Adoptive T Cell Therapy Strategies for Viral Infections in Patients Receiving Haematopoietic Stem Cell Transplantation

 , ,
, , 
Abstract
1. Introduction
2. Refractory Viral Infections Following HSCT
3. From DLI to Virus-Specific T Cells and Beyond
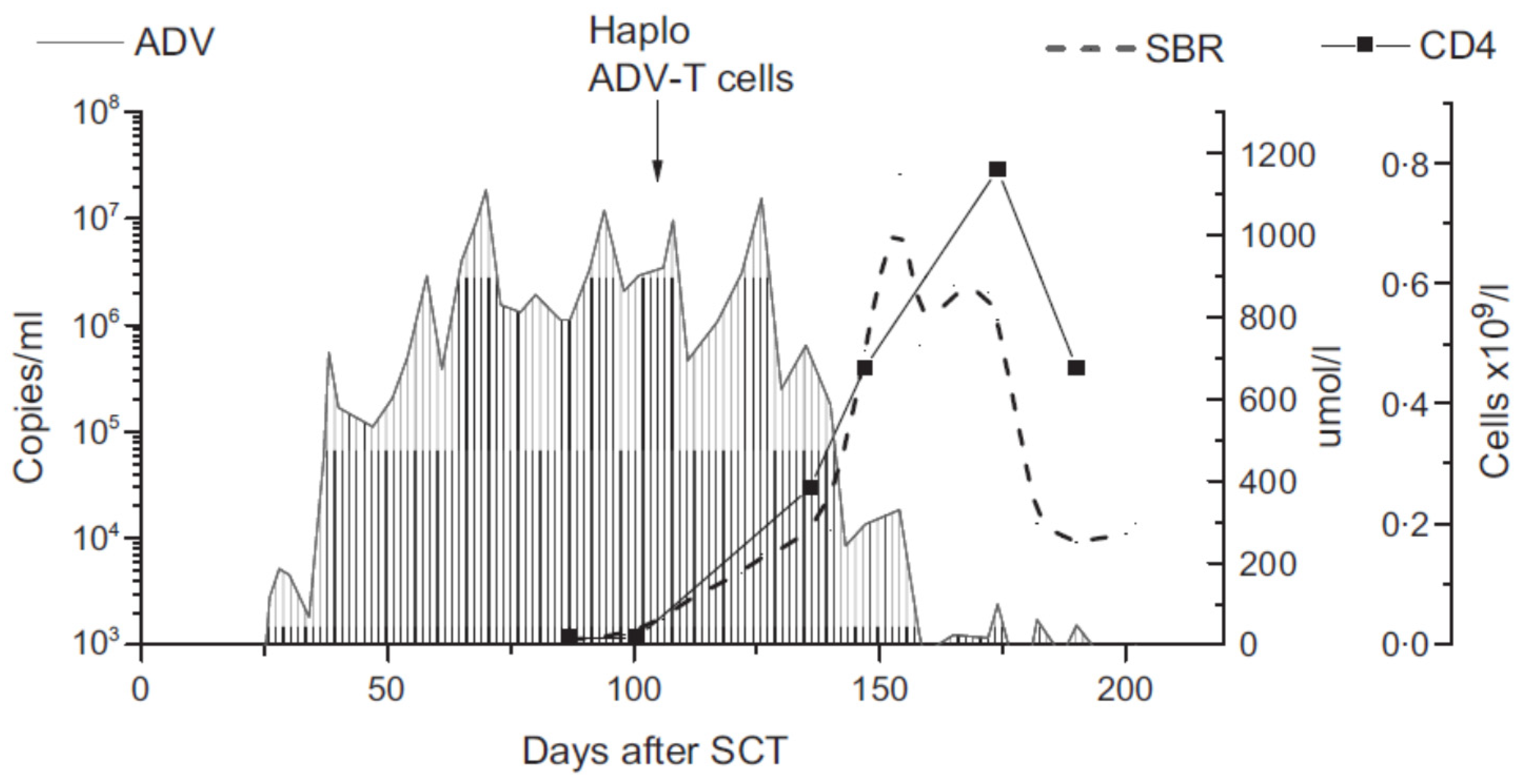
4. Efficacy of Donor-Derived T Cells: Promising Potential and Pitfalls
5. Third Party CTLs
6. Conclusions
Funding
Conflicts of Interest
References
- Hiwarkar, P.; Gaspar, H.B.; Gilmour, K.; Jagani, M.; Chiesa, R.; Bennett-Rees, N.; Breuer, J.; Rao, K.; Cale, C.; Goulden, N.; et al. Impact of viral reactivations in the era of pre-emptive antiviral drug therapy following allogeneic haematopoietic SCT in paediatric recipients. Bone Marrow Transplant. 2013, 48, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Lazzarotto, T.; Chiereghin, A.; Piralla, A.; Piccirilli, G.; Girello, A.; Campanini, G.; Gabrielli, L.; Costa, C.; Prete, A.; Bonifazi, F.; et al. Cytomegalovirus and Epstein-Barr virus DNA kinetics in whole blood and plasma of allogeneic hematopoietic stem cell transplantation recipients. Biol. Blood Marrow Transplant. 2018, 24, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Rustia, E.; Violago, L.; Jin, Z.; Foca, M.D.; Kahn, J.M.; Arnold, S.; Sosna, J.; Bhatia, M.; Kung, A.L.; George, D.; et al. Risk factors and utility of a risk-based algorithm for monitoring cytomegalovirus, Epstein-Barr virus, and adenovirus infections in pediatric recipients after allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2016, 22, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Atay, D.; Akcay, A.; Erbey, F.; Ozturk, G. The impact of alternative donor types on viral infections in pediatric hematopoietic stem cell transplantation. Pediatr. Transplant. 2018, 22, e13109. [Google Scholar] [CrossRef] [PubMed]
- Sedláček, P.; Petterson, T.; Robin, M.; Sivaprakasam, P.; Vainorius, E.; Brundage, T.; Chandak, A.; Mozaffari, E.; Nichols, G.; Voigt, S. Incidence of adenovirus infection in hematopoietic stem cell transplant recipients: Findings from the AdVance study. Biol. Blood Marrow Transplant. 2018. [Google Scholar] [CrossRef]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Matthes-Martin, S.; Feuchtinger, T.; Shaw, P.J.; Engelhard, D.; Hirsch, H.H.; Cordonnier, C.; Ljungman, P. The fourth European conference on infections in Leukemia European guidelines for diagnosis and treatment of adenovirus infection in leukemia and stem cell transplantation: Summary of ECIL-4 (2011). Transpl. Infect. Dis. 2012, 14, 555–563. [Google Scholar] [CrossRef]
- Sivaprakasam, P.; Carr, T.F.; Coussons, M.; Khalid, T.; Bailey, A.S.; Guiver, M.; Mutton, K.J.; Turner, A.J.; Grainger, J.D.; Wynn, R.F. Improved outcome from invasive adenovirus infection in pediatric patients after hemopoietic stem cell transplantation using intensive clinical surveillance and early intervention. J. Pediatr. Hematol. Oncol. 2007, 29, 81–85. [Google Scholar] [CrossRef]
- Walls, T.; Hawrami, K.; Ushiro-Lumb, I.; Shingadia, D.; Saha, V.; Shankar, A.G. Adenovirus infection after pediatric bone marrow transplantation: Is treatment always necessary? Clin. Infect. