Autophagic- and Lysosomal-Related Biomarkers for Parkinson’s Disease: Lights and Shadows


Abstract
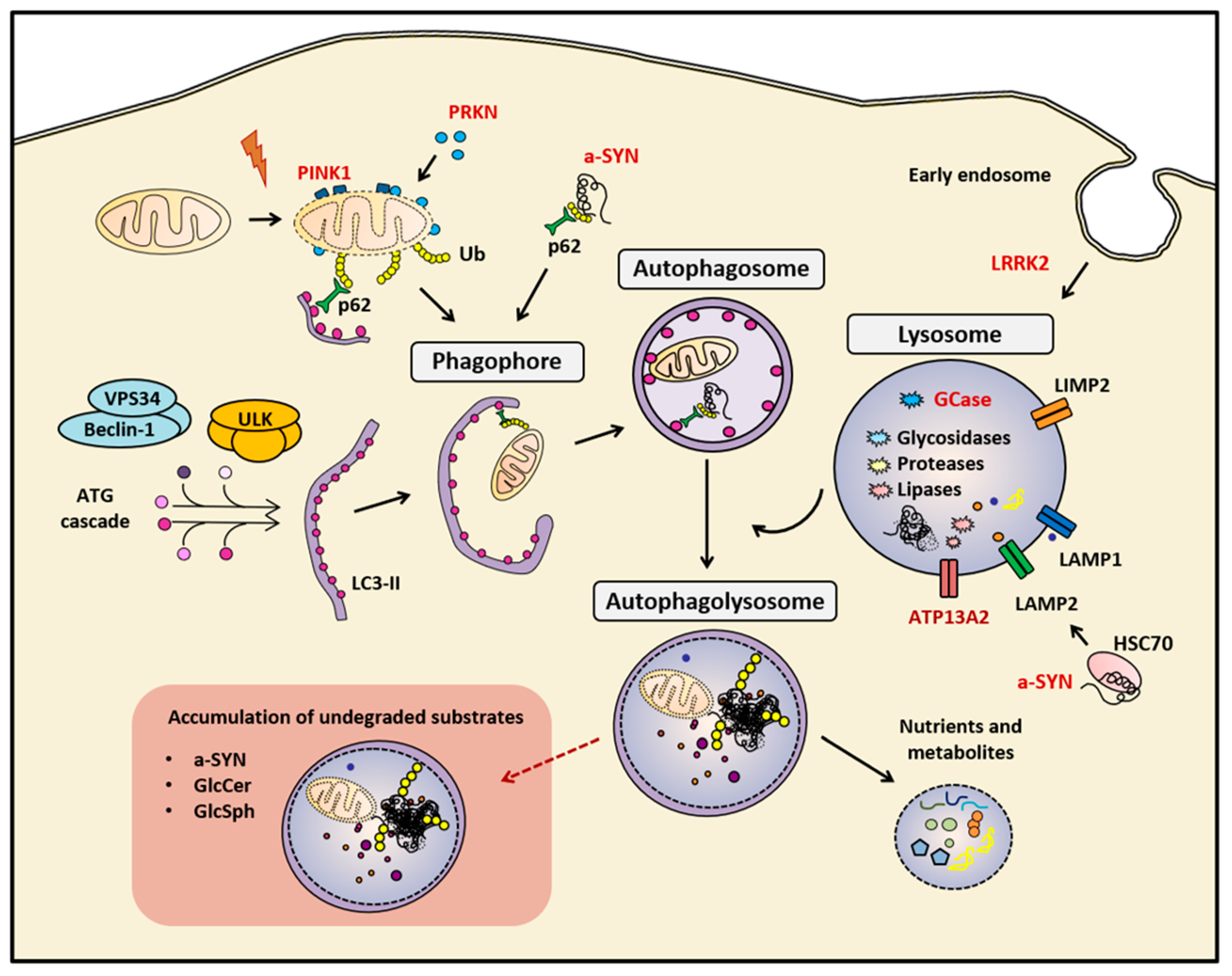
1. Introduction
2. Expression of Autophagic-Lysosomal Proteins
2.1. Autophagosome Formation–Related Proteins
2.2. Autophagic Cargo Delivery–Related Proteins
2.3. Lysosomal Proteins
2.3.1. Structural Membrane Proteins
2.3.2. Enzymes
3. Direct Markers of Autophagic-Lysosomal Function
3.1. Enzymatic Activities
3.1.1. Glucocerebrosidase
3.1.2. Galactosidases
3.1.3. Other Lysosomal Enzymes
3.2. Non-Enzymatic Assays
4. Indirect Markers of Autophagic-Lysosomal Function
4.1. A-SYN Levels
4.2. Mitophagy Markers
4.3. Lipid Levels Influenced by Autophagy-Related Proteins
5. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Kowal, S.L.; Dall, T.M.; Chakrabarti, R.; Storm, M.V.; Jain, A. The current and projected economic burden of Parkinson’s disease in the United States. Mov. Disord. 2013, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Mao, Z.-H. Progression of motor symptoms in Parkinson’s disease. Neurosci. Bull. 2012, 28, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Schapira, A.H. V Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef]
- Postuma, R.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chiasserini, D.; Beccari, T.; Parnetti, L. Glucocerebrosidase in Parkinson’s disease: Insights into pathogenesis and prospects for treatment. Mov. Disord. 2016, 31, 830–835. [Google Scholar] [CrossRef]
- Eusebi, P.; Giannandrea, D.; Biscetti, L.; Abraha, I.; Chiasserini, D.; Orso, M.; Calabresi, P.; Parnetti, L. Diagnostic utility of cerebrospinal fluid α-synuclein in Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2017, 32, 1389–1400. [Google Scholar] [CrossRef]
- Johar, I.; Mollenhauer, B.; Aarsland, D. Cerebrospinal Fluid Biomarkers of Cognitive Decline in Parkinson’s Disease. In International Review of Neurobiology; Academic Press Inc.: New York, NY, USA, 2017; Volume 132, pp. 275–294. [Google Scholar]
- Magdalinou, N.; Lees, A.J.; Zetterberg, H. Cerebrospinal fluid biomarkers in parkinsonian conditions: An update and future directions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1065–1075. [Google Scholar] [CrossRef]
- Parnetti, L.; Gaetani, L.; Eusebi, P.; Paciotti, S.; Hansson, O.; El-Agnaf, O.; Mollenhauer, B.; Blennow, K.; Calabresi, P. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 2019, 18, 573–586. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P. Parkinson disease: from pathology to molecular disease mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.J.H.M.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Park. Dis. 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.; Morris, H.; Schapira, A. Chaperone-mediated autophagy as a therapeutic target for Parkinson disease. Expert Opin. Ther. Targets 2018, 22, 823–832. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Vila, M. Alpha-synuclein and protein degradation pathways in Parkinson’s disease: A pathological feed-back loop. Exp. Neurol. 2013, 247, 308–313. [Google Scholar] [CrossRef]
- Moors, T.; Paciotti, S.; Chiasserini, D.; Calabresi, P.; Parnetti, L.; Beccari, T.; van de Berg, W.D.J. Lysosomal Dysfunction and α-Synuclein Aggregation in Parkinson’s Disease: Diagnostic Links. Mov. Disord. 2016, 31, 791–801. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Arotcarena, M.-L.; Teil, M.; Dehay, B. Autophagy in Synucleinopathy: The Overwhelmed and Defective Machinery. Cells 2019, 8, 565. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Platt, F.M.; Futerman, A.H. Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem. 2010, 285, 20423–20427. [Google Scholar] [CrossRef] [PubMed]
