |
| AP | Action potential |
| APD | Action potential duration |
| ATM | Atomic force microscopy |
| AV Node | Atrioventricular node |
| HCM | Hypertrophic cardiomyopathy |
| Ca2+ | Calcium |
| CiPA | Comprehensive in vitro proarrhythmia assay |
| CM | Cardiomyocytes |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| CSAHI | Consortium for Safety Assessment using Human iPS cells |
| CVD | Cardiovascular disease |
| DCM | Familial dilated cardiomyopathy |
| FP | Field potential |
| FPDc | Field potential duration (corrected) |
| FRET | Fluorescence resonance energy transfer |
| GL-3 | Basic helix-loop-helix proteins, GLABRA3 |
| gpAPD | Guinea pig papillary muscle action potential assay |
| hERG | Human ether-a-go-go Related gene-voltage sensitive potassium channel |
| hiPSC | Human induced pluripotent stem cell |
| K+ | Potassium |
| LIMP-2 | Lysosomal integral membrane protein 2 |
| LMNA | Lamin A/C |
| MEA | Multi-/micro-electrode array |
| MYH7 | Myosin heavy chain 7 |
| Na+ | Sodium |
| PKD1/2 | Polycystic kidney disease 1 |
| RNA | Ribonucleic acid |
| SCN5A | Sodium voltage-gated channel alpha subunit 5 |
| TBX5 | T-box transcription factor |
| TdP | Torsade-de-Pointes-Tachycardia |
| TnnT | TroponinT |
| VSO | Voltage-sensitive optical system |
| WT | Wild type |
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Yamanaka, S. Induced Pluripotent Stem Cells 10 Years Later. Circ. Res. 2017, 120, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Zuppinger, C. 3D Cardiac Cell Culture: A Critical Review of Current Technologies and Applications. Front. Cardiovasc. Med. 2019, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Rojas, S.V.; Kensah, G.; Rotaermel, A.; Baraki, H.; Kutschka, I.; Zweigerdt, R.; Martin, U.; Haverich, A.; Gruh, I.; Martens, A. Transplantation of purified iPSC-derived cardiomyocytes in myocardial infarction. PLoS ONE 2017, 12, e0173222. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gregorich, Z.R.; Zhu, W.; Mattapally, S.; Oduk, Y.; Lou, X.; Kannappan, R.; Borovjagin, A.V.; Walcott, G.P.; Pollard, A.E.; et al. Large Cardiac Muscle Patches Engineered From Human Induced-Pluripotent Stem Cell–Derived Cardiac Cells Improve Recovery From Myocardial Infarction in Swine. Circulation 2018, 137, 1712–1730. [Google Scholar] [CrossRef]
- Shadrin, I.Y.; Allen, B.W.; Qian, Y.; Jackman, C.P.; Carlson, A.L.; Juhas, M.E.; Bursac, N. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat. Commun. 2017, 8, 1825. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, H.; Xiao, D.; Yang, H.; Itzhaki, I.; Qin, X.; Chour, T.; Aguirre, A.; Lehmann, K.; Kim, Y.; et al. Comparison of Non-human Primate versus Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Treatment of Myocardial Infarction. Stem Cell Rep. 2018, 10, 422–435. [Google Scholar] [CrossRef]
- Pfeiffer-Kaushik, E.R.; Smith, G.L.; Cai, B.; Dempsey, G.T.; Hortigon-Vinagre, M.P.; Zamora, V.; Feng, S.; Ingermanson, R.; Zhu, R.; Hariharan, V.; et al. Electrophysiological characterization of drug response in hSC-derived cardiomyocytes using voltage-sensitive optical platforms. J. Pharmacol. Toxicol. Methods 2018, 99, 106612. [Google Scholar] [CrossRef]
- Huo, J.; Wei, F.; Cai, C.; Lyn-Cook, B.; Pang, L. Sex-Related Differences in Drug-Induced QT Prolongation and Torsades de Pointes: A New Model System with Human iPSC-CMs. Toxicol. Sci. 2018, 167, 360–374. [Google Scholar] [CrossRef]
- Izumi-Nakaseko, H.; Hagiwara-Nagasawa, M.; Naito, A.T.; Goto, A.; Chiba, K.; Sekino, Y.; Kanda, Y.; Sugiyama, A. Application of human induced pluripotent stem cell-derived cardiomyocytes sheets with microelectrode array system to estimate antiarrhythmic properties of multi-ion channel blockers. J. Pharmacol. Sci. 2018, 137, 372–378. [Google Scholar] [CrossRef]
- Edwards, S.L.; Zlochiver, V.; Conrad, D.B.; Vaidyanathan, R.; Valiquette, A.M.; Joshi-Mukherjee, R. A Multiwell Cardiac μGMEA Platform for Action Potential Recordings from Human iPSC-Derived Cardiomyocyte Constructs. Stem Cell Rep. 2018, 11, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Braam, S.R.; Tertoolen, L.; van de Stolpe, A.; Meyer, T.; Passier, R.; Mummery, C.L. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 2010, 4, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Goineau, S.; Castagné, V. Electrophysiological characteristics and pharmacological sensitivity of two lines of human induced pluripotent stem cell derived cardiomyocytes coming from two different suppliers. J. Pharmacol. Toxicol. Methods 2018, 90, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Grimm, F.A.; Blanchette, A.; House, J.S.; Ferguson, K.; Hsieh, N.-H.; Dalaijamts, C.; Wright, A.A.; Anson, B.; Wright, F.A.; Chiu, W.A.; et al. A human population-based organotypic in vitro model for cardiotoxicity screening. ALTEX 2018, 35, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Goversen, B.; van der Heyden, M.A.G.; van Veen, T.A.B.; de Boer, T.P. The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: Special focus on IK1. Pharm. Ther. 2018, 183, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Paci, M.; Hyttinen, J.; Rodriguez, B.; Severi, S. Human induced pluripotent stem cell-derived versus adult cardiomyocytes: An\textlessi\textgreaterin silico\textless/i\textgreater electrophysiological study on effects of ionic current block. Br. J. Pharmacol. 2015, 172, 5147–5160. [Google Scholar] [CrossRef]
- Tan, S.H.; Ye, L. Maturation of Pluripotent Stem Cell-Derived Cardiomyocytes: A Critical Step for Drug Development and Cell Therapy. J. Cardiovasc. Transl. Res. 2018, 11, 375–392. [Google Scholar] [CrossRef]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Insights into Molecular, Cellular, and Functional Phenotypes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef]
- Hausburg, F.; Jung, J.J.; David, R. Specific cell (re-)programming: Approaches and perspectives. In Advances in Biochemical Engineering/Biotechnology; Springer: Basel, Switzerland, 2017; Volume 163, pp. 71–115. [Google Scholar]
- Zhang, J.Z.; Termglinchan, V.; Shao, N.-Y.; Itzhaki, I.; Liu, C.; Ma, N.; Tian, L.; Wang, V.Y.; Chang, A.C.Y.; Guo, H.; et al. A Human iPSC Double-Reporter System Enables Purification of Cardiac Lineage Subpopulations with Distinct Function and Drug Response Profiles. Cell Stem. Cell 2019, 24, 802–811. [Google Scholar] [CrossRef]
- Obergrussberger, A.; Stölzle-Feix, S.; Becker, N.; Brüggemann, A.; Fertig, N.; Möller, C. Novel Screening Techniques For Ion Channel Targeting Drugs. Channels 2015, 9, 367–375. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Casini, S.; Verkerk, A.O.; Remme, C.A. Human iPSC-Derived Cardiomyocytes for Investigation of Disease Mechanisms and Therapeutic Strategies in Inherited Arrhythmia Syndromes: Strengths and Limitations. Cardiovasc. Drugs Ther. 2017, 31, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.P.; Casini, S.; van den Berg, C.W.; Hoekstra, M.; Remme, C.A.; Dambrot, C.; Salvatori, D.; Oostwaard, D.W.; Wilde, A.A.M.; Bezzina, C.R.; et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 2012, 125, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Sallam, K.; Li, Y.; Sager, P.T.; Houser, S.R.; Wu, J.C. Finding the Rhythm of Sudden Cardiac Death. Circ. Res. 2015, 116, 1989–2004. [Google Scholar] [CrossRef] [PubMed]
- Scheel, O.; Frech, S.; Amuzescu, B.; Eisfeld, J.; Lin, K.-H.; Knott, T. Action Potential Characterization of Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes Using Automated Patch-Clamp Technology. Assay Drug Dev. Technol. 2014, 12, 457–469. [Google Scholar] [CrossRef]
- Obergrussberger, A.; Goetze, T.A.; Brinkwirth, N.; Becker, N.; Friis, S.; Rapedius, M.; Haarmann, C.; Rinke-Weiß, I.; Stölzle-Feix, S.; Brüggemann, A.; et al. An update on the advancing high-throughput screening techniques for patch clamp-based ion channel screens: Implications for drug discovery. Expert Opin. Drug Discov. 2018, 13, 269–277. [Google Scholar] [CrossRef]
- Franz, D.; Olsen, H.L.; Klink, O.; Gimsa, J. Automated and manual patch clamp data of human induced pluripotent stem cell-derived dopaminergic neurons. Sci. Data 2017, 4, 170056. [Google Scholar] [CrossRef]
- Yajuan, X.; Xin, L.; Zhiyuan, L. A Comparison of the Performance and Application Differences Between Manual and Automated Patch-Clamp Techniques. Curr. Chem. Geno. 2012, 6, 87. [Google Scholar] [CrossRef]
- Bell, D.C.; Dallas, M.L. Using automated patch clamp electrophysiology platforms in pain-related ion channel research: Insights from industry and academia. Br. J. Pharmacol. 2018, 175, 2312–2321. [Google Scholar] [CrossRef]
- Huang, H.-L.; Hsing, H.-W.; Lai, T.-C.; Chen, Y.-W.; Lee, T.-R.; Chan, H.-T.; Lyu, P.-C.; Wu, C.-L.; Lu, Y.-C.; Lin, S.-T.; et al. Trypsin-induced proteome alteration during cell subculture in mammalian cells. J. Biomed. Sci. 2010, 17, 36. [Google Scholar] [CrossRef]
- Rajamohan, D.; Kalra, S.; Duc Hoang, M.; George, V.; Staniforth, A.; Russell, H.; Yang, X.; Denning, C. Automated Electrophysiological and Pharmacological Evaluation of Human Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Dev. 2016, 25, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Storace, D.; Rad, M.S.; Han, Z.; Jin, L.; Cohen, L.B.; Hughes, T.; Baker, B.J.; Sung, U. Genetically encoded protein sensors of membrane potential. In Membrane Potential Imaging in the Nervous System and Heart; Springer: Basel, Switzerland, 2015; pp. 493–509. [Google Scholar]
- del Álamo, J.C.; Lemons, D.; Serrano, R.; Savchenko, A.; Cerignoli, F.; Bodmer, R.; Mercola, M. High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim. Et Biophys. Acta Mol. Cell Res. 2016, 1863, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Bedut, S.; Seminatore-Nole, C.; Lamamy, V.; Caignard, S.; Boutin, J.A.; Nosjean, O.; Stephan, J.-P.; Coge, F. High-throughput drug profiling with voltage- and calcium-sensitive fluorescent probes in human iPSC-derived cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, 44–53. [Google Scholar] [CrossRef]
- Hortigon-Vinagre, M.P.; Zamora, V.; Burton, F.L.; Green, J.; Gintant, G.A.; Smith, G.L. The use of ratiometric fluorescence measurements of the voltage sensitive dye Di-4-ANEPPS to examine action potential characteristics and drug effects on human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Sci. 2016, 154, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Takaki, T.; Inagaki, A.; Chonabayashi, K.; Inoue, K.; Miki, K.; Ohno, S.; Makiyama, T.; Horie, M.; Yoshida, Y. Optical Recording of Action Potentials in Human Induced Pluripotent Stem Cell-Derived Cardiac Single Cells and Monolayers Generated from Long QT Syndrome Type 1 Patients. Stem Cells Int. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- St-Pierre, F.; Chavarha, M.; Lin, M.Z. Designs and sensing mechanisms of genetically encoded fluorescent voltage indicators. Curr. Opin. Chem. Biol. 2015, 27, 31–38. [Google Scholar] [CrossRef]
- Han, Z.; Jin, L.; Platisa, J.; Cohen, L.B.; Baker, B.J.; Pieribone, V.A. Fluorescent Protein Voltage Probes Derived from ArcLight that Respond to Membrane Voltage Changes with Fast Kinetics. PLoS ONE 2013, 8, e81295. [Google Scholar] [CrossRef]
- Shaheen, N.; Shiti, A.; Huber, I.; Shinnawi, R.; Arbel, G.; Gepstein, A.; Setter, N.; Goldfracht, I.; Gruber, A.; Chorna, S.V.; et al. Human Induced Pluripotent Stem Cell-Derived Cardiac Cell Sheets Expressing Genetically Encoded Voltage Indicator for Pharmacological and Arrhythmia Studies. Stem Cell Rep. 2018, 10, 1879–1894. [Google Scholar] [CrossRef] [PubMed]
- Leyton-Mange, J.S.; Mills, R.W.; Macri, V.S.; Jang, M.Y.; Butte, F.N.; Ellinor, P.T.; Milan, D.J. Rapid cellular phenotyping of human pluripotent stem cell-derived cardiomyocytes using a genetically encoded fluorescent voltage sensor. Stem Cell Rep. 2014, 2, 163–170. [Google Scholar] [CrossRef]
- Shinnawi, R.; Huber, I.; Maizels, L.; Shaheen, N.; Gepstein, A.; Arbel, G.; Tijsen, A.J.; Gepstein, L. Monitoring human-induced pluripotent stem cell-derived cardiomyocytes with genetically encoded calcium and voltage fluorescent reporters. Stem Cell Rep. 2015, 5, 582–596. [Google Scholar] [CrossRef]
- Song, L.; Awari, D.W.; Han, E.Y.; Uche-Anya, E.; Park, S.-H.E.; Yabe, Y.A.; Chung, W.K.; Yazawa, M. Dual optical recordings for action potentials and calcium handling in induced pluripotent stem cell models of cardiac arrhythmias using genetically encoded fluorescent indicators. Stem Cells Transl. Med. 2015, 4, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Herron, T.J. Calcium and voltage mapping in hiPSC-CM monolayers. Cell Calcium. 2016, 59, 84–90. [Google Scholar]
- Herron, T.J.; Lee, P.; Jalife, J. Optical Imaging of Voltage and Calcium in Cardiac Cells & Tissues. Circ. Res. 2012, 110, 609–623. [Google Scholar] [PubMed]
- Li, X.; Zhang, R.; Zhao, B.; Lossin, C.; Cao, Z. Cardiotoxicity screening: A review of rapid-throughput in vitro approaches. Arch. Toxicol. 2016, 90, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, W.; Asakura, K.; Ando, H.; Taniguchi, T.; Ojima, A.; Uda, T.; Osada, T.; Hayashi, S.; Kasai, C.; Miyamoto, N.; et al. Electrophysiological characteristics of human iPSC-derived cardiomyocytes for the assessment of drug-induced proarrhythmic potential. PLoS ONE 2016, 11, e0167348. [Google Scholar] [CrossRef]
- Sala, L.; Ward-van Oostwaard, D.; Tertoolen, L.G.J.; Mummery, C.L.; Bellin, M. Electrophysiological Analysis of human Pluripotent Stem Cell-derived Cardiomyocytes (hPSC-CMs) Using Multi-electrode Arrays (MEAs). J. Vis. Exp. 2017, 123, e55587. [Google Scholar] [CrossRef]
- Asakura, K.; Hayashi, S.; Ojima, A.; Taniguchi, T.; Miyamoto, N.; Nakamori, C.; Nagasawa, C.; Kitamura, T.; Osada, T.; Honda, Y.; et al. Improvement of acquisition and analysis methods in multi-electrode array experiments with iPS cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 2015, 75, 17–26. [Google Scholar] [CrossRef]
- Zhu, H.; Scharnhorst, K.S.; Stieg, A.Z.; Gimzewski, J.K.; Minami, I.; Nakatsuji, N.; Nakano, H.; Nakano, A. Two dimensional electrophysiological characterization of human pluripotent stem cell-derived cardiomyocyte system. Sci. Rep. 2017, 7, 43120. [Google Scholar] [CrossRef]
- Ryynänen, T.; Pekkanen-Mattila, M.; Shah, D.; Kreutzer, J.; Kallio, P.; Lekkala, J.; Aalto-Setälä, K. Microelectrode array for noninvasive analysis of cardiomyocytes at the single-cell level. Jpn. J. Appl. Phys. 2018, 57, 117001. [Google Scholar] [CrossRef]
- Kaneko, T.; Toriumi, H.; Shimada, J.; Nomura, F. Extracellular field potential recording of single cardiomyocytes in agarose microchambers using microelectrode array. J. Appl. Phys. 2018, 57, 03EB03. [Google Scholar] [CrossRef]
- Kang, C.; Qiao, Y.; Li, G.; Baechle, K.; Camelliti, P.; Rentschler, S.; Efimov, I.R. Human Organotypic Cultured Cardiac Slices: New Platform For High Throughput Preclinical Human Trials. Sci. Rep. 2016, 6, 28798. [Google Scholar] [CrossRef]
- Lane, J.D.; Montaigne, D.; Tinker, A. Tissue-Level Cardiac Electrophysiology Studied in Murine Myocardium Using a Microelectrode Array: Autonomic and Thermal Modulation. J. Membr. Biol. 2017, 250, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.A.; Tzortzis, K.N.; Dupont, E.; Selvadurai, S.; Perbellini, F.; Cantwell, C.D.; Ng, F.S.; Simon, A.R.; Terracciano, C.M.; Peters, N.S. Concurrent micro-to macro-cardiac electrophysiology in myocyte cultures and human heart slices. Sci. Rep. 2018, 8, 6947. [Google Scholar] [CrossRef] [PubMed]
- Doerr, L.; Thomas, U.; Guinot, D.R.; Bot, C.T.; Stoelzle-Feix, S.; Beckler, M.; George, M.; Fertig, N. New Easy-to-Use Hybrid System for Extracellular Potential and Impedance Recordings. J. Lab. Autom. 2015, 20, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Huang, C.; Lin, Y.D.; Ivanovskaya, A.N.; O’Hara, T.J.; Booth, R.H.; Creek, C.J.; Enright, H.A.; Soscia, D.A.; Belle, A.M.; et al. Simultaneous electrical recording of cardiac electrophysiology and contraction on chip. Lab. A Chip 2017, 17, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Takasuna, K.; Asakura, K.; Araki, S.; Ando, H.; Kazusa, K.; Kitaguchi, T.; Kunimatsu, T.; Suzuki, S.; Miyamoto, N. Comprehensive in Vitro Cardiac Safety Assessment Using Human Stem Cell Technology: Overview of Csahi Heart Initiative. J. Pharma. Toxico. Meth. 2017, 83, 42–54. [Google Scholar] [CrossRef]
- Hayakawa, T.; Kunihiro, T.; Ando, T.; Kobayashi, S.; Matsui, E.; Yada, H.; Kanda, Y.; Kurokawa, J.; Furukawa, T. Image-based evaluation of contraction-relaxation kinetics of human-induced pluripotent stem cell-derived cardiomyocytes: Correlation and complementarity with extracellular electrophysiology. J. Mol. Cell. Cardiol. 2014, 77, 178–191. [Google Scholar] [CrossRef]
- Cools, J.; Jin, Q.; Yoon, E.; Alba Burbano, D.; Luo, Z.; Cuypers, D.; Callewaert, G.; Braeken, D.; Gracias, D.H. A Micropatterned Multielectrode Shell for 3D Spatiotemporal Recording from Live Cells. Adv. Sci. 2018, 5, 1700731. [Google Scholar] [CrossRef]
- Nagarah, J.M.; Stowasser, A.; Parker, R.L.; Asari, H.; Wagenaar, D.A. Optically transparent multi-suction electrode arrays. Front. Neurosci. 2015, 9, 384. [Google Scholar] [CrossRef]
- Simeonov, S.; Schäffer, T.E. Ultrafast imaging of cardiomyocyte contractions by combining scanning ion conductance microscopy with a microelectrode array. Anal. Chem. 2019, 91, 9648–9655. [Google Scholar] [CrossRef]
- Kanda, Y.; Yamazaki, D.; Kurokawa, J.; Inutsuka, T.; Sekino, Y. Points to consider for a validation study of iPS cell-derived cardiomyocytes using a multi-electrode array system. J. Pharmacol. Toxicol. Methods 2016, 81, 196–200. [Google Scholar] [CrossRef]
- Uesugi, M.; Ojima, A.; Taniguchi, T.; Miyamoto, N.; Sawada, K. Low-density plating is sufficient to induce cardiac hypertrophy and electrical remodeling in highly purified human iPS cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 2014, 69, 177–188. [Google Scholar] [CrossRef]
- Amin, A.S.; Tan, H.L.; Wilde, A.A.M. Cardiac ion channels in health and disease. Heart Rhythm. 2010, 7, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.J.; Husse, B.; Rimmbach, C.; Krebs, S.; Stieber, J.; Steinhoff, G.; Dendorfer, A.; Franz, W.-M.; David, R. Programming and Isolation of Highly Pure Physiologically and Pharmacologically Functional Sinus-Nodal Bodies from Pluripotent Stem Cells. Stem Cell Rep. 2014, 2, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Protze, S.I.; Liu, J.; Nussinovitch, U.; Ohana, L.; Backx, P.H.; Gepstein, L.; Keller, G.M. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat. Biotechnol. 2017, 35, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Laksman, Z.; Backx, P.H. The electrophysiological development of cardiomyocytes. Adv. Drug Deliv. Rev. 2016, 96, 253–273. [Google Scholar]
