Impact of Autophagy of Innate Immune Cells on Inflammatory Bowel Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
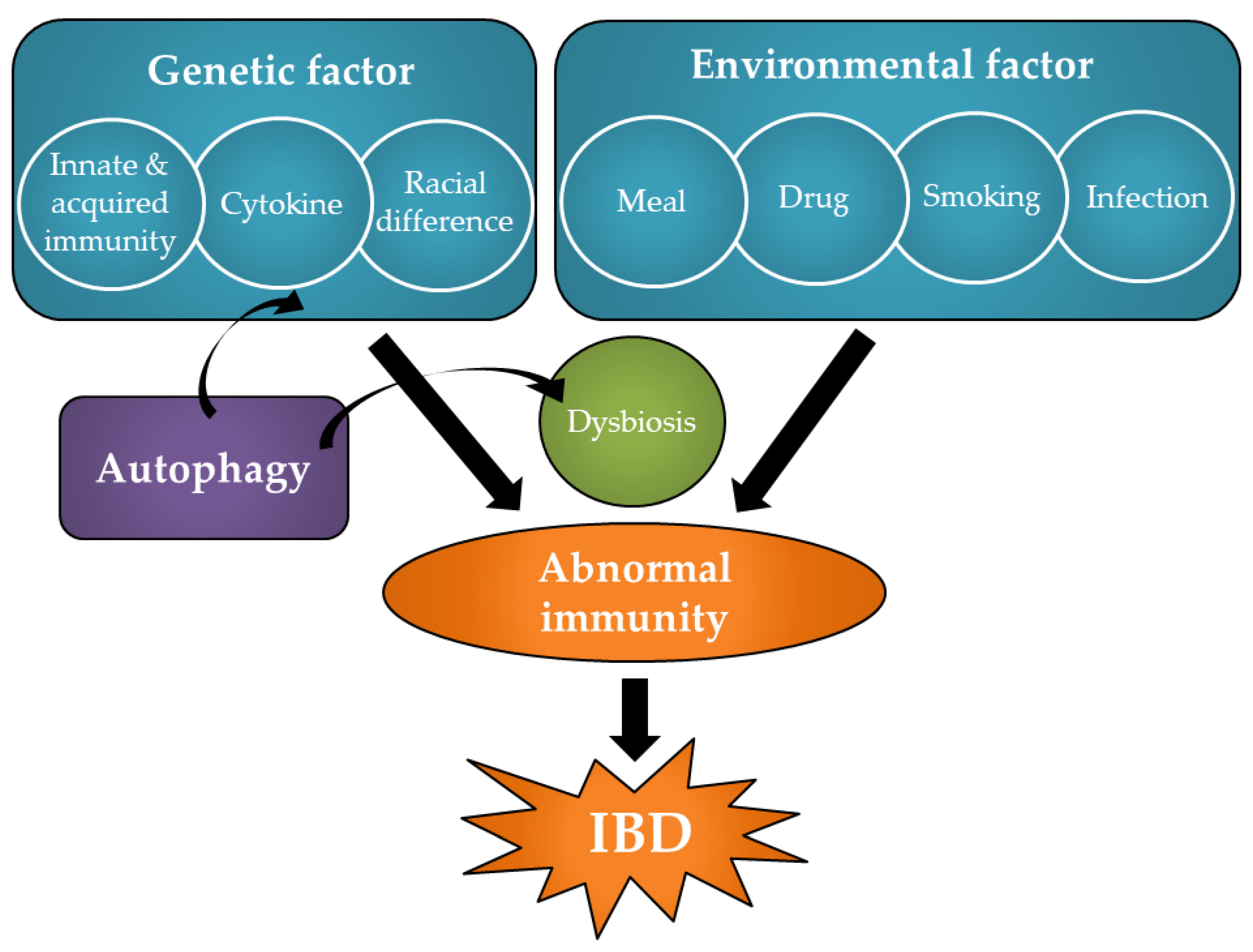
1. Introduction
2. Pathology and Pathogenesis of IBD
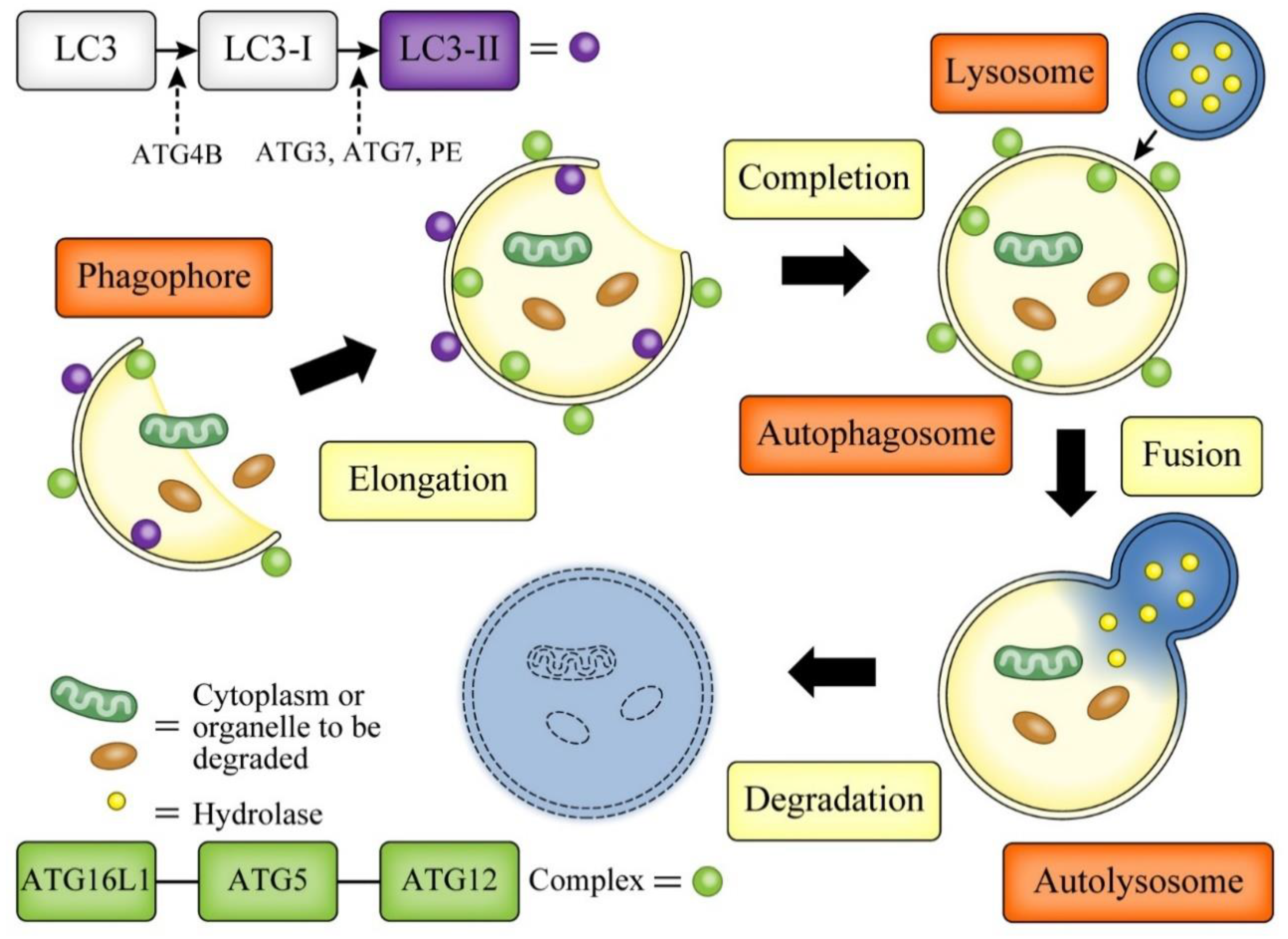
3. Autophagy
4. Role of Autophagy in Innate Immunity
4.1. Xenophagy, Mitophagy
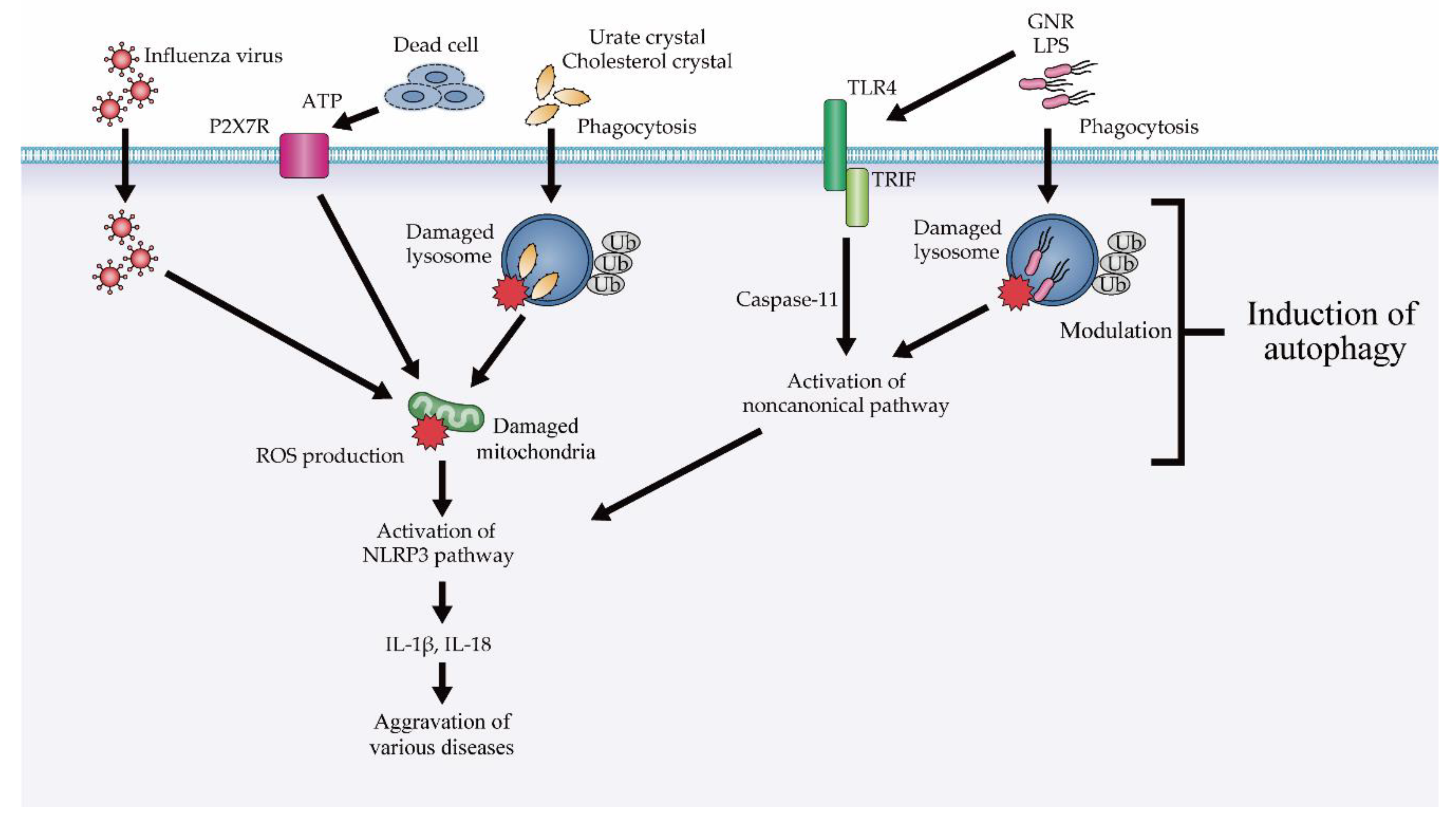
4.2. The Role of Autophagy in Inflammasomal and Type I Interferon Response
4.2.1. Modulation of NLRP3 Inflammasome Suppression via Autophagy
4.2.2. Modulation of Type I IFN Responses via Autophagy
5. Role of Autophagy of Innate Immune Cells in IBD
5.1. Hematopoietic Cells
5.1.1. Macrophages and DCs
Pathogen Degradation
Suppressing Inflammatory Cytokine Secretion
Antigen Presentation
5.1.2. Neutrophils
5.1.3. Innate Lymphoid Cells (ILCs)
5.1.4. NKT Cells (NKTs)
5.2. Intestinal Epithelial Cells
5.2.1. Paneth Cells
ERS Stress (ERS)
ROS
Gut Microbiota
5.2.2. Goblet Cells
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ebbo, M.; Crinier, A.; Vély, F.; Vivier, E. Innate lymphoid cells: Major players in inflammatory diseases. Nat. Rev. Immunol. 2017, 17, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Choy, M.C.; Visvanathan, K.; De Cruz, P. An overview of the innate and adaptive immune system in inflammatory bowel disease. Inflamm. Bowel Dis. 2017, 23, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.E.; Cho, M.L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.M.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for IBD and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Brant, S.R. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 2011, 140, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Ek, W.E.; D’Amato, M.; Halfvarson, J. The history of genetics in inflammatory bowel disease. Ann. Gastroenterol. 2014, 27, 294–303. [Google Scholar] [PubMed]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Böck, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Patman, G. Crohn’s disease. TCF1 regulates Paneth cell α-defensins. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 517. [Google Scholar] [CrossRef]
- Eriguchi, Y.; Nakamura, K.; Yokoi, Y.; Sugimoto, R.; Takahashi, S.; Hashimoto, D.; Teshima, T.; Ayabe, T.; Selsted, M.E.; Ouellette, A.J. Essential role of IFN-γ in T cell-associated intestinal inflammation. JCI. Insight. 2018, 20, pii:121886. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Blumberg, R.S. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology 2011, 140, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef]
- Weterman, I.T.; Peña, A.S. Familial incidence of Crohn’s disease in the Netherlands and a review of the literature. Gastroenterology 1984, 86, 449–452. [Google Scholar]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef]
- Goldstein, D.B. Common genetic variation and human traits. N. Engl. J. Med. 2009, 360, 1696–1698. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–325. [Google Scholar] [CrossRef]
- Hooper, K.M.; Barlow, P.G.; Stevens, C.; Henderson, P. Inflammatory bowel disease drugs: A focus on autophagy. J. Crohn’s Colitis 2017, 11, 118–127. [Google Scholar] [CrossRef]
- Lassen, K.G.; Xavier, R.J. Mechanisms and function of autophagy in intestinal disease. Autophagy 2018, 14, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Zeki, A.A.; Yeganeh, B.; Kenyon, N.J.; Post, M.; Ghavami, S. Autophagy in airway diseases: A new frontier in human asthma? Allergy 2016, 71, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.H.; Choy, D.F.; Chouiali, F.; Ramakrishnan, R.K.; Mahboub, B.; Audusseau, S.; Mogas, A.; Harris, J.M.; Arron, J.R.; Laprise, C.; et al. Increased autophagy-related 5 gene expression is associated with collagen expression in the airways of refractory asthmatics. Front. Immunol. 2017, 8, 355. [Google Scholar] [CrossRef] [PubMed]
- Mabalirajan, U. A possible differential role of autophagy in asthma? J. Allergy Clin. Immunol. 2017, 139, 712. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, A.C.; Kikkers, S.A.; Choi, A.M.K.; Cloonan, S.M. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.J. Systemic lupus erythematosus: Defective noncanonical autophagy in SLE-like disease. Nat. Rev. Rheumatol. 2016, 12, 311. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.; Kaplan, M.J. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin. Immunol. 2017, 185, 59–73. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association between autophagy and neurodegenerative diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Iida, T.; Onodera, K.; Nakase, H. Role of autophagy in the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2017, 23, 1944–1953. [Google Scholar] [CrossRef]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Levine, B. Eating oneself and uninvited guests: Autophagy-related pathways in cellular defense. Cell 2005, 120, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, T.; Minowa-Nozawa, A.; Aikawa, C.; Nakagawa, I. The STX6-VTI1B-VAMP3 complex facilitates xenophagy by regulating the fusion between recycling endosomes and autophagosomes. Autophagy 2017, 13, 57–69. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Yao, Z.; Klionsky, D.J. An unconventional pathway for mitochondrial protein degradation. Autophagy 2016, 12, 1971–1972. [Google Scholar] [CrossRef][Green Version]
- Kimura, T.; Mandell, M.; Deretic, V. Precision autophagy directed by receptor regulators-emerging examples within the TRIM family. J. Cell Sci. 2016, 129, 881–891. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Chan, F.K. Inflammasome, Inflammation, and Tissue Homeostasis. Trends Mol. Med. 2018, 24, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Watson, A.J.; Neurath, M.F. Complex roles of caspases in the pathogenesis of inflammatory bowel disease. Gastroenterology 2013, 144, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Opipari, A.; Franchi, L. Role of inflammasomes in intestinal inflammation and Crohn’s disease. Inflamm. Bowel Dis. 2015, 21, 173–181. [Google Scholar] [CrossRef]
- de Souza, H.S.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- de Souza, H.S.P. Etiopathogenesis of inflammatory bowel disease: Today and tomorrow. Curr. Opin. Gastroenterol. 2017, 33, 222–229. [Google Scholar] [CrossRef]
- Lazaridis, L.D.; Pistiki, A.; Giamarellos-Bourboulis, E.J.; Georgitsi, M.; Damoraki, G.; Polymeros, D.; Dimitriadis, G.D.; Triantafyllou, K. Activation of NLRP3 inflammasome in inflammatory bowel disease: Differences between Crohn’s disease and ulcerative colitis. Dig. Dis. Sci. 2017, 62, 2348–2356. [Google Scholar] [CrossRef]
- Mao, L.; Kitani, A.; Similuk, M.; Oler, A.J.; Albenberg, L.; Kelsen, J.; Aktay, A.; Quezado, M.; Yao, M.; Montgomery-Recht, K.; et al. Loss-of-function CARD8 mutation causes NLRP3 inflammasome activation and Crohn’s disease. J. Clin. Investig. 2018, 128, 1793–1806. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Giles, E.M.; Sanders, T.J.; McCarthy, N.E.; Lung, J.; Pathak, M.; MacDonald, T.T.; Lindsay, J.O.; Stagg, A.J. Regulation of human intestinal T-cell responses by type 1 interferon-STAT1 signaling is disrupted in inflammatory bowel disease. Mucosal Immunol. 2017, 10, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.l.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Lupfer, C.; Thomas, P.G.; Anand, P.K.; Vogel, P.; Milasta, S.; Martinez, J.; Huang, G.; Green, M.; Kundu, M.; Chi, H.; et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 2013, 14, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.Y.; Yang, C.H.; Chou, C.C.; Chiang, Y.P.; Chuang, T.H.; Hsu, L.C. TLR-induced PAI-2 expression suppresses IL-1β processing via increasing autophagy and NLRP3 degradation. Proc. Natl. Acad. Sci. USA 2013, 110, 16079–16084. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef]
- Deretic, V.; Kimura, T.; Timmins, G.; Moseley, P.; Chauhan, S.; Mandell, M. Immunologic manifestations of autophagy. J. Clin. Investig. 2015, 125, 75–84. [Google Scholar] [CrossRef]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Schroder, K.; Johansen, T.; Deretic, V. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 2015, 210, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kenny, S.J.; Ge, L.; Xu, K.; Schekman, R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife 2015, 4. [Google Scholar] [CrossRef]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Homer, C.R.; Richmond, A.L.; Rebert, N.A.; Achkar, J.P.; McDonald, C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 2010, 139, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef]
- Negroni, A.; Colantoni, E.; Vitali, R.; Palone, F.; Pierdomenico, M.; Costanzo, M.; Cesi, V.; Cucchiara, S.; Stronati, L. NOD2 induces autophagy to control AIEC bacteria infectiveness in intestinal epithelial cells. Inflamm. Res. 2016, 65, 803–813. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef]
- Prescott, N.J.; Fisher, S.A.; Franke, A.; Hampe, J.; Onnie, C.M.; Soars, D.; Bagnall, R.; Mirza, M.M.; Sanderson, J.; Forbes, A.; et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology 2007, 132, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Li, Y.; Peng, I.; Reichelt, M.; Katakam, A.K.; Noubade, R.; Roose-Girma, M.; DeVoss, J.; Diehl, L.; Graham, R.R.; et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014, 506, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Khosravi, A.; Kusumawardhani, I.P.; Kwon, A.H.; Vasconcelos, A.C.; Cunha, L.D.; Mayer, A.E.; Shen, Y.; Wu, W.L.; Kambal, A.; et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 2016, 352, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Yang, X.; Li, J.; Chen, X.; Zhao, X.; Chen, Y.; Wen, Y. Trimethylamine N-oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem. Biophys. Res. Commun. 