Depletion of Bone Marrow-Derived Fibrocytes Attenuates TAA-Induced Liver Fibrosis in Mice

 ,
,  ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
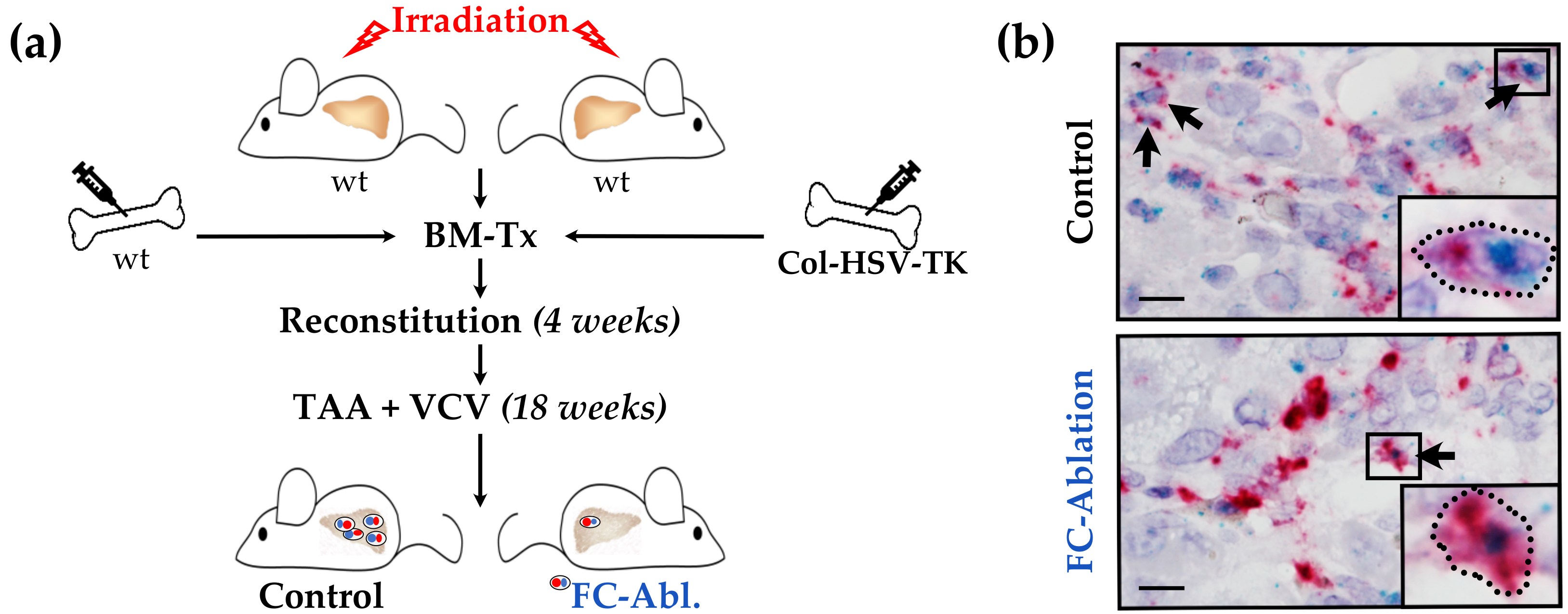
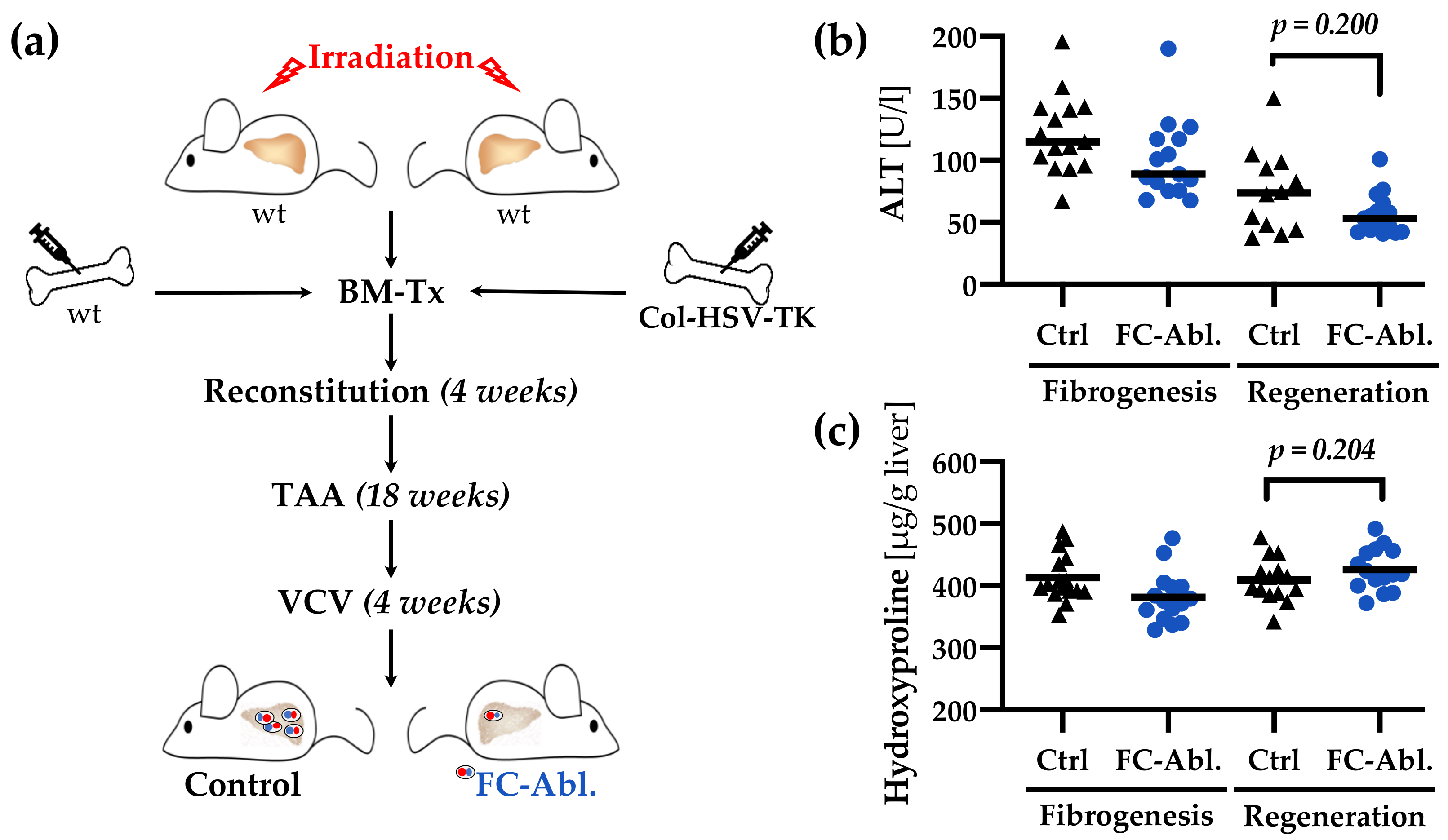
2.1. Animal Experiments
2.2. RNA in Situ Hybridization Assay
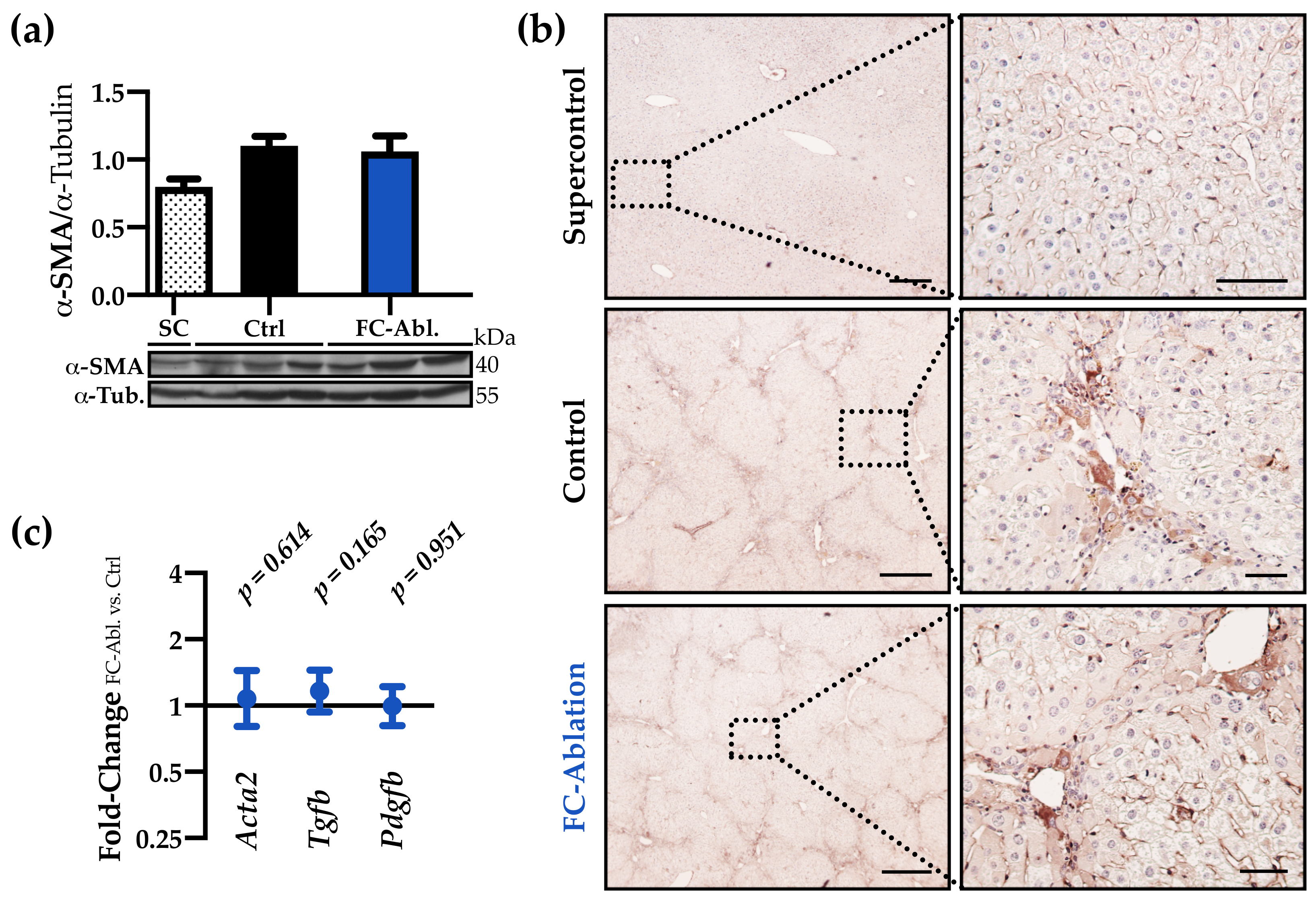
2.3. Histology and Immunohistochemistry
2.4. Pathological Staging and Grading
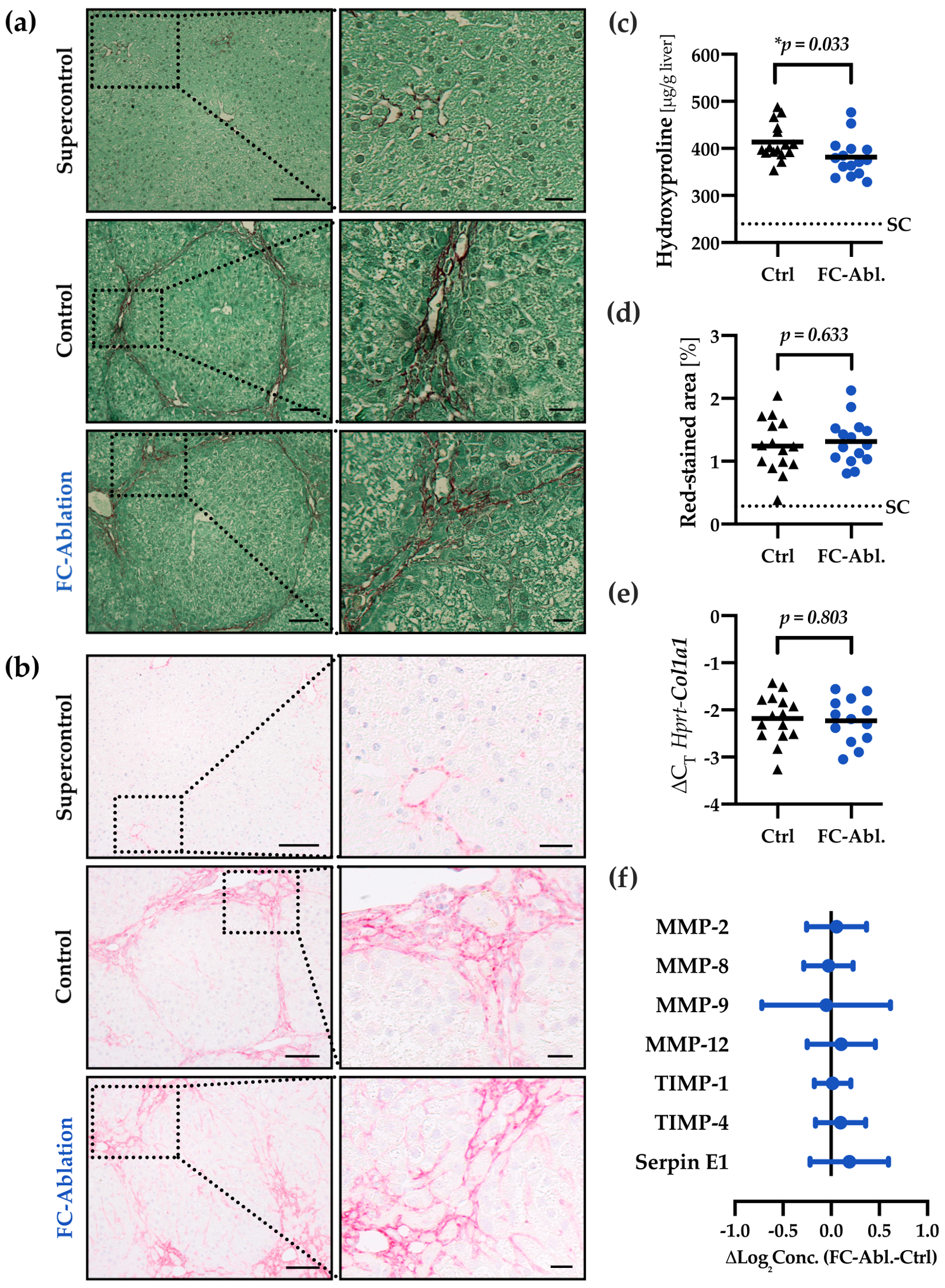
2.5. Morphometric Analysis
2.6. Hydroxyproline Assay
2.7. Quantitative Real-Time PCR
2.8. Gene Expression Array
2.9. Western Blot Analysis
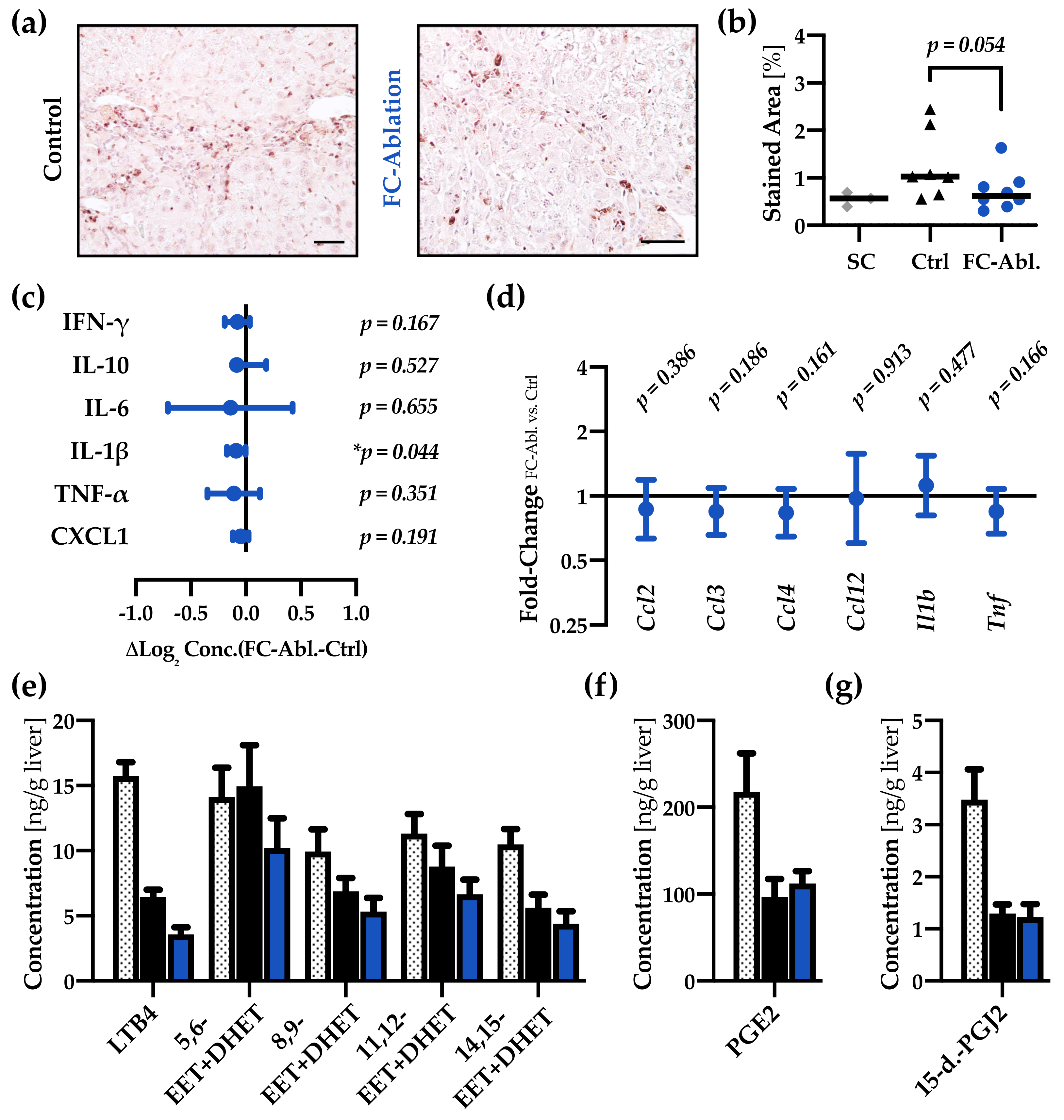
2.10. Multiplex ELISA
2.11. Proteome Profiling
2.12. Eicosanoid Profiling
2.13. Statistical Analysis
3. Results
3.1. Suicide Gene Strategy Enabled the Depletion of Bone Marrow-Derived Fibrocytes
3.2. Depletion of Fibrocytes Attenuated Hepatic Fibrogenesis
3.3. The Antifibrotic Effect was Not Accompanied by A Reduction of Myofibroblasts
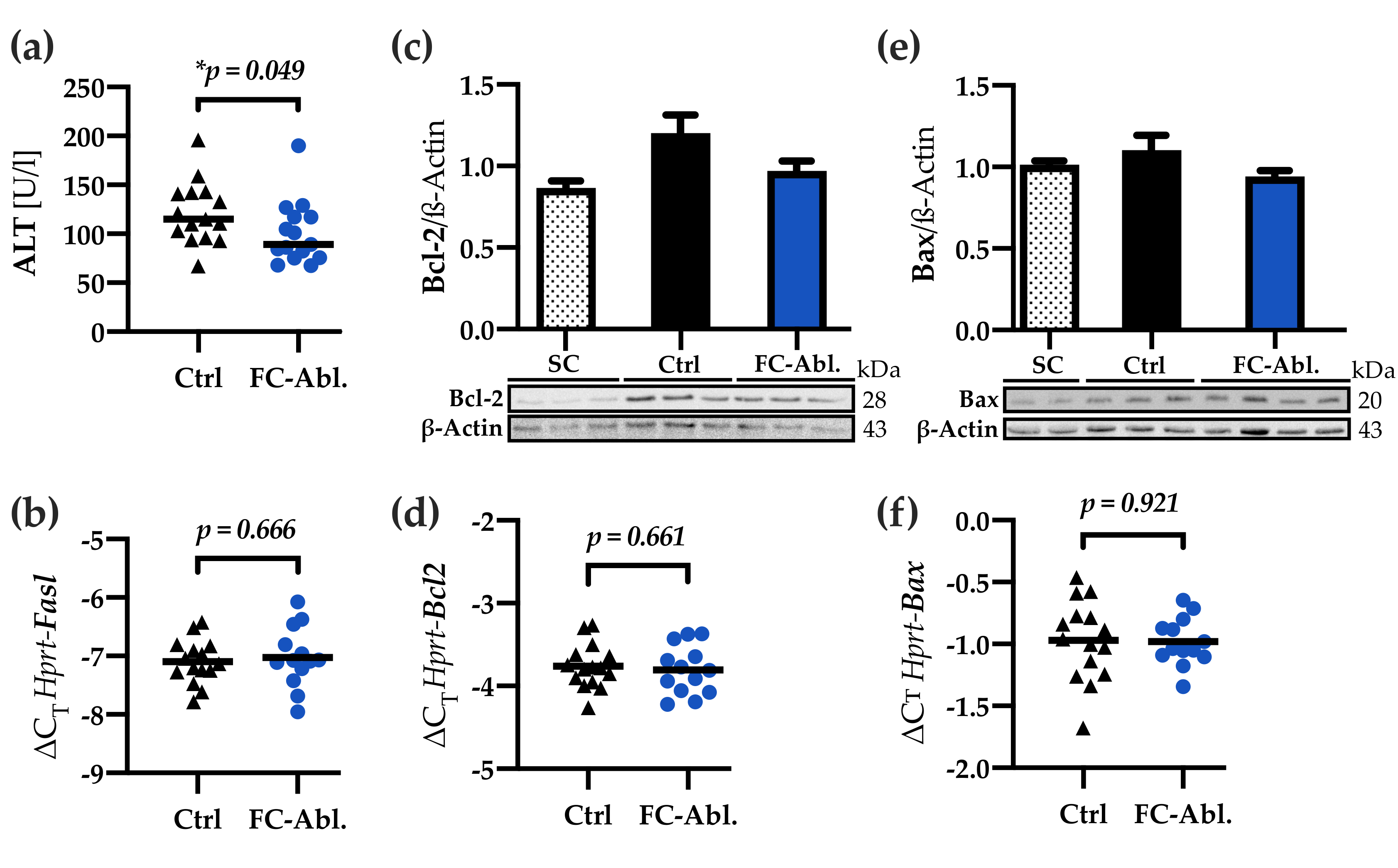
3.4. Fibrocyte Ablation Lead to A Reduction of Hepatic IL-1β Levels
3.5. Liver Integrity was Ameliorated by Fibrocyte Depletion
3.6. A Four-Week Period is Not Sufficient to Provoke Regression of TAA-Induced Liver Fibrosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Spiegel, L.A.; Chesney, J.; Hogan, M.; Cerami, A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994, 1, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Pilling, D.; Fan, T.; Huang, D.; Kaul, B.; Gomer, R.H. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS ONE 2009, 4, e7475. [Google Scholar] [CrossRef] [PubMed]
- Suga, H.; Rennert, R.C.; Rodrigues, M.; Sorkin, M.; Glotzbach, J.P.; Januszyk, M.; Fujiwara, T.; Longaker, M.T.; Gurtner, G.C. Tracking the elusive fibrocyte: Identification and characterization of collagen-producing hematopoietic lineage cells during murine wound healing. Stem Cells 2014, 32, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Scott, P.G.; Dodd, C.; Medina, A.; Jiao, H.; Shankowsky, H.A.; Ghahary, A.; Tredget, E.E. Identification of fibrocytes in postburn hypertrophic scar. Wound Repair Regen. 