- Feher, J. The Cardiac Action Potential. In Quantitative Human Physiology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 528–536. [Google Scholar]
- Du, D.T.M.; Hellen, N.; Kane, C.; Terracciano, C.M.N. Action potential morphology of human induced pluripotent stem cell-derived cardiomyocytes does not predict cardiac chamber specificity and is dependent on cell density. Biophys. J. 2015, 108, 1–4. [Google Scholar] [CrossRef]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Markandeya, Y.S.; Kamp, T.J.; Makielski, J.C.; January, C.T.; Eckhardt, L.L. IK1-enhanced human-induced pluripotent stem cell-derived cardiomyocytes: An improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, 1611–1621. [Google Scholar]
- Goversen, B.; Becker, N.; Stoelzle-Feix, S.; Obergrussberger, A.; Vos, M.A.; van Veen, T.A.B.; Fertig, N.; de Boer, T.P. A Hybrid Model for Safety Pharmacology on an Automated Patch Clamp Platform: Using Dynamic Clamp to Join iPSC-Derived Cardiomyocytes and Simulations of Ik1 Ion Channels in Real-Time. Front. Physiol. 2018, 8, 1094. [Google Scholar] [CrossRef]
- Jonsson, M.K.B.; Vos, M.A.; Mirams, G.R.; Duker, G.; Sartipy, P.; De Boer, T.P.; Van Veen, T.A.B. Application of human stem cell-derived cardiomyocytes in safety pharmacology requires caution beyond hERG. J. Mol. Cell. Cardiol. 2012, 52, 998–1008. [Google Scholar] [CrossRef]
- Tertoolen, L.G.J.; Braam, S.R.; van Meer, B.J.; Passier, R.; Mummery, C.L. Interpretation of field potentials measured on a multi electrode array in pharmacological toxicity screening on primary and human pluripotent stem cell-derived cardiomyocytes. Biochem. Biophys. Res. Commun. 2018, 497, 1135–1141. [Google Scholar] [CrossRef]
- Clements, M. Multielectrode array (MEA) assay for unit 22.4 profiling electrophysiological drug effects in human stem cell-derived cardiomyocytes. Curr. Protoc. Toxicol. 2016, 2016, 1–32. [Google Scholar]
- Buzsáki, G.; Anastassiou, C.A.; Koch, C. The origin of extracellular fields and currents — EEG, ECoG, LFP and spikes. Nat. Rev. Neurosci. 2012, 13, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Halbach, M.D.; Egert, U.; Hescheler, J.; Banach, K. Estimation of action potential changes from field potential recordings in multicellular mouse cardiac myocyte cultures. Cell. Physiol. Biochem. 2003, 13, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Hayes, H.B.; Nicolini, A.M.; Arrowood, C.A.; Chvatal, S.A.; Wolfson, D.W.; Cho, H.C.; Sullivan, D.D.; Chal, J.; Fermini, B.; Clements, M.; et al. Novel method for action potential measurements from intact cardiac monolayers with multiwell microelectrode array technology. Sci. Rep. 2019, 9, 11893. [Google Scholar] [CrossRef] [PubMed]
- Jans, D.; Callewaert, G.; Krylychkina, O.; Hoffman, L.; Gullo, F.; Prodanov, D.; Braeken, D. Action potential-based MEA platform for in vitro screening of drug-induced cardiotoxicity using human iPSCs and rat neonatal myocytes. J. Pharmacol. Toxicol. Methods 2017, 87, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Lin, Z.; Hanson, L.; Cui, Y.; Cui, B. Intracellular recording of action potentials by nanopillar electroporation. Nat. Nanotechnol. 2012, 7, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Fendyur, A.; Spira, M.E. Toward on-chip, in-cell recordings from cultured cardiomyocytes by arrays of gold mushroom-shaped microelectrodes. Front. Neuroeng. 2012, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- WHO. Cardiovascular diseases. 2019. Available online: https://www.who.int/health-topics/cardiovascular-diseases (accessed on 28 October 2019).
- Fermini, B.; Hancox, J.C.; Abi-Gerges, N.; Bridgland-Taylor, M.; Chaudhary, K.W.; Colatsky, T.; Correll, K.; Crumb, W.; Damiano, B.; Erdemli, G.; et al. A New Perspective in the Field of Cardiac Safety Testing through the Comprehensive In Vitro Proarrhythmia Assay Paradigm. J. Biomol. Screen 2016, 21, 1–11. [Google Scholar] [CrossRef]
- Millard, D.; Dang, Q.; Shi, H.; Zhang, X.; Strock, C.; Kraushaar, U.; Zeng, H.; Levesque, P.; Lu, H.-R.; Guillon, J.-M.; et al. Cross-Site Reliability of Human Induced Pluripotent stem cell-derived Cardiomyocyte Based Safety Assays Using Microelectrode Arrays: Results from a Blinded CiPA Pilot Study. Toxicol. Sci. 2018, 164, 550–562. [Google Scholar] [CrossRef]
- CiPA Initiative. 2019. Available online: https://cipaproject.org (accessed on 28 October 2019).
- Kitaguchi, T.; Moriyama, Y.; Taniguchi, T.; Maeda, S.; Ando, H.; Uda, T.; Otabe, K.; Oguchi, M.; Shimizu, S.; Saito, H.; et al. CSAHi study: Detection of drug-induced ion channel/receptor responses, QT prolongation, and arrhythmia using multi-electrode arrays in combination with human induced pluripotent stem cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 2017, 85, 73–81. [Google Scholar] [CrossRef]
- Nozaki, Y.; Honda, Y.; Watanabe, H.; Saiki, S.; Koyabu, K.; Itoh, T.; Nagasawa, C.; Nakamori, C.; Nakayama, C.; Iwasaki, H.; et al. CSAHi study-2: Validation of multi-electrode array systems (MEA60/2100) for prediction of drug-induced proarrhythmia using human iPS cell-derived cardiomyocytes: Assessment of reference compounds and comparison with non-clinical studies and clinical information. Regul. Toxicol. Pharmacol. 2017, 88, 238–251. [Google Scholar]
- Sager, P.T.; Gintant, G.; Turner, J.R.; Pettit, S.; Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: A meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 2014, 167, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Kitaguchi, T.; Moriyama, Y.; Taniguchi, T.; Ojima, A.; Ando, H.; Uda, T.; Otabe, K.; Oguchi, M.; Shimizu, S.; Saito, H.; et al. CSAHi study: Evaluation of multi-electrode array in combination with human iPS cell-derived cardiomyocytes to predict drug-induced QT prolongation and arrhythmia — Effects of 7 reference compounds at 10 facilities. J. Pharmacol. Toxicol. Methods 2016, 78, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Guo, L.; Fiene, S.J.; Anson, B.D.; Thomson, J.A.; Kamp, T.J.; Kolaja, K.L.; Swanson, B.J.; January, C.T. High purity human-induced pluripotent stem cell-derived cardiomyocytes: Electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, N.H.L.; van Veen, T.A.B.; Lim, B.; Jonsson, M.K.B. Lessons from the heart: Mirroring electrophysiological characteristics during cardiac development to in vitro differentiation of stem cell derived cardiomyocytes. J. Mol. Cell. Cardiol. 2014, 67, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Blinova, K.; Dang, Q.; Millard, D.; Smith, G.; Pierson, J.; Guo, L.; Brock, M.; Lu, H.R.; Kraushaar, U.; Zeng, H.; et al. International Multisite Study of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Drug Proarrhythmic Potential Assessment. Cell Rep. 2018, 24, 3582–3592. [Google Scholar] [CrossRef]
- Schocken, D.; Stohlman, J.; Vicente, J.; Chan, D.; Patel, D.; Matta, M.K.; Patel, V.; Brock, M.; Millard, D.; Ross, J.; et al. Comparative analysis of media effects on human induced pluripotent stem cell-derived cardiomyocytes in proarrhythmia risk assessment. J. Pharmacol. Toxicol. Methods 2018, 90, 39–47. [Google Scholar] [CrossRef]
- Zeng, H.; Wang, J.; Clouse, H.; Lagrutta, A.; Sannajust, F. HiPSC-CMs from different sex and ethnic origin donors exhibit qualitatively different responses to several classes of pharmacological challenges. J. Pharmacol. Toxicol. Methods 2019, 99, 106598. [Google Scholar] [CrossRef]
- Burnett, S.D.; Blanchette, A.D.; Grimm, F.A.; House, J.S.; Reif, D.M.; Wright, F.A.; Chiu, W.A.; Rusyn, I. Population-based toxicity screening in human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Appl. Pharmacol. 2019, 381, 114711. [Google Scholar] [CrossRef]
- Egashira, T.; Yuasa, S.; Suzuki, T.; Aizawa, Y.; Yamakawa, H.; Matsuhashi, T.; Ohno, Y.; Tohyama, S.; Okata, S.; Seki, T.; et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc. Res. 2012, 95, 419–429. [Google Scholar] [CrossRef]
- Harris, K.; Aylott, M.; Cui, Y.; Louttit, J.B.; McMahon, N.C.; Sridhar, A. Comparison of Electrophysiological Data From Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes to Functional Preclinical Safety Assays. Toxicol. Sci. 2013, 134, 412–426. [Google Scholar] [CrossRef]
- Liang, P.; Lan, F.; Lee, A.S.; Gong, T.; Sanchez-Freire, V.; Wang, Y.; Diecke, S.; Sallam, K.; Knowles, J.W.; Wang, P.J.; et al. Drug Screening Using a Library of Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes Reveals Disease-Specific Patterns of Cardiotoxicity. Circulation 2013, 127, 1677–1691. [Google Scholar] [CrossRef] [PubMed]
- Maillet, A.; Tan, K.; Chai, X.; Sadananda, S.N.; Mehta, A.; Ooi, J.; Hayden, M.R.; Pouladi, M.A.; Ghosh, S.; Shim, W.; et al. Modeling Doxorubicin-Induced Cardiotoxicity in Human Pluripotent Stem Cell Derived-Cardiomyocytes. Sci. Rep. 2016, 6, 25333. [Google Scholar] [CrossRef]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.-G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.; de Korte, T.; Dragicevic, E.; Kraushaar, U.; Printemps, R.; Vlaming, M.L.H.; Braam, S.R.; Valentin, J.-P. Predicting cardiac safety using human induced pluripotent stem cell-derived cardiomyocytes combined with multi-electrode array (MEA) technology: A conference report. J. Pharmacol. Toxicol. Methods 2018, 91, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Acimovic, I.; Refaat, M.; Moreau, A.; Salykin, A.; Reiken, S.; Sleiman, Y.; Souidi, M.; Přibyl, J.; Kajava, A.; Richard, S.; et al. Post-Translational Modifications and Diastolic Calcium Leak Associated to the Novel RyR2-D3638A Mutation Lead to CPVT in Patient-Specific hiPSC-Derived Cardiomyocytes. JCM 2018, 7, 423. [Google Scholar] [CrossRef]