2017, 490, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Pott, J.; Kabat, A.M.; Maloy, K.J. Intestinal epithelial cell autophagy is required to protect against TNF-induced apoptosis during chronic colitis in mice. Cell Host Microbe 2018, 23, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Davis, A.S.; Taylor, G.A.; Deretic, V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006, 313, 1438–1441. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, S.A.; Huett, A.; Kuballa, P.; Chilewski, S.D.; Landry, A.; Goyette, P.; Zody, M.C.; Hall, J.L.; Brant, S.R.; Cho, J.H.; et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat. Genet. 2008, 40, 1107–1112. [Google Scholar] [CrossRef]
- Rufini, S.; Ciccacci, C.; Di Fusco, D.; Ruffa, A.; Pallone, F.; Novelli, G.; Biancone, L.; Borgiani, P. Autophagy and inflammatory bowel disease: Association between variants of the autophagy related IRGM gene and susceptibility to Crohn’s disease. Dig. Liver Dis. 2015, 47, 744–750. [Google Scholar] [CrossRef]
- Chauhan, S.; Mandell, M.A.; Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 2015, 58, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Crook, N.E.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol. 1993, 67, 2168–2174. [Google Scholar] [PubMed]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, Z.; Mei, Y.; Wu, M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013, 32, 2204–2216. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, Y.; Petersen, B.S.; Milutinovic, S.; Bosse, E.; Mayr, G.; Peuker, K.; Hartwig, J.; Keller, A.; Kohl, M.; Laass, M.W.; et al. XIAP variants in male Crohn’s disease. Gut 2015, 64, 66–76. [Google Scholar] [CrossRef]
- Schwerd, T.; Pandey, S.; Yang, H.T.; Bagola, K.; Jameson, E.; Jung, J.; Lachmann, R.H.; Shah, N.; Patel, S.Y.; Booth, C.; et al. Impaired antibacterial autophagy links granulomatous intestinal inflammation in Niemann-Pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn’s disease. Gut 2017, 66, 1060–1073. [Google Scholar] [CrossRef]
- Elinav, E.; Strowig, T.; Henao-Mejia, J.; Flavell, R.A. Regulation of the antimicrobial response by NLR proteins. Immunity 2011, 34, 665–679. [Google Scholar] [CrossRef]
- Yano, T.; Kurata, S. Intracellular recognition of pathogens and autophagy as an innate immune host defence. J. Biochem. 2011, 150, 143–149. [Google Scholar] [CrossRef]
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1⁺ macrophages to CD103⁺ dendritic cells. Immunity 2014, 40, 248–261. [Google Scholar] [CrossRef]
- Martin, B.; Hirota, K.; Cua, D.J.; Stockinger, B.; Veldhoen, M. Interleukin-17-producing gamma delta T cells selectively expand in response topathogen products and environmental signals. Immunity 2009, 31, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Talero, E.; Garcia-Maurino, S.; Motilva, V. Melatonin, autophagy and intestinal bowel disease. Curr. Pharm. Des. 2014, 20, 4816–4827. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Autophagy beyond intracellular MHC class II antigen presentation. Trends. Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Strisciuglio, C.; Duijvestein, M.; Verhaar, A.P.; Vos, A.C.; van den Brink, G.R.; Hommes, D.W.; Wildenberg, M.E. Impaired autophagy leads to abnormal dendritic cell-epithelial cell interactions. J. Crohns. Colitis. 2013, 7, 534–541. [Google Scholar] [CrossRef]