2005, 13, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.J.; Burdick, M.D.; Hong, K.; Lutz, M.A.; Murray, L.A.; Xue, Y.Y.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Investig. 2004, 114, 438–446. [Google Scholar] [CrossRef]
- Sakai, N.; Wada, T.; Yokoyama, H.; Lipp, M.; Ueha, S.; Matsushima, K.; Kaneko, S. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 14098–14103. [Google Scholar] [CrossRef]
- Haudek, S.B.; Xia, Y.; Huebener, P.; Lee, J.M.; Carlson, S.; Crawford, J.R.; Pilling, D.; Gomer, R.H.; Trial, J.; Frangogiannis, N.G.; et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 18284–18289. [Google Scholar] [CrossRef]
- Sazuka, S.; Katsuno, T.; Nakagawa, T.; Saito, M.; Saito, K.; Maruoka, D.; Matsumura, T.; Arai, M.; Miyauchi, H.; Matsubara, H.; et al. Fibrocytes are involved in inflammation as well as fibrosis in the pathogenesis of crohn’s disease. Dig. Dis. Sci. 2014, 59, 760–768. [Google Scholar] [CrossRef]
- Kuroda, N.; Masuya, M.; Tawara, I.; Tsuboi, J.; Yoneda, M.; Nishikawa, K.; Kageyama, Y.; Hachiya, K.; Ohishi, K.; Miwa, H.; et al. Infiltrating CCR2(+) monocytes and their progenies, fibrocytes, contribute to colon fibrosis by inhibiting collagen degradation through the production of TIMP-1. Sci. Rep. 2019, 9, 8568. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Sun, G.; Stacey, M.A.; Mori, L.; Mattoli, S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J. Immunol. 2003, 171, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Uehara, H.; Nakagawa, T.; Katsuno, T.; Sato, T.; Isono, A.; Noguchi, Y.; Saito, Y. Emergence of fibrocytes showing morphological changes in the inflamed colonic mucosa. Dig. Dis. Sci. 2010, 55, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, K.; Zhao, H.; He, Y. The emerging role of fibrocytes in ocular disorders. Stem Cell Res. Ther. 2018, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–E3305. [Google Scholar] [CrossRef] [PubMed]