- Van Der Brug, M.P.; Singleton, A.; Gasser, T.; Lewis, P.A. Parkinson’s disease: From human genetics to clinical trials. Sci. Transl. Med. 2015, 7, 305ps20. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; Nalls, M.A.; Plagnol, V.; et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Mullin, S.; Schapira, A. The genetics of Parkinson’s disease. Br. Med. Bull. 2015, 114, 39–52. [Google Scholar] [CrossRef]
- Neumann, J.; Bras, J.; Deas, E.; O’sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef]
- Lunati, A.; Lesage, S.; Brice, A. The genetic landscape of Parkinson’s disease. Rev. Neurol. 2018, 174, 628–643. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sekine, T.; Funayama, M.; Li, L.; Yoshino, H.; Nishioka, K.; Tomiyama, H.; Hattori, N. Clinicogenetic study of GBA mutations in patients with familial Parkinson’s disease. Neurobiol. Aging 2014, 35, 935.e3–935.e8. [Google Scholar] [CrossRef]
- Sidransky, E. Gaucher disease and parkinsonism. Mol. Genet. Metab. 2005, 84, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Rana, H.Q.; Balwani, M.; Bier, L.; Alcalay, R.N. Age-specific Parkinson disease risk in GBA mutation carriers: Information for genetic counseling. Genet. Med. 2013, 15, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaïti, B.; Bonaïti-Pellié, C.; Brice, A.; et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012, 78, 417–420. [Google Scholar] [CrossRef]
- Rosenbloom, B.; Balwani, M.; Bronstein, J.M.; Kolodny, E.; Sathe, S.; Gwosdow, A.R.; Taylor, J.S.; Cole, J.A.; Zimran, A.; Weinreb, N.J. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher Registry. Blood Cells, Mol. Dis. 2011, 46, 95–102. [Google Scholar] [CrossRef]
- Bultron, G.; Kacena, K.; Pearson, D.; Boxer, M.; Yang, R.; Sathe, S.; Pastores, G.; Mistry, P.K. The risk of Parkinson’s disease in type 1 Gaucher disease. J. Inherit. Metab. Dis. 2010, 33, 167–173. [Google Scholar] [CrossRef]
- Clark, L.N.; Ross, B.M.; Wang, Y.; Mejia-Santana, H.; Harris, J.; Louis, E.D.; Cote, L.J.; Andrews, H.; Fahn, S.; Waters, C.; et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 2007, 69, 1270–1277. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Winder-Rhodes, S.E.; Evans, J.R.; Ban, M.; Mason, S.L.; Williams-Gray, C.H.; Foltynie, T.; Duran, R.; Mencacci, N.E.; Sawcer, S.J.; Barker, R.A. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013, 136, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.K.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. 2015, 30, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Adler, C.H.; Beach, T.G.; Shill, H.A.; Caviness, J.N.; Driver-Dunckley, E.; Sabbagh, M.N.; Patel, A.; Sue, L.I.; Serrano, G.; Jacobson, S.A.; et al. GBA mutations in Parkinson disease: earlier death but similar neuropathological features. Eur. J. Neurol. 2017, 24, 1363–1368. [Google Scholar] [CrossRef]
- Mullin, S.; Beavan, M.; Bestwick, J.; McNeill, A.; Proukakis, C.; Cox, T.; Hughes, D.; Mehta, A.; Zetterberg, H.; Schapira, A.H.V. Evolution and clustering of prodromal parkinsonian features in GBA1 carriers. Mov. Disord. 2019, 34, 1365–1373. [Google Scholar] [CrossRef]
- Thaler, A.; Bregman, N.; Gurevich, T.; Shiner, T.; Dror, Y.; Zmira, O.; Gan-Or, Z.; Bar-Shira, A.; Gana-Weisz, M.; Orr-Urtreger, A.; et al. Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat. Disord. 2018, 55, 45–49. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.-H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P.; et al. Cerebrospinal fluid β-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. 2017, 32, 1423–1431. [Google Scholar] [CrossRef]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Reports 2016, 6, 342–356. [Google Scholar] [CrossRef]