- Navarrete, E.G.; Liang, P.; Lan, F.; Sanchez-Freire, V.; Simmons, C.; Gong, T.; Sharma, A.; Burridge, P.W.; Patlolla, B.; Lee, A.S.; et al. Screening Drug-Induced Arrhythmia Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Low-Impedance Microelectrode Arrays. Circulation 2013, 128, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Svendsen, C.N. Human stem cells and drug screening: Opportunities and challenges. Nat. Rev. Drug Discov. 2010, 9, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Wallace, E.; Howard, L.; Liu, M.; O’Brien, T.; Ward, D.; Shen, S.; Prendiville, T. Long QT Syndrome: Genetics and Future Perspective. Pediatr Cardiol 2019, 40, 1–12. [Google Scholar] [CrossRef]
- Bellin, M.; Casini, S.; Davis, R.P.; D’Aniello, C.; Haas, J.; Ward-van Oostwaard, D.; Tertoolen, L.G.J.; Jung, C.B.; Elliott, D.A.; Welling, A.; et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome: Isogenic pairs of LQT2 pluripotent stem cells. Embo J. 2013, 32, 3161–3175. [Google Scholar] [CrossRef]
- Brandão, K.O.; Tabel, V.A.; Atsma, D.E.; Mummery, C.L.; Davis, R.P. Human pluripotent stem cell models of cardiac disease: From mechanisms to therapies. Dis. Model. Mech. 2017, 10, 1039–1059. [Google Scholar] [CrossRef]
- Zhang, M.; D’Aniello, C.; Verkerk, A.O.; Wrobel, E.; Frank, S.; Ward-van Oostwaard, D.; Piccini, I.; Freund, C.; Rao, J.; Seebohm, G.; et al. Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: Disease mechanisms and pharmacological rescue. Proc. Natl. Acad. Sci. USA 2014, 111, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-Specific Induced Pluripotent Stem-Cell Models for Long-QT Syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; Gnecchi, M.; Schwartz, P.J. Derived from Human-induced Pluripotent Stem Cells. Arrhythmia Electrophysiol. Rev. 2019, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Ruan, Y.; Priori, S.G. Catecholaminergic Polymorphic Ventricular Tachycardia. Prog. Cardiovasc. Dis. 2008, 51, 23–30. [Google Scholar] [CrossRef]
- Sasaki, K.; Makiyama, T.; Yoshida, Y.; Wuriyanghai, Y.; Kamakura, T.; Nishiuchi, S.; Hayano, M.; Harita, T.; Yamamoto, Y.; Kohjitani, H.; et al. Patient-Specific Human Induced Pluripotent Stem Cell Model Assessed with Electrical Pacing Validates S107 as a Potential Therapeutic Agent for Catecholaminergic Polymorphic Ventricular Tachycardia. PLoS ONE 2016, 11, e0164795. [Google Scholar] [CrossRef]
- Carvajal-Vergara, X.; Sevilla, A.; D’Souza, S.L.; Ang, Y.-S.; Schaniel, C.; Lee, D.-F.; Yang, L.; Kaplan, A.D.; Adler, E.D.; Rozov, R.; et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010, 465, 808–812. [Google Scholar] [CrossRef]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal Calcium Handling Properties Underlie Familial Hypertrophic Cardiomyopathy Pathology in Patient-Specific Induced Pluripotent Stem Cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef]
- Yang, K.-C.; Breitbart, A.; De Lange, W.J.; Hofsteen, P.; Futakuchi-Tsuchida, A.; Xu, J.; Schopf, C.; Razumova, M.V.; Jiao, A.; Boucek, R.; et al. Novel Adult-Onset Systolic Cardiomyopathy Due to MYH7 E848G Mutation in Patient-Derived Induced Pluripotent Stem Cells. JACC Basic Transl. Sci. 2018, 3, 728–740. [Google Scholar] [CrossRef]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-Specific Induced Pluripotent Stem Cells as a Model for Familial Dilated Cardiomyopathy. Sci. Transl. Med. 2012, 4, 47–130. [Google Scholar] [CrossRef]
- Siu, C.-W.; Lee, Y.-K.; Ho, J.C.-Y.; Lai, W.-H.; Chan, Y.-C.; Ng, K.-M.; Wong, L.-Y.; Au, K.-W.; Lau, Y.-M.; Zhang, J.; et al. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging 2012, 4, 803–822. [Google Scholar] [CrossRef]
- Tse, H.-F.; Ho, J.C.Y.; Choi, S.-W.; Lee, Y.-K.; Butler, A.W.; Ng, K.-M.; Siu, C.-W.; Simpson, M.A.; Lai, W.-H.; Chan, Y.-C.; et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum. Mol. Genet. 2013, 22, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Streckfuss-Bömeke, K.; Tiburcy, M.; Fomin, A.; Luo, X.; Li, W.; Fischer, C.; Özcelik, C.; Perrot, A.; Sossalla, S.; Haas, J.; et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2017, 113, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Wyles, S.P.; Li, X.; Hrstka, S.C.; Reyes, S.; Oommen, S.; Beraldi, R.; Edwards, J.; Terzic, A.; Olson, T.M.; Nelson, T.J. Modeling structural and functional deficiencies of RBM20 familial dilated cardiomyopathy using human induced pluripotent stem cells. Hum. Mol. Genet. 2016, 25, 254–265. [Google Scholar] [CrossRef]
- Giacomelli, E.; Mummery, C.L.; Bellin, M. Human heart disease: Lessons from human pluripotent stem cell-derived cardiomyocytes. Cell. Mol. Life Sci. 2017, 74, 3711–3739. [Google Scholar] [CrossRef]
- Lin, B.; Li, Y.; Han, L.; Kaplan, A.D.; Ao, Y.; Kalra, S.; Bett, G.C.L.; Rasmusson, R.L.; Denning, C.; Yang, L. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis. Models Mech. 2015, 8, 457–466. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Carrier, L. Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes—a systematic review. Pflug. Arch. Eur. J. Physiol. 2019, 471, 755–768. [Google Scholar] [CrossRef]
- de la Roche, J.; Angsutararux, P.; Kempf, H.; Janan, M.; Bolesani, E.; Thiemann, S.; Wojciechowski, D.; Coffee, M.; Franke, A.; Schwanke, K.; et al. Comparing human iPSC-cardiomyocytes versus HEK293T cells unveils disease-causing effects of Brugada mutation A735V of NaV1.5 sodium channels. Sci. Rep. 2019, 9, 11173. [Google Scholar] [CrossRef]
- Bozzi, A.; Sayed, N.; Matsa, E.; Sass, G.; Neofytou, E.; Clemons, K.V.; Correa-Oliveira, R.; Stevens, D.A.; Wu, J.C. Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes as a Model to Study Trypanosoma cruzi Infection. Stem Cell Rep. 2019, 12, 1232–1241. [Google Scholar] [CrossRef]
- Lee, J.-J.; Cheng, S.-J.; Huang, C.-Y.; Chen, C.-Y.; Feng, L.; Hwang, D.-Y.; Kamp, T.J.; Chen, H.-C.; Hsieh, P.C.H. Primary cardiac manifestation of autosomal dominant polycystic kidney disease revealed by patient induced pluripotent stem cell-derived cardiomyocytes. EBioMedicine 2019, 40, 675–684. [Google Scholar] [CrossRef]
- Birket, M.J.; Raibaud, S.; Lettieri, M.; Adamson, A.D.; Letang, V.; Cervello, P.; Redon, N.; Ret, G.; Viale, S.; Wang, B.; et al. A Human Stem Cell Model of Fabry Disease Implicates LIMP-2 Accumulation in Cardiomyocyte Pathology. Stem Cell Rep. 2019, 13, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Caluori, G.; Pribyl, J.; Pesl, M.; Jelinkova, S.; Rotrekl, V.; Skladal, P.; Raiteri, R. Non-invasive electromechanical cell-based biosensors for improved investigation of 3D cardiac models. Biosens. Bioelectron. 2019, 124, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Huebsch, N.; Koo, S.; Mandegar, M.A.; Siemons, B.; Boggess, S.; Conklin, B.R.; Grigoropoulos, C.P.; Healy, K.E. Contractile deficits in engineered cardiac microtissues as a result of MYBPC3 deficiency and mechanical overload. Nat. Biomed. Eng. 2018, 2, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Zhang, D.; Qi, Y.; Li, Y.; Lee, K.Y.; Bezzerides, V.J.; Yang, P.; Xia, S.; Kim, S.L.; Liu, X.; et al. Insights Into the Pathogenesis of Catecholaminergic Polymorphic Ventricular Tachycardia From Engineered Human Heart Tissue. Circulation 2019, 140, 390–404. [Google Scholar] [CrossRef]
- Paci, M.; Casini, S.; Bellin, M.; Hyttinen, J.; Severi, S. Large-Scale Simulation of the Phenotypical Variability Induced by Loss-of-Function Long QT Mutations in Human Induced Pluripotent Stem Cell Cardiomyocytes. Int. J. Mol. Sci. 2018, 19, 3583. [Google Scholar] [CrossRef]
- Satsuka, A.; Kanda, Y. Cardiotoxicity assessment of drugs using human iPS cell-derived cardiomyocytes: From proarrhythmia risk to cardiooncology. Curr. Pharm. Biotechnol. 2019. [Google Scholar] [CrossRef]
- Zhang, B.; Korolj, A.; Lai, B.F.L.; Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 2018, 3, 257–278. [Google Scholar] [CrossRef]
- Maoz, B.M.; Herland, A.; Henry, O.Y.F.; Leineweber, W.D.; Yadid, M.; Doyle, J.; Mannix, R.; Kujala, V.J.; FitzGerald, E.A.; Parker, K.K.; et al. Organs-on-Chips with combined multi-electrode array and transepithelial electrical resistance measurement capabilities. Lab. Chip 2017, 17, 2294–2302. [Google Scholar] [CrossRef]
- Di Sanzo, M.; Cipolloni, L.; Borro, M.; La Russa, R.; Santurro, A.; Scopetti, M.; Simmaco, M.; Frati, P. Clinical Applications of Personalized Medicine: A New Paradigm and Challenge. Curr. Pharm. Biotechnol. 2017, 18, 194–203. [Google Scholar] [CrossRef]
- Chen, I.Y.; Matsa, E.; Wu, J.C. Induced pluripotent stem cells: At the heart of cardiovascular precision medicine. Nat. Rev. Cardiol. 2016, 13, 333–349. [Google Scholar] [CrossRef]
- Hamazaki, T.; El Rouby, N.; Fredette, N.C.; Santostefano, K.E.; Terada, N. Concise Review: Induced Pluripotent Stem Cell Research in the Era of Precision Medicine. Stem Cells 2017, 35, 545–550. [Google Scholar] [CrossRef] [PubMed]



| FP Morphology | Physiological Parameter |
|---|---|
| Spatiotemporal Assessment | Propagation velocity, direction origin of AP spread |
| FP Duration | QT interval of AP |
| FPs Over Time | Beating frequency |
| Spike Amplitude | Na+ current |
| Spike Plateau | Ca2+/K+ current |
| Beat-to-Beat Interval | AP duration |
| High TdP Risk | Intermediate TdP Risk | No or Very Low TdP Risk |
|---|---|---|
| Astemizole | ||
| Chlorpromazine | Diltiazem | |
| Azimilide | Cisapride | Loratadine |
| Bepridil | Clarithromycin | Metoprolol |
| Dofetilide | Clozapine | Mexiletine |
| Ibutilide | Domperidone | Nifedipine |
| Quinidine | Droperidol | Nitrendipine |
| Vandetanib | Terfenadine | Ranolazine |
| Disopyramide | Pimozide | Tamoxifen |
| D,l Sotalol | Risperidone | Verapamil |
| Ondansetron |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kussauer, S.; David, R.; Lemcke, H. hiPSCs Derived Cardiac Cells for Drug and Toxicity Screening and Disease Modeling: What Micro- Electrode-Array Analyses Can Tell Us. Cells 2019, 8, 1331. https://doi.org/10.3390/cells8111331
Kussauer S, David R, Lemcke H. hiPSCs Derived Cardiac Cells for Drug and Toxicity Screening and Disease Modeling: What Micro- Electrode-Array Analyses Can Tell Us. Cells. 2019; 8(11):1331. https://doi.org/10.3390/cells8111331
Chicago/Turabian StyleKussauer, Sophie, Robert David, and Heiko Lemcke. 2019. "hiPSCs Derived Cardiac Cells for Drug and Toxicity Screening and Disease Modeling: What Micro- Electrode-Array Analyses Can Tell Us" Cells 8, no. 11: 1331. https://doi.org/10.3390/cells8111331
APA StyleKussauer, S., David, R., & Lemcke, H. (2019). hiPSCs Derived Cardiac Cells for Drug and Toxicity Screening and Disease Modeling: What Micro- Electrode-Array Analyses Can Tell Us. Cells, 8(11), 1331. https://doi.org/10.3390/cells8111331