- Nishio, T.; Hu, R.; Koyama, Y.; Liang, S.; Rosenthal, S.B.; Yamamoto, G.; Karin, D.; Baglieri, J.; Ma, H.Y.; Xu, J.; et al. Activated hepatic stellate cells and portal fibroblasts contribute to cholestatic liver fibrosis in MDR2 knockout mice. J. Hepatol. 2019, 71, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Yuan, W.G.; He, P.; Lei, J.H.; Wang, C.X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Scholten, D.; Reichart, D.; Paik, Y.H.; Lindert, J.; Bhattacharya, J.; Glass, C.K.; Brenner, D.A.; Kisseleva, T. Migration of fibrocytes in fibrogenic liver injury. Am. J. Pathol. 2011, 179, 189–198. [Google Scholar] [CrossRef]
- Kisseleva, T.; Uchinami, H.; Feirt, N.; Quintana-Bustamante, O.; Segovia, J.C.; Schwabe, R.F.; Brenner, D.A. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J. Hepatol. 2006, 45, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Roderfeld, M.; Rath, T.; Voswinckel, R.; Dierkes, C.; Dietrich, H.; Zahner, D.; Graf, J.; Roeb, E. Bone marrow transplantation demonstrates medullar origin of CD34+ fibrocytes and ameliorates hepatic fibrosis in Abcb4-/- mice. Hepatology 2010, 51, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Donnelly, S.C.; Peng, T.; Bucala, R.; Metz, C.N. Peripheral blood fibrocytes: Differentiation pathway and migration to wound sites. J. Immunol. 2001, 166, 7556–7562. [Google Scholar] [CrossRef] [PubMed]
- Reilkoff, R.A.; Bucala, R.; Herzog, E.L. Fibrocytes: Emerging effector cells in chronic inflammation. Nat. Rev. Immunol. 2011, 11, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Kleaveland, K.R.; Moore, B.B.; Kim, K.K. Paracrine functions of fibrocytes to promote lung fibrosis. Expert Rev. Respir. Med. 2014, 8, 163–172. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chesney, J.; Metz, C.; Stavitsky, A.B.; Bacher, M.; Bucala, R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J. Immunol. 1998, 160, 419–425. [Google Scholar] [PubMed]
