Retinal Neuron Is More Sensitive to Blue Light-Induced Damage than Glia Cell Due to DNA Double-Strand Breaks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Ethics Statement
2.2. In Vitro Blue Light Exposure
2.3. Cell Treatment
2.4. In Vivo Blue Light Exposure
2.5. Immunofluorescence Assay
2.6. Cell Counting kit-8 (CCK-8) Assay
2.7. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
2.8. Western Blotting
2.9. Statistical Analysis
3. Results
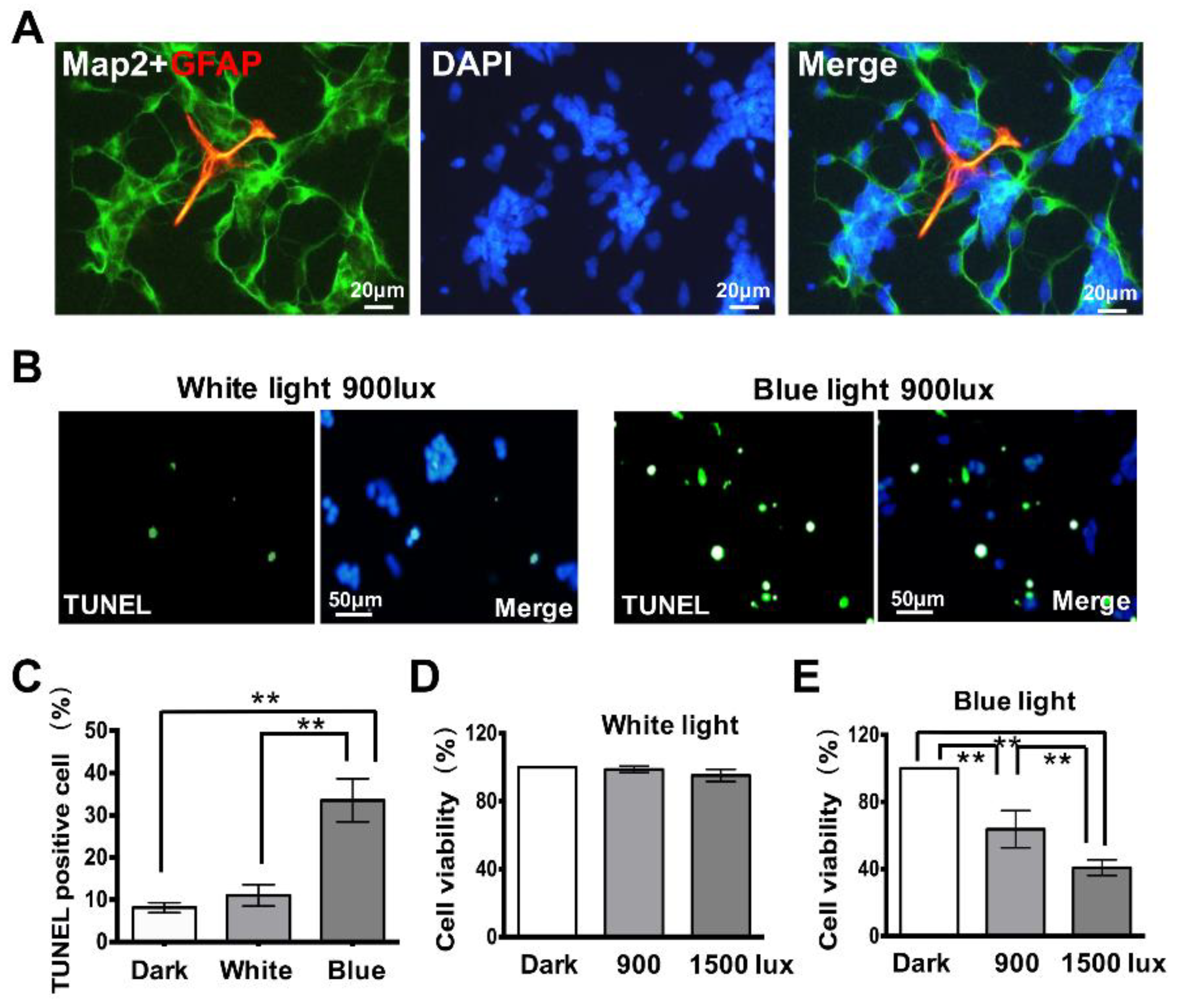
3.1. Exposure to Blue Light Induces Cell Apoptosis in Retinal Neurocytes
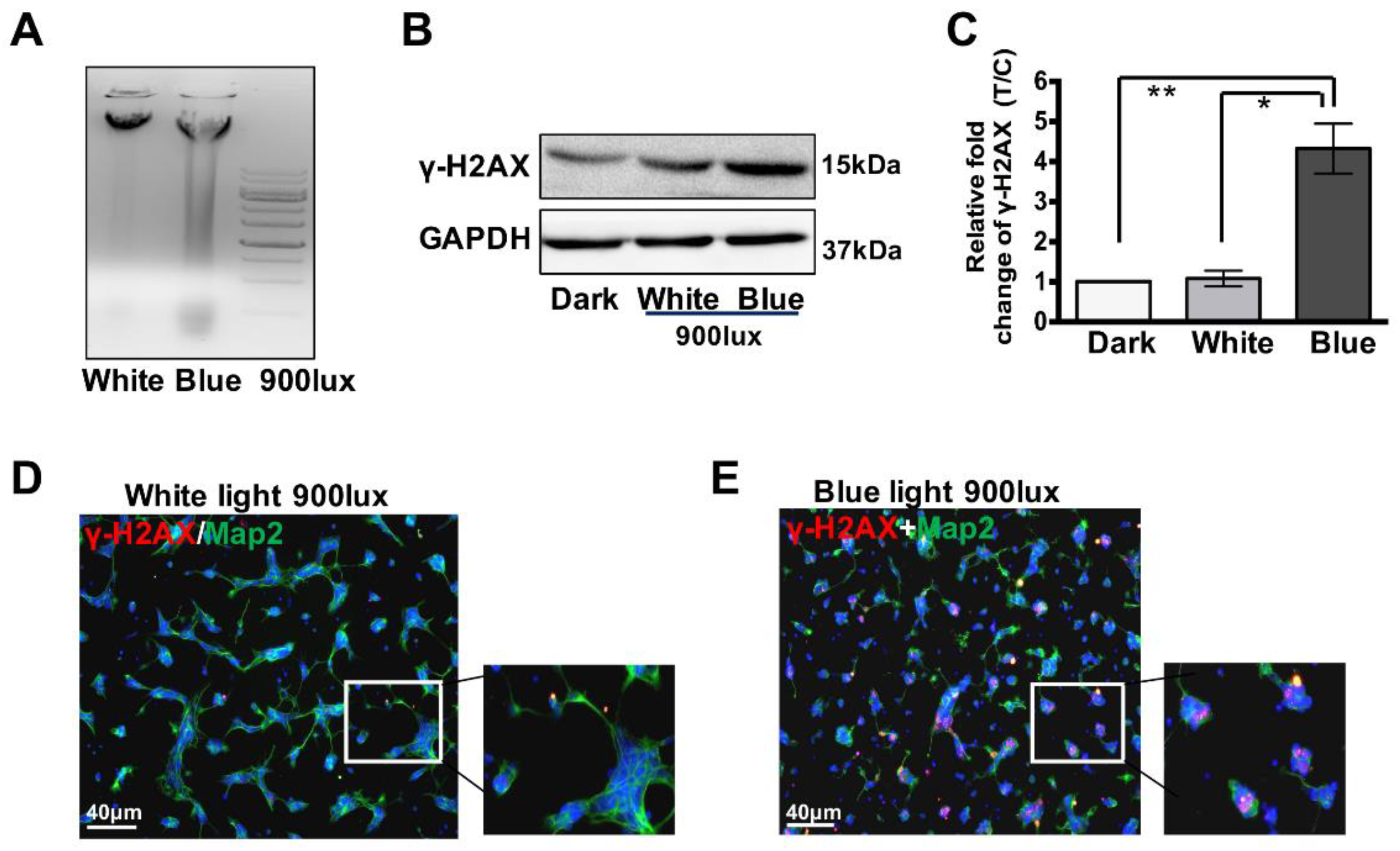
3.2. Blue Light Induces DNA Double-Strand Breaks (DSBs) in Retinal Neurocytes
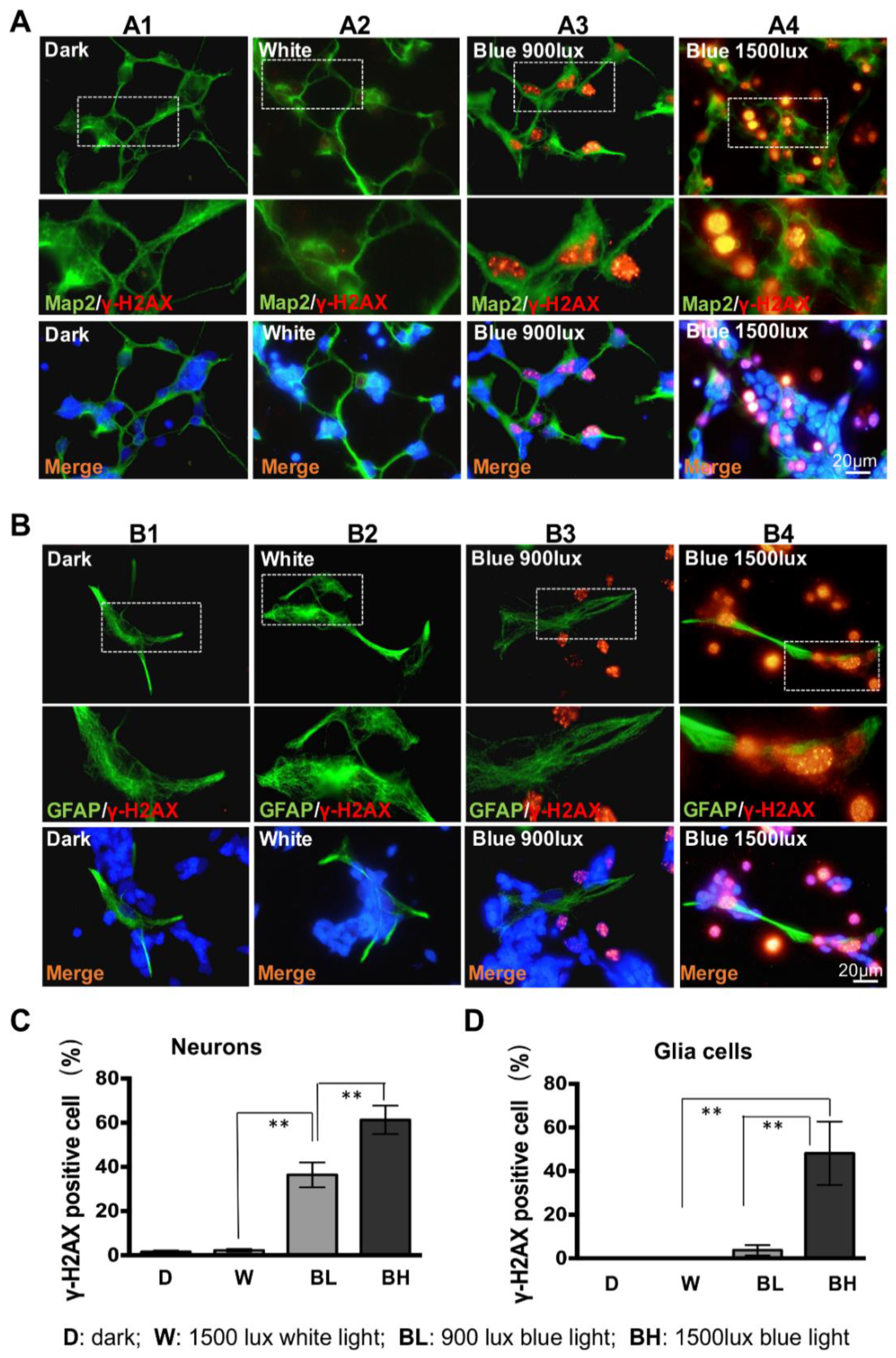
3.3. Retinal Neuron Is More Sensitive to Blue Light Exposure than Glia Cells
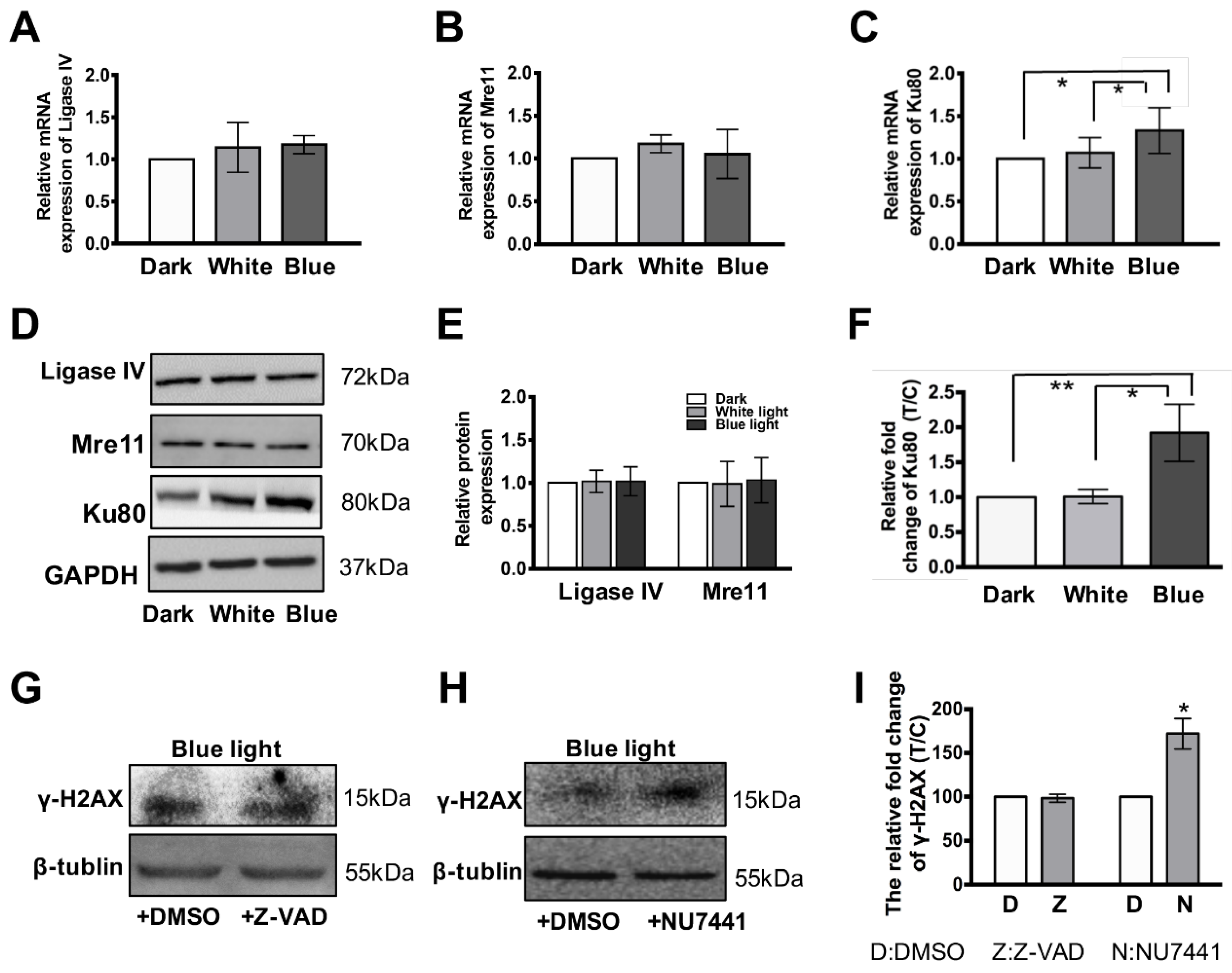
3.4. Ku80 Is Up-Regulated in Glia Cells but not in Retinal Neurons
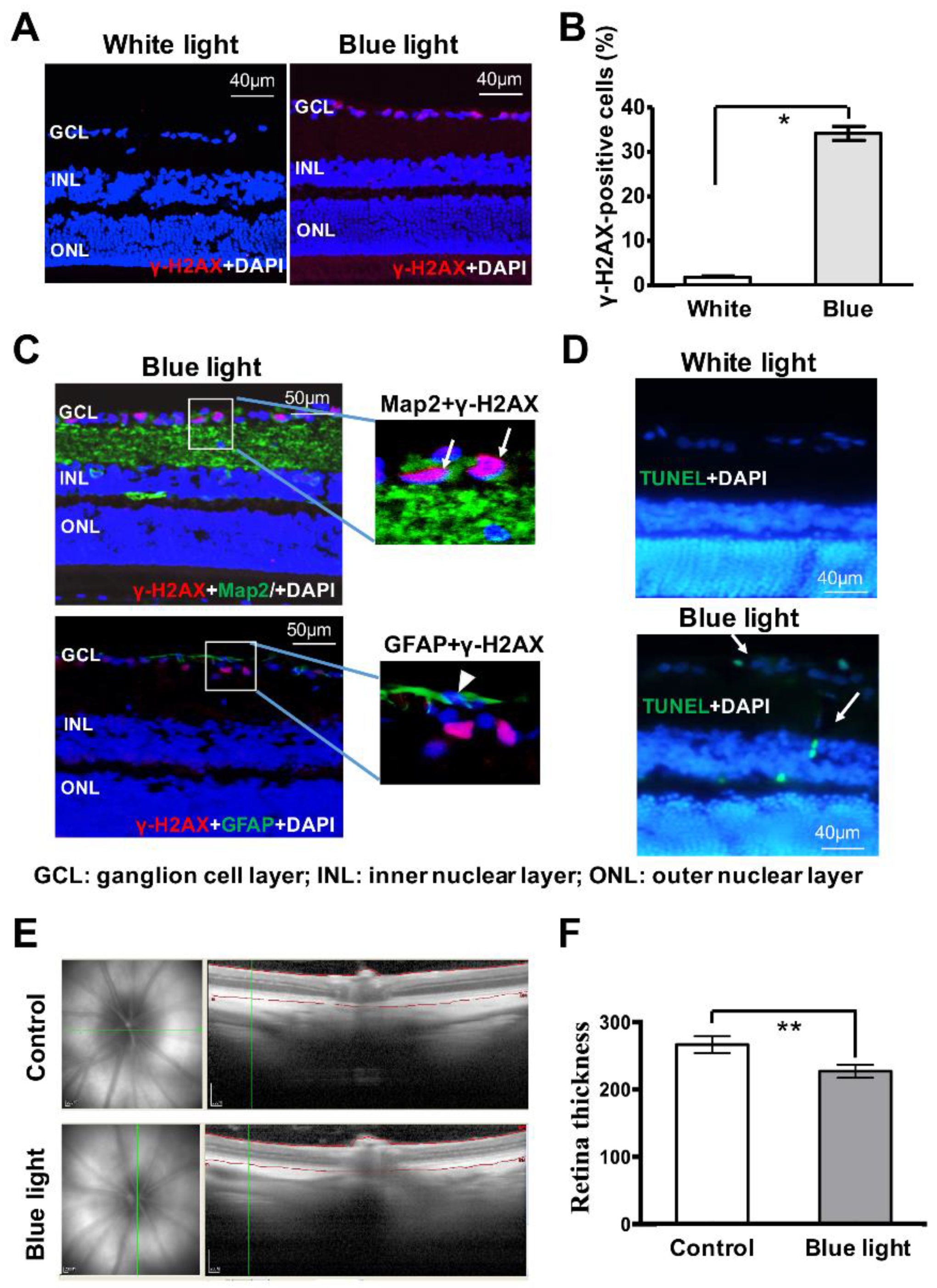
3.5. Blue Light Induces Damages in Retina In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Grimm, C.; Wenzel, A.; Williams, T.; Rol, P.; Hafezi, F.; Remé, C. Rhodopsin-mediated blue-light damage to the rat retina: Effect of photoreversal of bleaching. Investig. Ophthalmol. Vis. Sci. 2001, 42, 497–505. [Google Scholar]