- Lapaquette, P.; Bringer, M.A.; Darfeuille-Michaud, A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell. Microbiol. 2012, 14, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Lassen, K.G.; McKenzie, C.I.; Mari, M.; Murano, T.; Begun, J.; Baxt, L.A.; Goel, G.; Villablanca, E.J.; Kuo, S.Y.; Huang, H.; et al. Genetic coding variant in GPR65 alters lysosomal pH and links lysosomal dysfunction with colitis risk. Immunity 2016, 44, 1392–1405. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Dick, M.S.; Dreier, R.F.; Schürmann, N.; Kenzelmann Broz, D.; Warming, S.; Roose-Girma, M.; Bumann, D.; Kayagaki, N.; Takeda, K.; et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 2014, 509, 366–370. [Google Scholar] [CrossRef]
- Ke, P.; Shao, B.Z.; Xu, Z.Q.; Wei, W.; Han, B.Z.; Chen, X.W.; Su, D.F.; Liu, C. Activation of cannabinoid receptor 2 ameliorates DSS-induced colitis through inhibiting NLRP3 inflammasome in macrophages. PLoS ONE 2016, 11, e0155076. [Google Scholar] [CrossRef]
- Samie, M.; Lim, J.; Verschueren, E.; Baughman, J.M.; Peng, I.; Wong, A.; Kwon, Y.; Senbabaoglu, Y.; Hackney, J.A.; Keir, M.; et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat. Immunol. 2018, 19, 246–254. [Google Scholar] [CrossRef]
- Fan, K.; Lin, L.; Ai, Q.; Wan, J.; Dai, J.; Liu, G.; Tang, L.; Yang, Y.; Ge, P.; Jiang, R.; et al. Lipopolysaccharide-induced dephosphorylation of AMPK-activated protein kinase potentiates inflammatory injury via repression of ULK1-dependent autophagy. Front. Immunol. 2018, 9, 1464. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, L.; Chen, J.; Fukata, M.; Ichikawa, R.; Shih, D.Q.; Zhang, X. The protection role of Atg16l1 in CD11c+dendritic cells in murine colitis. Immunobiology 2017, 222, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Baxt, L.A.; Xavier, R.J. Role of autophagy in the maintenance of intestinal homeostasis. Gastroenterology 2015, 149, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Van Grol, J.; Subauste, C.S. Atg5 but not Atg7 in dendritic cells enhances IL-2 and IFN-γ production by Toxoplasma gondii-reactive CD4+ T cells. Microbes Infect. 2015, 17, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Wéra, O.; Lancellotti, P.; Oury, C. The dual role of neutrophils in inflammatory bowel diseases. Clin. Med. 2016, 5. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, L.; McGovern, D.P.; Hamill, A.M.; Ichikawa, R.; Kanazawa, Y.; Luu, J.; Kumagai, K.; Cilluffo, M.; Fukata, M.; et al. Myeloid ATG16L1 facilitates host-bacteria interactions in maintaining intestinal homeostasis. J. Immunol. 2017, 198, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Chew, T.S.; O’Shea, N.R.; Sewell, G.W.; Oehlers, S.H.; Mulvey, C.M.; Crosier, P.S.; Godovac-Zimmermann, J.; Bloom, S.L.; Smith, A.M.; Segal, A.W. Optineurin deficiency in mice contributes to impaired cytokine secretion and neutrophil recruitment in bacteria-driven colitis. Dis. Model. Mech. 2015, 8, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.; Prescott, N.; Lord, G.M.; MacDonald, T.T.; Powell, N. The unusual suspects--innate lymphoid cells as novel therapeutic targets in IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 271–283. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.E.; Geary, C.D.; Weizman, O.E.; Geiger, T.L.; Rapp, M.; Dorn, G.W., 2nd; Overholtzer, M.; Sun, J.C. Atg5 is essential for the development and survival of innate lymphocytes. Cell Rep. 2016, 15, 1910–1919. [Google Scholar] [CrossRef]
- Wang, S.; Xia, P.; Huang, G.; Zhu, P.; Liu, J.; Ye, B.; Du, Y.; Fan, Z. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat. Commun. 2016, 7, 11023. [Google Scholar] [CrossRef] [PubMed]
- López-Soto, A.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L.; Gonzalez, S. Involvement of autophagy in NK cell development and function. Autophagy 2017, 13, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Heller, F.; Fuss, I.J.; Nieuwenhuis, E.E.; Blumberg, R.S.; Strober, W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity 2002, 17, 629–638. [Google Scholar] [CrossRef]
- Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Salio, M.; Puleston, D.J.; Mathan, T.S.; Shepherd, D.; Stranks, A.J.; Adamopoulou, E.; Veerapen, N.; Besra, G.S.; Hollander, G.A.; Simon, A.K. Essential role for autophagy during invariant NKT cell development. Proc. Natl. Acad. Sci. USA 2014, 111, E5678–E5687. [Google Scholar] [CrossRef] [PubMed]
- Pei, B.; Zhao, M.; Miller, B.C.; Véla, J.L.; Bruinsma, M.W.; Virgin, H.W.; Kronenberg, M. Invariant NKT cells require autophagy to coordinate proliferation and survival signals during differentiation. J. Immunol. 2015, 194, 5872–5884. [Google Scholar] [CrossRef]
- Zhu, L.; Xie, X.; Zhang, L.; Wang, H.; Jie, Z.; Zhou, X.; Shi, J.; Zhao, S.; Zhang, B.; Cheng, X. TBK-binding protein 1 regulates IL-15-induced autophagy and NKT cell survival. Nat. Commun. 2018, 9, 2812. [Google Scholar] [CrossRef]
- Ouellette, A.J.; Hsieh, M.M.; Nosek, M.T.; Cano-Gauci, D.F.; Huttner, K.M.; Buick, R.N.; Selsted, M.E. Mouse Paneth cell defensins: Primary structures and antibacterial activities of numerous cryptdin isoforms. Infect. Immun. 1994, 62, 5040–5047. [Google Scholar]
- Ayabe, T.; Satchell, D.P.; Wilson, C.L.; Parks, W.C.; Selsted, M.E.; Ouellette, A.J. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat. Immunol. 2000, 1, 113–118. [Google Scholar] [CrossRef]
- Bar Shira, E.; Friedman, A. Innate immune functions of avian intestinal epithelial cells: Response to bacterial stimuli and localization of responding cells in the developing avian digestive tract. PLoS ONE 2018, 13, e0200393. [Google Scholar] [CrossRef]
- Cobo, E.R.; Holani, R.; Moreau, F.; Nakamura, K.; Ayabe, T.; Mastroianni, J.R.; Ouellette, A.; Chadee, K. Entamoeba histolytica alters ileal Paneth cell functions in intact and Muc2 mucin deficiency. Infect. Immun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bel, S.; Pendse, M.; Wang, Y.; Li, Y.; Ruhn, K.A.; Hassell, B.; Leal, T.; Winter, S.E.; Xavier, R.J.; Hooper, L.V. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 2017, 357, 1047–1052. [Google Scholar] [CrossRef]
- Stappenbeck, T.S.; McGovern, D.P.B. Paneth cell alterations in the development and phenotype of Crohn’s disease. Gastroenterology 2017, 152, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Niinuma, Y.; Nomura, Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol. Pharm. Bull. 2003, 26, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Fritz, T.; Niederreiter, L.; Adolph, T.; Blumberg, R.S.; Kaser, A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut 2011, 60, 1580–1588. [Google Scholar] [CrossRef]
- Kaser, A.; Blumberg, R.S. ATG16L1 Crohn’s disease risk stresses the endoplasmic reticulum of Paneth cells. Gut 2014, 63, 1038–1039. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matsuzawa-Ishimoto, Y.; Shono, Y.; Gomez, L.E.; Hubbard-Lucey, V.M.; Cammer, M.; Neil, J.; Dewan, M.Z.; Lieberman, S.R.; Lazrak, A.; Marinis, J.M.; et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 2017, 214, 3687–3705. [Google Scholar] [CrossRef]
- Balmus, I.M.; Ciobica, A.; Trifan, A.; Stanciu, C. The implications of oxidative stress and antioxidant therapies in Inflammatory Bowel Disease: Clinical aspects and animal models. Saudi J. Gastroenterol. 2016, 22, 3–17. [Google Scholar] [CrossRef]
- Chen, L.; You, Q.; Hu, L.; Gao, J.; Meng, Q.; Liu, W.; Wu, X.; Xu, Q. The antioxidant procyanidin reduces reactive oxygen species signaling in macrophages and ameliorates experimental colitis in mice. Front. Immunol. 2018, 8, 1910. [Google Scholar] [CrossRef] [PubMed]
- Denson, L.A.; Jurickova, I.; Karns, R.; Shaw, K.A.; Cutler, D.J.; Okou, D.T.; Dodd, A.; Quinn, K.; Mondal, K.; Aronow, B.J.; et al. Clinical and genomic correlates of neutrophil reactive oxygen species production in pediatric patients with Crohn’s disease. Gastroenterology 2018, 154, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, P.; Zhang, S.; Lei, R.; Li, B.; Wu, X.; Jiang, C.; Zhang, X.; Ma, R.; Yang, L.; et al. Oxidative stress-elicited autophagosome accumulation contributes to human neuroblastoma SH-SY5Y cell death induced by PBDE-47. Environ. Toxicol. Pharmacol. 2017, 56, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Ludwig, R.L.; Vousden, K.H. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 20491–20496. [Google Scholar] [CrossRef] [PubMed]
- Mahalingaiah, P.K.; Singh, K.P. Chronic oxidative stress increases growth and tumorigenic potential of MCF-7 breast cancer cells. PLoS ONE 2014, 9, e87371. [Google Scholar] [CrossRef] [PubMed]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 770–775. [Google Scholar] [CrossRef]
- Adolph, T.E.; Mayr, L.; Grabherr, F.; Tilg, H. Paneth cells and their antimicrobials in intestinal immunity. Curr. Pharm. Des. 2018, 24, 1121–1129. [Google Scholar] [CrossRef]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef]
- Sun, J. VDR/vitamin D receptor regulates autophagic activity through ATG16L1. Autophagy 2016, 12, 1057–1058. [Google Scholar] [CrossRef]
- Burger, E.; Araujo, A.; López-Yglesias, A.; Rajala, M.W.; Geng, L.; Levine, B.; Hooper, L.V.; Burstein, E.; Yarovinsky, F.; et al. Loss of Paneth cell autophagy causes acute susceptibility to Toxoplasma gondii-mediated Inflammation. Cell Host Microbe 2018, 23, 177–190. [Google Scholar] [CrossRef]