- Seki, E.; Brenner, D.A. Recent advancement of molecular mechanisms of liver fibrosis. J. Hepatobiliary Pancreat. Sci. 2015, 22, 512–518. [Google Scholar] [CrossRef]
- Vannella, K.M.; McMillan, T.R.; Charbeneau, R.P.; Wilke, C.A.; Thomas, P.E.; Toews, G.B.; Peters-Golden, M.; Moore, B.B. Cysteinyl leukotrienes are autocrine and paracrine regulators of fibrocyte function. J. Immunol. 2007, 179, 7883–7890. [Google Scholar] [CrossRef]
- Chesney, J.; Bacher, M.; Bender, A.; Bucala, R. The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc. Natl. Acad. Sci. USA 1997, 94, 6307–6312. [Google Scholar] [CrossRef]
- Higashiyama, R.; Inagaki, Y.; Hong, Y.Y.; Kushida, M.; Nakao, S.; Niioka, M.; Watanabe, T.; Okano, H.; Matsuzaki, Y.; Shiota, G.; et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology 2007, 45, 213–222. [Google Scholar] [CrossRef]
- Garcia-de-Alba, C.; Becerril, C.; Ruiz, V.; Gonzalez, Y.; Reyes, S.; Garcia-Alvarez, J.; Selman, M.; Pardo, A. Expression of matrix metalloproteases by fibrocytes: Possible role in migration and homing. Am. J. Respir Crit. Care Med. 2010, 182, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Hartlapp, I.; Abe, R.; Saeed, R.W.; Peng, T.; Voelter, W.; Bucala, R.; Metz, C.N. Fibrocytes induce an angiogenic phenotype in cultured endothelial cells and promote angiogenesis in vivo. FASEB J. 2001, 15, 2215–2224. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tan, H.; Wang, X.; Li, Y.; Samuelson, L.; Li, X.; Cui, C.; Gerber, D.A. Circulating fibrocytes stabilize blood vessels during angiogenesis in a paracrine manner. Am. J. Pathol. 2014, 184, 556–571. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; von Kockritz-Blickwede, M.; Reichart, D.; McGillvray, S.M.; Wingender, G.; Kronenberg, M.; Glass, C.K.; Nizet, V.; Brenner, D.A. Fibrocyte-like cells recruited to the spleen support innate and adaptive immune responses to acute injury or infection. J. Mol. Med. (Berl) 2011, 89, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, E.; Heyman, R.; Hsi, M.; Evans, R.M. Targeting of an inducible toxic phenotype in animal cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7572–7576. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, E.; Heyman, R.A.; Arias, C.; Sawchenko, P.E.; Evans, R.M. Transgenic mice with inducible dwarfism. Nature 1989, 339, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Heyman, R.A.; Borrelli, E.; Lesley, J.; Anderson, D.; Richman, D.D.; Baird, S.M.; Hyman, R.; Evans, R.M. Thymidine kinase obliteration: Creation of transgenic mice with controlled immune deficiency. Proc. Natl. Acad. Sci. USA 1989, 86, 2698–2702. [Google Scholar] [CrossRef]
- Tian, B.; Han, L.; Kleidon, J.; Henke, C. An HSV-TK transgenic mouse model to evaluate elimination of fibroblasts for fibrosis therapy. Am. J. Pathol. 2003, 163, 789–801. [Google Scholar] [CrossRef]
- Ozono, Y.; Shide, K.; Toyoshima, F.; Takaishi, Y.; Tsuchimochi, M.; Kamiunten, A.; Kameda, T.; Nakamura, K.; Miike, T.; Kusumoto, K.; et al. Monocyte-derived fibrocytes elimination had little contribution on liver fibrosis. Hepatobiliary Pancreat. Dis. Int. 2019, 18, 348–353. [Google Scholar] [CrossRef]
- Mederacke, I. Liver fibrosis - mouse models and relevance in human liver diseases. Z Gastroenterol. 2013, 51, 55–62. [Google Scholar] [CrossRef]
- Salguero Palacios, R.; Roderfeld, M.; Hemmann, S.; Rath, T.; Atanasova, S.; Tschuschner, A.; Gressner, O.A.; Weiskirchen, R.; Graf, J.; Roeb, E. Activation of hepatic stellate cells is associated with cytokine expression in thioacetamide-induced hepatic fibrosis in mice. Lab. Investig. 