- Foulds, W.S.; Barathi, V.A.; Luu, C.D. Progressive myopia or hyperopia can be induced in chicks and reversed by manipulation of the chromaticity of ambient light. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8004–8012. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schumann, U.; Ader, M.; Knels, L.; Funk, R.H. Influence of blue light on photoreceptors in a live retinal explant system. Mol. Vis. 2011, 17, 876–884. [Google Scholar] [PubMed]
- Narimatsu, T.; Ozawa, Y.; Miyake, S.; Kubota, S.; Yuki, K.; Nagai, N.; Tsubota, K. Biological effects of blocking blue and other visible light on the mouse retina. Clin. Exp. Ophthalmol. 2014, 42, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Cai, B. Blue light-induced apoptosis of A2E-containing RPE: Involvement of caspase-3 and protection by Bcl-2. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1356–1362. [Google Scholar]
- Downie, L.E.; Busija, L.; Keller, P.R. Blue-light filtering intraocular lenses (IOLs) for protecting macular health. Cochrane Database Syst. Rev. 2018, 5, CD011977. [Google Scholar] [CrossRef] [PubMed]
- Algvere, P.V.; Marshall, J.; Seregard, S. Age-related maculopathy and the impact of blue light hazard. Acta Ophthalmol. Scand. 2006, 84, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Del Olmo-Aguado, S.; Manso, A.G.; Osborne, N.N. Light might directly affect retinal ganglion cell mitochondria to potentially influence function. Photochem. Photobiol. 2012, 88, 1346–1355. [Google Scholar] [CrossRef]
- Knels, L.; Valtink, M.; Roehlecke, C.; Lupp, A.; de la Vega, J.; Mehner, M.; Funk, R.H. Blue light stress in retinal neurocytesal (R28) cells is dependent on wavelength range and irradiance. Eur. J. Neurosci. 2011, 34, 548–558. [Google Scholar] [CrossRef]
- Nakanishi-Ueda, T.; Majima, H.J.; Watanabe, K.; Ueda, T.; Indo, H.P.; Suenaga, S.; Hisamitsu, T.; Ozawa, T.; Yasuhara, H.; Koide, R. Blue LED light exposure develops intracellular reactive oxygen species, lipid peroxidation, and subsequent cellular injuries in cultured bovine retinal pigment epithelial cells. Free Radic Res. 2013, 47, 774–880. [Google Scholar] [CrossRef]
- Bennet, D.; Kim, M.G.; Kim, S. Light-induced anatomical alterations in retinal cells. Anal. Biochem. 2013, 436, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.Y.; Chou, B.R.; Cullen, A.P.; Sivak, J.G. Effects of 400 nm, 420 nm, and 435.8 nm radiations on cultured human retinal pigment epithelial cells. J. Photochem. Photobiol. B 2009, 95, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, A.; Grimm, C.; Samardzija, M.; Remé, C.E. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog. Retin. Eye Res. 2005, 24, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Kuse, Y.; Ogawa, K.; Tsuruma, K.; Shimazawa, M.; Hara, H. Damage of photoreceptor-derived cells in culture induced by light emitting diode-derived blue light. Sci. Rep. 2014, 4, 5223. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, P.; Wang, W.; Xu, Y.; Wang, M.; Chen, X.; Dong, X. Long-term blue light exposure induces RGC-5 cell death in vitro: Involvement of mitochondria-dependent apoptosis, oxidative stress, and MAPK signaling pathways. Apoptosis 2014, 19, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.; Kamalden, T.A.; del Olmo-Aguado, S.; Osborne, N.N. Light- and sodium azide-induced death of RGC-5 cells in culture occurs via different mechanisms. Apoptosis 2011, 16, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Szaflik, J.P.; Janik-Papis, K.; Synowiec, E.; Ksiazek, D.; Zaras, M.; Wozniak, K.; Szaflik, J.; Blasiak, J. DNA damage and repair in age-related macular degeneration. Mutat. Res. 2009, 669, 169–176. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA Repair Deficiency in Neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef]
- Barzilai, A.; Biton, S.; Shiloh, Y. The role of the DNA damage response in neurocytesal development, organization and maintenance. DNA Repair (Amst). 2008, 7, 1010–1027. [Google Scholar] [CrossRef]
- Zhuang, J.; Li, F.; Liu, X.; Li, F.; Liu, X.; Liu, Z.; Lin, J.; Ge, Y.; Kaminski, J.M.; Summers, J.B.; et al. Lithium chloride protects retinal neurocytes from nutrient deprivation by promoting DNA non-homologous end-joining. Biochem. Biophys. Res. Commun. 2009, 380, 650–654. [Google Scholar] [CrossRef]
- Sasaki, M.; Yuki, K.; Kurihara, T.; Noda, K.; Kobayashi, S.; Ishida, S.; Tsubota, K.; Ozawa, Y. Biological role of lutein in the light-induced retinal degeneration. J. Nutr. Biochem. 2012, 23, 423–429. [Google Scholar]
- Gordon, W.C.; Casey, D.M.; Lukiw, W.J.; Bazan, N.G. DNA damage and repair in light-induced photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3511–3521. [Google Scholar]
- Osborne, N.N.; Li, G.Y.; Ji, D.; Mortiboys, H.J.; Jackson, S. Light affects mitochondria to cause apoptosis to cultured cells: Possible relevance to ganglion cell death in certain optic neuropathies. J. Neurochem. 2008, 105, 2013–2028. [Google Scholar] [CrossRef]