- Garrett, W.S.; Gordon, J.I.; Glimcher, L.H. Homeostasis and inflammation in the intestine. Cell 2010, 140, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, S.; Lee, J.H.; Kim, J.H.; Che, X.; Ma, H.W.; Seo, D.H.; Kim, T.I.; Kim, W.H.; Kim, S.W.; et al. Lactobacillus acidophilus suppresses intestinal inflammation by inhibiting endoplasmic reticulum stress. J. Gastroenterol. Hepatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ganji-Arjenaki, M.; Rafieian-Kopaei, M. Probiotics are a good choice in remission of inflammatory bowel diseases: A meta analysis and systematic review. J. Cell. Physiol. 2018, 233, 2091–2103. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, P. Goblet cells secretion and mucogenesis. Annu. Rev. Physiol. 1990, 52, 157–176. [Google Scholar] [CrossRef] [PubMed]
- Specian, R.D.; Oliver, M.G. Functional biology of intestinal goblet cells. Am. J. Physiol. 1991, 260, C183–C193. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Beck, P.L.; Inoue, N.; Xavier, R.; Podolsky, D.K. A paradoxical reduction in susceptibility to colonic injury upon targeted transgenic ablation of goblet cells. J. Clin. Investig. 1999, 104, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Loening-Baucke, V.; Herber, A. Mucosal flora in Crohn’s disease and ulcerative colitis–An overview. J. Physiol. Pharmacol. 2009, 60, 61–71. [Google Scholar] [PubMed]
- Patel, K.K.; Miyoshi, H.; Beatty, W.L.; Head, R.D.; Malvin, N.P.; Cadwell, K.; Guan, J.L.; Saitoh, T.; Akira, S.; Seglen, P.O.; et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013, 32, 3130–3144. [Google Scholar] [CrossRef]
- Johansson, M.E.; Hansson, G.C. Is the intestinal goblet cell a major immune cell? Cell Host Microbe 2014, 15, 251–252. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Gagné-Sansfaçon, J.; Allaire, J.M.; Jones, C.; Boudreau, F.; Perreault, N. Loss of Sonic hedgehog leads to alterations in intestinal secretory cell maturation and autophagy. PLoS ONE 2014, 9, e98751. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Q.; Zang, Y.; Zhao, Y.; Liu, N.; Wang, Y.; Xu, X.; Liu, L.; Mei, Q. Apple Polysaccharide inhibits microbial dysbiosis and chronic inflammation and modulates gut permeability in HFD-fed rats. Int. J. Biol. Macromol. 2017, 99, 282–292. [Google Scholar] [CrossRef] [PubMed]
- El-Khider, F.; McDonald, C. Links of Autophagy Dysfunction to Inflammatory Bowel Disease Onset. Dig. Dis. 2016, 34, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.; Shao, B.Z.; Xu, Z.Q.; Chen, X.W.; Liu, C. Intestinal Autophagy and Its Pharmacological Control in Inflammatory Bowel Disease. Front. Immunol. 2017, 7, 695. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iida, T.; Yokoyama, Y.; Wagatsuma, K.; Hirayama, D.; Nakase, H. Impact of Autophagy of Innate Immune Cells on Inflammatory Bowel Disease. Cells 2019, 8, 7. https://doi.org/10.3390/cells8010007
Iida T, Yokoyama Y, Wagatsuma K, Hirayama D, Nakase H. Impact of Autophagy of Innate Immune Cells on Inflammatory Bowel Disease. Cells. 2019; 8(1):7. https://doi.org/10.3390/cells8010007
Chicago/Turabian StyleIida, Tomoya, Yoshihiro Yokoyama, Kohei Wagatsuma, Daisuke Hirayama, and Hiroshi Nakase. 2019. "Impact of Autophagy of Innate Immune Cells on Inflammatory Bowel Disease" Cells 8, no. 1: 7. https://doi.org/10.3390/cells8010007
APA StyleIida, T., Yokoyama, Y., Wagatsuma, K., Hirayama, D., & Nakase, H. (2019). Impact of Autophagy of Innate Immune Cells on Inflammatory Bowel Disease. Cells, 8(1), 7. https://doi.org/10.3390/cells8010007