2008, 88, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.C.; Hamesch, K.; Lunova, M.; Kim, Y.; Weiskirchen, R.; Strnad, P.; Friedman, S.L. Standard operating procedures in experimental liver research: Thioacetamide model in mice and rats. Lab. Anim. 2015, 49, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.T.; Ma, X.J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Ishak, K.; Baptista, A.; Bianchi, L.; Callea, F.; De Groote, J.; Gudat, F.; Denk, H.; Desmet, V.; Korb, G.; MacSween, R.N.; et al. Histological grading and staging of chronic hepatitis. J. Hepatol. 1995, 22, 696–699. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Roderfeld, M.; Padem, S.; Lichtenberger, J.; Quack, T.; Weiskirchen, R.; Longerich, T.; Schramm, G.; Churin, Y.; Irungbam, K.; Tschuschner, A.; et al. Schistosoma mansoni egg-secreted antigens activate hepatocellular carcinoma-associated transcription factors c-Jun and STAT3 in hamster and human hepatocytes. Hepatology 2019. [Google Scholar] [CrossRef]
- Kiss, L.; Roder, Y.; Bier, J.; Weissmann, N.; Seeger, W.; Grimminger, F. Direct eicosanoid profiling of the hypoxic lung by comprehensive analysis via capillary liquid chromatography with dual online photodiode-array and tandem mass-spectrometric detection. Anal. Bioanal. Chem. 2008, 390, 697–714. [Google Scholar] [CrossRef]
- Roderfeld, M.; Rath, T.; Pasupuleti, S.; Zimmermann, M.; Neumann, C.; Churin, Y.; Dierkes, C.; Voswinckel, R.; Barth, P.J.; Zahner, D.; et al. Bone marrow transplantation improves hepatic fibrosis in Abcb4-/- mice via Th1 response and matrix metalloproteinase activity. Gut 2012, 61, 907–916. [Google Scholar] [CrossRef]
- Lefebvre, E.; Moyle, G.; Reshef, R.; Richman, L.P.; Thompson, M.; Hong, F.; Chou, H.L.; Hashiguchi, T.; Plato, C.; Poulin, D.; et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS ONE 2016, 11, e0158156. [Google Scholar] [CrossRef]
- Puengel, T.; Krenkel, O.; Mossanen, J.; Longerich, T.; Lefebvre, E.; Trautwein, C.; Tacke, F. The dual CCR2/CCR5 antagonist cenicriviroc ameliorates steatohepatitis and fibrosis in vivo by inhibiting the infiltration of inflammatory monocytes into injured liver. J. Hepatol. 2016, 64, S159–S182. [Google Scholar] [CrossRef]
- Moore, B.B.; Kolodsick, J.E.; Thannickal, V.J.; Cooke, K.; Moore, T.A.; Hogaboam, C.; Wilke, C.A.; Toews, G.B. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am. J. Pathol. 2005, 166, 675–684. [Google Scholar] [CrossRef]
- Moore, B.B.; Murray, L.; Das, A.; Wilke, C.A.; Herrygers, A.B.; Toews, G.B. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am. J. Respir Cell Mol. Biol. 2006, 35, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Puche, J.E.; Lee, Y.A.; Jiao, J.; Aloman, C.; Fiel, M.I.; Munoz, U.; Kraus, T.; Lee, T.; Yee, H.F., Jr.; Friedman, S.L. A novel murine model to deplete hepatic stellate cells uncovers their role in amplifying liver damage in mice. Hepatology 2013, 57, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cong, M.; Park, T.J.; Scholten, D.; Brenner, D.A.; Kisseleva, T. Contribution of bone marrow-derived fibrocytes to liver fibrosis. Hepatobiliary Surg. Nutr. 2015, 4, 34–47. [Google Scholar] [PubMed]
- Kleaveland, K.R.; Velikoff, M.; Yang, J.; Agarwal, M.; Rippe, R.A.; Moore, B.B.; Kim, K.K. Fibrocytes are not an essential source of type I collagen during lung fibrosis. J. Immunol. 2014, 193, 5229–5239. [Google Scholar] [CrossRef]
- Hemmann, S.; Graf, J.; Roderfeld, M.; Roeb, E. Expression of MMPs and TIMPs in liver fibrosis - a systematic review with special emphasis on anti-fibrotic strategies. J. Hepatol. 2007, 46, 955–975. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Reilkoff, R.; Peng, X.; Russell, T.; Chen, Q.; Mathai, S.K.; Homer, R.; Gulati, M.; Siner, J.; Elias, J.; et al. Role of semaphorin 7a signaling in transforming growth factor beta1-induced lung fibrosis and scleroderma-related interstitial lung disease. Arthritis Rheum 2011, 63, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Pilling, D.; Buckley, C.D.; Salmon, M.; Gomer, R.H. Inhibition of fibrocyte differentiation by serum amyloid P. J. Immunol. 2003, 171, 5537–5546. [Google Scholar] [CrossRef]
- Cong, M.; Zhao, W.; Liu, T.; Wang, P.; Fan, X.; Zhai, Q.; Bao, X.; Zhang, D.; You, H.; Kisseleva, T.; et al. Protective effect of human serum amyloid P on CCl4-induced acute liver injury in mice. Int. J. Mol. Med. 2017, 40, 454–464. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | 0 | 1 | 2 | 3 | 4 | 5 | 6 | Median |
|---|---|---|---|---|---|---|---|---|
| Supercontrol | 3 | 5 | 0 | 0 | 0 | 0 | 0 | 1 |
| Control | 0 | 0 | 0 | 8 | 6 | 2 | 0 | 3.5 1 |
| FC-Ablation | 0 | 0 | 0 | 6 | 5 | 4 | 0 | 4 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hempel, F.; Roderfeld, M.; Savai, R.; Sydykov, A.; Irungbam, K.; Schermuly, R.; Voswinckel, R.; Köhler, K.; Churin, Y.; Kiss, L.; et al. Depletion of Bone Marrow-Derived Fibrocytes Attenuates TAA-Induced Liver Fibrosis in Mice. Cells 2019, 8, 1210. https://doi.org/10.3390/cells8101210
Hempel F, Roderfeld M, Savai R, Sydykov A, Irungbam K, Schermuly R, Voswinckel R, Köhler K, Churin Y, Kiss L, et al. Depletion of Bone Marrow-Derived Fibrocytes Attenuates TAA-Induced Liver Fibrosis in Mice. Cells. 2019; 8(10):1210. https://doi.org/10.3390/cells8101210
Chicago/Turabian StyleHempel, Felix, Martin Roderfeld, Rajkumar Savai, Akylbek Sydykov, Karuna Irungbam, Ralph Schermuly, Robert Voswinckel, Kernt Köhler, Yury Churin, Ladislau Kiss, and et al. 2019. "Depletion of Bone Marrow-Derived Fibrocytes Attenuates TAA-Induced Liver Fibrosis in Mice" Cells 8, no. 10: 1210. https://doi.org/10.3390/cells8101210
APA StyleHempel, F., Roderfeld, M., Savai, R., Sydykov, A., Irungbam, K., Schermuly, R., Voswinckel, R., Köhler, K., Churin, Y., Kiss, L., Bier, J., Pons-Kühnemann, J., & Roeb, E. (2019). Depletion of Bone Marrow-Derived Fibrocytes Attenuates TAA-Induced Liver Fibrosis in Mice. Cells, 8(10), 1210. https://doi.org/10.3390/cells8101210