- Marc, R.E.; Jones, B.W.; Watt, C.B.; Vazquez-Chona, F.; Vaughan, D.K.; Organisciak, D.T. Extreme retinal remodeling triggered by light damage: Implications for age related macular degeneration. Mol. Vis. 2008, 14, 782–806. [Google Scholar]
- Wu, J.; Seregard, S.; Algvere, P.V. Photochemical damage of the retina. Surv. Ophthalmol. 2006, 51, 461–481. [Google Scholar] [CrossRef]
- Roehlecke, C.; Schaller, A.; Knels, L.; Funk, R.H. The influence of sublethal blue light exposure on human RPE cells. Mol. Vis. 2009, 15, 1929–1938. [Google Scholar]
- Geiger, P.; Barben, M.; Grimm, C.; Samardzija, M. Blue light-induced retinal lesions, intraretinal vascular leakage and edema formation in the all-cone mouse retina. Cell Death Dis. 2015, 6, e1985. [Google Scholar] [CrossRef]
- Shang, Y.M.; Wang, G.S.; Sliney, D.; Yang, C.H.; Lee, L.L. White light-emitting diodes (LEDs) at domestic lighting levels and retinal injury in a rat model. Environ. Health Perspect. 2014, 122, 269–276. [Google Scholar] [CrossRef]
- Chen, P.; Hu, H.; Chen, Z.; Cai, X.; Zhang, Z.; Yang, Y.; Yu, N.; Zhang, J.; Xia, L.; Ge, J.; et al. BRCA1 silencing is associated with failure of DNA repairing in retinal neurocytes. PLoS ONE 2014, 9, e99371. [Google Scholar]