- Sardi, S.P.; Cedarbaum, J.M.; Brundin, P. Targeted Therapies for Parkinson’s Disease: From Genetics to the Clinic. Mov. Disord. 2018, 33, 684–696. [Google Scholar] [CrossRef]
- Manzoni, C. The LRRK2-macroautophagy axis and its relevance to Parkinson’s disease. Biochem. Soc. Trans. 2017, 45, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Poehler, A.-M.; Xiang, W.; Spitzer, P.; May, V.E.L.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, E.; Seaman, M.N.J.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1-and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- McCoy, M.K.; Cookson, M.R. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy 2011, 7, 531–532. [Google Scholar] [CrossRef]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.-H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef]
- Zhou, Q.; Yen, A.; Rymarczyk, G.; Asai, H.; Trengrove, C.; Aziz, N.; Kirber, M.T.; Mostoslavsky, G.; Ikezu, T.; Wolozin, B.; et al. Impairment of PARK14-dependent Ca 2+ signalling is a novel determinant of Parkinson’s disease. Nat. Commun. 2016, 7, 10332. [Google Scholar] [CrossRef]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef]
- Edvardson, S.; Cinnamon, Y.; Ta-Shma, A.; Shaag, A.; Yim, Y.I.; Zenvirt, S.; Jalas, C.; Lesage, S.; Brice, A.; Taraboulos, A.; et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating Co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 2012, 7, e36458. [Google Scholar] [CrossRef] [PubMed]
- Varga, R.-E.; Khundadze, M.; Damme, M.; Nietzsche, S.; Hoffmann, B.; Stauber, T.; Koch, N.; Hennings, J.C.; Franzka, P.; Huebner, A.K.; et al. In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet. 2015, 11, e1005454. [Google Scholar] [CrossRef] [PubMed]
- George, A.A.; Hayden, S.; Stanton, G.R.; Brockerhoff, S.E. Arf6 and the 5’phosphatase of synaptojanin 1 regulate autophagy in cone photoreceptors. BioEssays 2016, 38, S119–S135. [Google Scholar] [CrossRef] [PubMed]
- Fasano, D.; Parisi, S.; Pierantoni, G.M.; De Rosa, A.; Picillo, M.; Amodio, G.; Pellecchia, M.T.; Barone, P.; Moltedo, O.; Bonifati, V.; et al. Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by SYNJ1 mutations. Cell Death Dis. 2018, 9, 385. [Google Scholar] [CrossRef]
- Muñoz-Braceras, S.; Calvo, R.; Escalante, R. TipC and the chorea-acanthocytosis protein VPS13A regulate autophagy in Dictyostelium and human HeLa cells. Autophagy 2015, 11, 918–927. [Google Scholar] [CrossRef]
- Sellier, C.; Campanari, M.-L.; Julie Corbier, C.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar]
- Youn, J.; Lee, S.-B.; Lee, H.S.; Yang, H.O.; Park, J.; Kim, J.S.; Oh, E.; Park, S.; Jang, W. Cerebrospinal Fluid Levels of Autophagy-related Proteins Represent Potentially Novel Biomarkers of Early-Stage Parkinson’s Disease. Sci. Rep. 2018, 8, 16866. [Google Scholar] [CrossRef]
- Boman, A.; Svensson, S.; Boxer, A.; Rojas, J.C.; Seeley, W.W.; Karydas, A.; Miller, B.; Kågedal, K.; Svenningsson, P. Distinct Lysosomal Network Protein Profiles in Parkinsonian Syndrome Cerebrospinal Fluid. J. Parkinsons. Dis. 2016, 6, 307–315. [Google Scholar] [CrossRef]
- Miki, Y.; Shimoyama, S.; Kon, T.; Ueno, T.; Hayakari, R.; Tanji, K.; Matsumiya, T.; Tsushima, E.; Mori, F.; Wakabayashi, K.; et al. Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. Neurobiol. Aging 2018, 63, 33–43. [Google Scholar] [CrossRef]
- Prigione, A.; Piazza, F.; Brighina, L.; Begni, B.; Galbussera, A.; DiFrancesco, J.C.; Andreoni, S.; Piolti, R.; Ferrarese, C. Alpha-synuclein nitration and autophagy response are induced in peripheral blood cells from patients with Parkinson disease. Neurosci. Lett. 2010, 477, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Wang, X.; Feng, X.; Zhang, A.; Li, J.; Gu, K.; Huang, J.; Pang, S.; Dong, H.; Gao, H.; et al. Altered expression of autophagic genes in the peripheral leukocytes of patients with sporadic Parkinson’s disease. Brain Res. 2011, 1394, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Teves, J.M.Y.; Bhargava, V.; Kirwan, K.R.; Corenblum, M.J.; Justiniano, R.; Wondrak, G.T.; Anandhan, A.; Flores, A.J.; Schipper, D.A.; Khalpey, Z.; et al. Parkinson’s Disease Skin Fibroblasts Display Signature Alterations in Growth, Redox Homeostasis, Mitochondrial Function, and Autophagy. Front. Neurosci. 2018, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.M.; Drouin-Ouellet, J.; Kuan, W.-L.; Cox, T.; Barker, R.A. Dermal fibroblasts from patients with Parkinson’s disease have normal GCase activity and autophagy compared to patients with PD and GBA mutations. F1000Research 2017, 6, 1751. [Google Scholar] [CrossRef]
- Ambrosi, G.; Ghezzi, C.; Sepe, S.; Milanese, C.; Payan-Gomez, C.; Bombardieri, C.R.; Armentero, M.-T.; Zangaglia, R.; Pacchetti, C.; Mastroberardino, P.G.; et al. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic Parkinson’s disease. Biochim. Biophys. Acta - Mol. Basis Dis. 2014, 1842, 1385–1394. [Google Scholar] [CrossRef]
- McNeill, A.; Magalhaes, J.; Shen, C.; Chau, K.-Y.; Hughes, D.; Mehta, A.; Foltynie, T.; Cooper, J.M.; Abramov, A.Y.; Gegg, M.; et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 2014, 137, 1481–1495. [Google Scholar] [CrossRef]
- Guerra, F.; Girolimetti, G.; Beli, R.; Mitruccio, M.; Pacelli, C.; Ferretta, A.; Gasparre, G.; Cocco, T.; Bucci, C. Synergistic Effect of Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Cells 2019, 8, 452. [Google Scholar] [CrossRef]
- González-Casacuberta, I.; Juárez-Flores, D.-L.; Ezquerra, M.; Fucho, R.; Catalán-García, M.; Guitart-Mampel, M.; Tobías, E.; García-Ruiz, C.; Fernández-Checa, J.C.; Tolosa, E.; et al. Mitochondrial and autophagic alterations in skin fibroblasts from Parkinson disease patients with Parkin mutations. Aging 2019, 11, 3750–3767. [Google Scholar] [CrossRef]
- Manzoni, C.; Mamais, A.; Dihanich, S.; McGoldrick, P.; Devine, M.J.; Zerle, J.; Kara, E.; Taanman, J.-W.; Healy, D.G.; Marti-Masso, J.-F.; et al. Pathogenic Parkinson’s disease mutations across the functional domains of LRRK2 alter the autophagic/lysosomal response to starvation. Biochem. Biophys. Res. Commun. 2013, 441, 862–866. [Google Scholar] [CrossRef]
- Grünewald, A.; Arns, B.; Meier, B.; Brockmann, K.; Tadic, V.; Klein, C. Does uncoupling protein 2 expression qualify as marker of disease status in LRRK2-associated Parkinson’s disease? Antioxid. Redox Signal. 2014, 20, 1955–1960. [Google Scholar] [CrossRef]
- Xiong, Y.; Dawson, T.M.; Dawson, V.L. Models of LRRK2-Associated Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 163–191. [Google Scholar] [PubMed]
- Papagiannakis, N.; Xilouri, M.; Koros, C.; Stamelou, M.; Antonelou, R.; Maniati, M.; Papadimitriou, D.; Moraitou, M.; Michelakakis, H.; Stefanis, L. Lysosomal alterations in peripheral blood mononuclear cells of Parkinson’s disease patients. Mov. Disord. 2015, 30, 1830–1834. [Google Scholar] [CrossRef] [PubMed]
- Sala, G.; Stefanoni, G.; Arosio, A.; Riva, C.; Melchionda, L.; Saracchi, E.; Fermi, S.; Brighina, L.; Ferrarese, C. Reduced expression of the chaperone-mediated autophagy carrier hsc70 protein in lymphomonocytes of patients with Parkinson’s disease. Brain Res. 2014, 1546, 46–52. [Google Scholar] [CrossRef]
- Eskelinen, E.-L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Aspects Med. 2006, 27, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Klaver, A.C.; Coffey, M.P.; Aasly, J.O.; Loeffler, D.A. CSF lamp2 concentrations are decreased in female Parkinson’s disease patients with LRRK2 mutations. Brain Res. 2018, 1683, 12–16. [Google Scholar] [CrossRef]
- Alieva, A.K.; Filatova, E.V.; Karabanov, A.V.; Illarioshkin, S.N.; Slominsky, P.A.; Shadrina, M.I. Potential Biomarkers of the Earliest Clinical Stages of Parkinson’s Disease. Parkinsons. Dis. 2015, 2015, 294396. [Google Scholar] [CrossRef]
- Ichinose, Y.; Ishiura, H.; Tanaka, M.; Yoshimura, J.; Doi, K.; Umeda, T.; Yamauchi, H.; Tsuchiya, M.; Koh, K.; Yamashiro, N.; et al. Neuroimaging, genetic, and enzymatic study in a Japanese family with a GBA gross deletion. Parkinsonism Relat. Disord. 2019, 61, 57–63. [Google Scholar] [CrossRef]
- Atashrazm, F.; Hammond, D.; Perera, G.; Dobson-Stone, C.; Mueller, N.; Pickford, R.; Kim, W.S.; Kwok, J.B.; Lewis, S.J.G.; Halliday, G.M.; et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci. Rep. 2018, 8, 15446. [Google Scholar] [CrossRef]
- Ishii, S.; Chang, H.-H.; Kawasaki, K.; Yasuda, K.; Wu, H.-L.; Garman, S.C.; Fan, J.-Q. Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem. J. 2007, 406, 285–295. [Google Scholar] [CrossRef]
- Pchelina, S.; Emelyanov, A.; Baydakova, G.; Andoskin, P.; Senkevich, K.; Nikolaev, M.; Miliukhina, I.; Yakimovskii, A.; Timofeeva, A.; Fedotova, E.; et al. Oligomeric α-synuclein and glucocerebrosidase activity levels in GBA-associated Parkinson’s disease. Neurosci. Lett. 2017, 636, 70–76. [Google Scholar] [CrossRef]
- Ortega, R.A.; Torres, P.A.; Swan, M.; Nichols, W.; Boschung, S.; Raymond, D.; Barrett, M.J.; Johannes, B.A.; Severt, L.; Shanker, V.; et al. Glucocerebrosidase enzyme activity in GBA mutation Parkinson’s disease. J. Clin. Neurosci. 2016, 28, 185–186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Pierguidi, L.; Persichetti, E.; Parnetti, L.; Sbaragli, M.; Tassi, C.; Orlacchio, A.; Calabresi, P.; Beccari, T.; Rossi, A. Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson’s disease. Mov. Disord. 2007, 22, 1481–1484. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, K.D.; Persichetti, E.; Chiasserini, D.; Eusebi, P.; Beccari, T.; Calabresi, P.; Berendse, H.W.; Parnetti, L.; van de Berg, W.D.J. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 2013, 28, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Cerri, S.; Ghezzi, C.; Sampieri, M.; Siani, F.; Avenali, M.; Dornini, G.; Zangaglia, R.; Minafra, B.; Blandini, F. The Exosomal/Total α-Synuclein Ratio in Plasma Is Associated With Glucocerebrosidase Activity and Correlates With Measures of Disease Severity in PD Patients. Front. Cell. Neurosci. 2018, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Lecourt, S.; Vanneaux, V.; Rapatel, C.; Boisgard, S.; Caillaud, C.; Boiret-Dupré, N.; Chomienne, C.; Marolleau, J.-P.; Larghero, J.; et al. Glucocerebrosidase deficiency dramatically impairs human bone marrow haematopoiesis in an in vitro model of Gaucher disease. Br. J. Haematol. 2010, 150, 93–101. [Google Scholar] [CrossRef]
- Kim, H.-J.; Jeon, B.; Song, J.; Lee, W.-W.; Park, H.; Shin, C.-W. Leukocyte glucocerebrosidase and β-hexosaminidase activity in sporadic and genetic Parkinson disease. Parkinsonism Relat. Disord. 2016, 23, 99–101. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Wolf, P.; Levy, O.A.; Kang, U.J.; Waters, C.; Fahn, S.; Ford, B.; Kuo, S.H.; Vanegas, N.; Shah, H.; et al. Alpha galactosidase A activity in Parkinson’s disease. Neurobiol. Dis. 2018, 112, 85–90. [Google Scholar] [CrossRef]
- Niimi, Y.; Ito, S.; Mizutani, Y.; Murate, K.; Shima, S.; Ueda, A.; Satake, W.; Hattori, N.; Toda, T.; Mutoh, T. Altered regulation of serum lysosomal acid hydrolase activities in Parkinson’s disease: A potential peripheral biomarker? Parkinsonism Relat. Disord. 2019, 61, 132–137. [Google Scholar] [CrossRef]
- Wu, G.; Yan, B.; Wang, X.; Feng, X.; Zhang, A.; Xu, X.; Dong, H. Decreased activities of lysosomal acid alpha-D-galactosidase A in the leukocytes of sporadic Parkinson’s disease. J. Neurol. Sci. 2008, 271, 168–173. [Google Scholar] [CrossRef]
- Papagiannakis, N.; Xilouri, M.; Koros, C.; Simitsi, A.-M.; Stamelou, M.; Maniati, M.; Stefanis, L. Autophagy dysfunction in peripheral blood mononuclear cells of Parkinson’s disease patients. Neurosci. Lett. 2019, 704, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Bolognin, S.; Antony, P.M.A.; Nickels, S.L.; Poovathingal, S.K.; Salamanca, L.; Magni, S.; Perfeito, R.; Hoel, F.; Qing, X.; et al. Neural Stem Cells of Parkinson’s Disease Patients Exhibit Aberrant Mitochondrial Morphology and Functionality. Stem Cell Reports 2019, 12, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The Role of Lipids in Parkinson’s Disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Burré, J. The Synaptic Function of α-Synuclein. J. Parkinsons. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef]
- Pchelina, S.N.; Nuzhnyi, E.P.; Emelyanov, A.K.; Boukina, T.M.; Usenko, T.S.; Nikolaev, M.A.; Salogub, G.N.; Yakimovskii, A.F.; Zakharova, E.Y. Increased plasma oligomeric alpha-synuclein in patients with lysosomal storage diseases. Neurosci. Lett. 2014, 583, 188–193. [Google Scholar] [CrossRef]
- Papagiannakis, N.; Koros, C.; Stamelou, M.; Simitsi, A.-M.; Maniati, M.; Antonelou, R.; Papadimitriou, D.; Dermentzaki, G.; Moraitou, M.; Michelakakis, H.; et al. Alpha-synuclein dimerization in erythrocytes of patients with genetic and non-genetic forms of Parkinson’s Disease. Neurosci. Lett. 2018, 672, 145–149. [Google Scholar] [CrossRef]
- Moraitou, M.; Dermentzaki, G.; Dimitriou, E.; Monopolis, I.; Dekker, N.; Aerts, H.; Stefanis, L.; Michelakakis, H. α-Synuclein dimerization in erythrocytes of Gaucher disease patients: correlation with lipid abnormalities and oxidative stress. Neurosci. Lett. 2016, 613, 1–5. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. The Parkinson Disease Mitochondrial Hypothesis: Where Are We at? Neuroscientist 2016, 22, 266–277. [Google Scholar] [CrossRef]
- Ding, W.-X.; Yin, X.-M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Smith, G.A.; Jansson, J.; Rocha, E.M.; Osborn, T.; Hallett, P.J.; Isacson, O. Fibroblast Biomarkers of Sporadic Parkinson’s Disease and LRRK2 Kinase Inhibition. Mol. Neurobiol. 2016, 53, 5161–5177. [Google Scholar] [CrossRef] [PubMed]
- Bonello, F.; Hassoun, S.-M.; Mouton-Liger, F.; Shin, Y.S.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.M.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1645–1660. [Google Scholar] [CrossRef] [PubMed]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced Basal Autophagy and Impaired Mitochondrial Dynamics Due to Loss of Parkinson’s Disease-Associated Protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed]
- van Echten-Deckert, G.; Herget, T. Sphingolipid metabolism in neural cells. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1978–1994. [Google Scholar] [CrossRef]
- Vitner, E.B.; Farfel-Becker, T.; Eilam, R.; Biton, I.; Futerman, A.H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain 2012, 135, 1724–1735. [Google Scholar] [CrossRef]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; Bandaru, V.V.R.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.-K.; Gräber-Sultan, S.; Schleicher, E.; et al. Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: a pilot study. PLoS ONE 2013, 8, e73094. [Google Scholar] [CrossRef]
- Pchelina, S.; Baydakova, G.; Nikolaev, M.; Senkevich, K.; Emelyanov, A.; Kopytova, A.; Miliukhina, I.; Yakimovskii, A.; Timofeeva, A.; Berkovich, O.; et al. Blood lysosphingolipids accumulation in patients with parkinson’s disease with glucocerebrosidase 1 mutations. Mov. Disord. 2018, 33, 1325–1330. [Google Scholar] [CrossRef]
- Guedes, L.C.; Chan, R.B.; Gomes, M.A.; Conceição, V.A.; Machado, R.B.; Soares, T.; Xu, Y.; Gaspar, P.; Carriço, J.A.; Alcalay, R.N.; et al. Serum lipid alterations in GBA-associated Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 44, 58–65. [Google Scholar] [CrossRef]
- Lobasso, S.; Tanzarella, P.; Vergara, D.; Maffia, M.; Cocco, T.; Corcelli, A. Lipid profiling of parkin-mutant human skin fibroblasts. J. Cell. Physiol. 2017, 232, 3540–3551. [Google Scholar] [CrossRef]
- Chiasserini, D.; Paciotti, S.; Eusebi, P.; Persichetti, E.; Tasegian, A.; Kurzawa-Akanbi, M.; Chinnery, P.F.; Morris, C.M.; Calabresi, P.; Parnetti, L.; et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 2015, 10, 15. [Google Scholar] [CrossRef]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurobiol. 2019, 56, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H. V Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- McGhee, D.J.M.; Royle, P.L.; Thompson, P.A.; Wright, D.E.; Zajicek, J.P.; Counsell, C.E. A systematic review of biomarkers for disease progression in Parkinson’s disease. BMC Neurol. 2013, 13, 35. [Google Scholar] [CrossRef]
- Paciotti, S.; Gatticchi, L.; Beccari, T.; Parnetti, L. Lysosomal enzyme activities as possible CSF biomarkers of synucleinopathies. Clin. Chim. Acta. 2019, 495, 13–24. [Google Scholar] [CrossRef]
- Farah, R.; Haraty, H.; Salame, Z.; Fares, Y.; Ojcius, D.M.; Said Sadier, N. Salivary biomarkers for the diagnosis and monitoring of neurological diseases. Biomed. J. 2018, 41, 63–87. [Google Scholar] [CrossRef]
- Wang, S.; Kojima, K.; Mobley, J.A.; West, A.B. Proteomic analysis of urinary extracellular vesicles reveal biomarkers for neurologic disease. EBioMedicine 2019, 45, 351–361. [Google Scholar] [CrossRef]
- Dong, H.; Czaja, M.J. Regulation of lipid droplets by autophagy. Trends Endocrinol. Metab. 2011, 22, 234–240. [Google Scholar] [CrossRef]
- Tolosa, E.; Wenning, G.; Poewe, W. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006, 5, 75–86. [Google Scholar] [CrossRef]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.; Hill, D.; Cedarbaum, J.M.; Tome, M.; Vamvakas, S.; Romero, K.; Conrado, D.J.; Dexter, D.T.; Seibyl, J.; Jennings, D.; et al. The Qualification of an Enrichment Biomarker for Clinical Trials Targeting Early Stages of Parkinson’s Disease. J. Parkinsons. Dis. 2019, 9, 553–563. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Symbol | Name | Aliases | Inheritance | ALP involvement |
|---|---|---|---|---|
| LRRK2 | Leucine rich repeat kinase 2 | PARK8; RIPK7; ROCO2; AURA17; DARDARIN | AD/AR | Yes [52] |
| SNCA | Synuclein alpha | PD1; NACP; PARK1; PARK4 | AD | Yes [53] |
| VPS35 | VPS35 retromer complex component | MEM3; PARK17 | AD | Yes [54] |
| ATXN2 | Ataxin 2 | ATX2, SCA2, TNRC13 | AD | Unknown |
| GCH1 | GTP cyclohydrolase 1 | DYT14, DYT5, DYT5a, GCH, GTP-CH-1, GTPCH1, HPABH4B | AD | Unknown |
| PRKN | Parkin RBR E3 ubiquitin protein ligase | AR-JP, LPRS2, PARK2, PDJ | AR | Yes [55] |
| PINK1 | PTEN induced kinase 1 | BRPK, PARK6 | AR | Yes [55,56] |
| PARK7 | Parkinsonism associated deglycase | DJ1; DJ-1; GATD2; HEL-S-67p | AR | Yes [57,58] |
| ATP13A2 | ATPase cation transporting 13A2 | CLN12; KRPPD; PARK9; SPG78; HSA9947 | AR | Yes [59] |
| PLA2G6 | Phospholipase A2 group VI | GVI; PLA2; INAD1; NBIA2; iPLA2; NBIA2A; NBIA2B; PARK14; PNPLA9; CaI-PLA2; IPLA2-VIA; iPLA2beta | AR | Yes [60] |
| FBXO7 | F-box protein 7 | FBX; FBX7; PKPS; FBX07; PARK15 | AR | Yes [61] |
| DNAJC6 | DnaJ heat shock protein family (Hsp40) member C6 | DJC6; PARK19 | AR | Yes [62] |
| SPG11 | SPG11 vesicle trafficking associated, spatacsin | ALS5; CMT2X; KIAA1840 | AR | Yes [63] |
| SYNJ1 | Synaptojanin 1 | EIEE53; INPP5G; PARK20 | AR | Yes [64,65] |
| VPS13C | Vacuolar protein sorting 13 homolog C | PARK23 | AR | Yes [66] |
| RAB39B | RAB39B, member RAS oncogene family | WSN; BGMR; WSMN; MRX72 | XL-D | Yes [67] |
| Biomarker | Measurement | Biospecimen | Directionality |
|---|---|---|---|
| alpha/beta-mannosidase | Activity | CSF | ↓ [94]/ND [93,95] |
| Fibroblasts | ND [75] | ||
| Leukocytes | ND [73] | ||
| Serum | ND [94] | ||
| alpha-galactosidase | Activity | Dried blood spots | ↓ [99]/ND [91,100] |
| Fibroblasts | ND [75,77] | ||
| Leukocytes | ↓ [73] | ||
| Protein | Leukocytes | ↓ [73] | |
| Beclin 1 | Protein | CSF | ↓ [69] |
| PBMCs | ↑ [71] | ||
| beta-galactosidase | Activity | CSF | ↑ [95]/ND [93,94] |
| Fibroblasts | ND [75,77] | ||
| Serum | ↑ [100]/ND [94] | ||
| beta-hexosaminidase | Activity | CSF | ↑ [93]/ND [49,94,95] |
| Fibroblasts | ND [75] | ||
| Leukocytes | ND [98,101] | ||
| Serum | ND [94,100] | ||
| Cathepsin D | Activity | CSF | ↓ [49]/ND [95] |
| Ceramides | Lipid | Plasma | ↑ [117]/ND [89] |
| GCase | Activity | CSF | ↓ [49,93,94]/ND [95] |
| Dried blood spots | ↓ [48]/ND [91] | ||
| Fibroblasts | ND [75,77] | ||
| Lymphocytes | ND [96] | ||
| Monocytes | ↓ [89] | ||
| PBMCs | ND [107] | ||
| Serum | ↓ [94] | ||
| HSC70 | mRNA | PBMCs | ↓ [83,84] |
| LAMP2 | mRNA | PBMCs | ND [84] |
| Protein | CSF | ↓ [70]/ND [86] | |
| PBMCs | ND [84] | ||
| LC3 | mRNA | Leukocytes | ↑ [73] |
| Protein | CSF | ↓ [69]/ND [70] | |
| Fibroblasts | ↓ [74]/ND [75,76] | ||
| Leukocytes | ↑ [73] | ||
| PBMCs | ↑ [72]/ND [71] | ||
| p62 | Protein | Fibroblasts | ↓ [74]/ND [76] |
| PRKN | Protein | Fibroblasts | ↑ [76,112] |
| GBA-PD | |||
|---|---|---|---|
| Biomarker | Measurement | Biospecimen | Directionality |
| a-SYN | Protein—Dimers | Erythrocyte membranes | ↑ [107] |
| Protein—EC | iPSC-derived DA neurons | ↑ [50] | |
| Protein—Oligomeric | Dried blood spots | ↑ [91] | |
| Cathepsin D | Protein | Fibroblasts | ↑ [77] |
| iPSC-derived DA neurons | ↑ [50] | ||
| GCase | Activity | Dried blood spots | ↓ [48,91] |
| Fibroblasts | ↓ [75,77] | ||
| iPSC-derived DA neurons | ↓ [50] | ||
| Monocytes | ↓ [88] | ||
| PBMCs | ↓ [83] | ||
| mRNA | Fibroblasts | ↓ [77] | |
| Leukocytes | ↓ [88] | ||
| Protein | Fibroblasts | ↓ [77]/ND [75] | |
| iPSC-derived DA neurons | ND [50] | ||
| LC3 | Protein | Fibroblasts | ND [77] |
| iPSC-derived DA neurons | ↑ [50] | ||
| p62 | Protein | Fibroblasts | ND [77] |
| iPSC-derived DA neurons | ↑ [50] | ||
| LRRK2-PD | |||
| LC3 | Protein | Fibroblasts | ↑ [78,81]/ND [80] |
| p62 | Protein | Fibroblasts | ↑ [81]/ND [80] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xicoy, H.; Peñuelas, N.; Vila, M.; Laguna, A. Autophagic- and Lysosomal-Related Biomarkers for Parkinson’s Disease: Lights and Shadows. Cells 2019, 8, 1317. https://doi.org/10.3390/cells8111317
Xicoy H, Peñuelas N, Vila M, Laguna A. Autophagic- and Lysosomal-Related Biomarkers for Parkinson’s Disease: Lights and Shadows. Cells. 2019; 8(11):1317. https://doi.org/10.3390/cells8111317
Chicago/Turabian StyleXicoy, Helena, Núria Peñuelas, Miquel Vila, and Ariadna Laguna. 2019. "Autophagic- and Lysosomal-Related Biomarkers for Parkinson’s Disease: Lights and Shadows" Cells 8, no. 11: 1317. https://doi.org/10.3390/cells8111317
APA StyleXicoy, H., Peñuelas, N., Vila, M., & Laguna, A. (2019). Autophagic- and Lysosomal-Related Biomarkers for Parkinson’s Disease: Lights and Shadows. Cells, 8(11), 1317. https://doi.org/10.3390/cells8111317