- Solier, S.; Pommier, Y. The apoptotic ring: A novel entity with phosphorylated histones H2AX and H2B and activated DNA damage response kinases. Cell Cycle 2009, 8, 1853–1859. [Google Scholar] [CrossRef]
- Khan, C.; Muliyil, S.; Ayyub, C.; Rao, B.J. The initiator caspase Dronc plays a non-apoptotic role in promoting DNA damage signalling in, D. melanogaster. J. Cell Sci. 2017, 130, 2984–2995. [Google Scholar] [CrossRef]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Hörl, D.; Chen, W.; et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nogawa, S.; Ito, D. Activated phosphorylation of cyclic AMP response element binding protein is associated with preservation of striatal neurocytess after focal cerebral ischemia in the rat. Neuroscience 2000, 100, 345–354. [Google Scholar] [CrossRef]
- Blomgren, K.; Hagberg, H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic. Biol. Med. 2006, 40, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Noshita, N.; Lewén, A.; Kim, G.W.; Chan, P.H. Neurocytesal expression of the DNA repair protein Ku 70 after ischemic preconditioning corresponds to tolerance to global cerebral ischemia. Stroke 2001, 32, 2388–2393. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lee, K.J.; Davis, A.J.; Chen, D.J. Human Ku70/80 protein blocks exonuclease 1-mediated DNA resection in the presence of human Mre11 or Mre11/Rad50 protein complex. J. Biol. Chem. 2012, 287, 4936–4945. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, P.; Lai, Z.; Wu, Y.; Xu, L.; Cai, X.; Qiu, J.; Yang, P.; Yang, M.; Zhou, P.; Zhuang, J.; et al. Retinal Neuron Is More Sensitive to Blue Light-Induced Damage than Glia Cell Due to DNA Double-Strand Breaks. Cells 2019, 8, 68. https://doi.org/10.3390/cells8010068
Chen P, Lai Z, Wu Y, Xu L, Cai X, Qiu J, Yang P, Yang M, Zhou P, Zhuang J, et al. Retinal Neuron Is More Sensitive to Blue Light-Induced Damage than Glia Cell Due to DNA Double-Strand Breaks. Cells. 2019; 8(1):68. https://doi.org/10.3390/cells8010068
Chicago/Turabian StyleChen, Pei, Zhipeng Lai, Yihui Wu, Lijun Xu, Xiaoxiao Cai, Jin Qiu, Panyang Yang, Meng Yang, Pan Zhou, Jiejie Zhuang, and et al. 2019. "Retinal Neuron Is More Sensitive to Blue Light-Induced Damage than Glia Cell Due to DNA Double-Strand Breaks" Cells 8, no. 1: 68. https://doi.org/10.3390/cells8010068
APA StyleChen, P., Lai, Z., Wu, Y., Xu, L., Cai, X., Qiu, J., Yang, P., Yang, M., Zhou, P., Zhuang, J., Ge, J., Yu, K., & Zhuang, J. (2019). Retinal Neuron Is More Sensitive to Blue Light-Induced Damage than Glia Cell Due to DNA Double-Strand Breaks. Cells, 8(1), 68. https://doi.org/10.3390/cells8010068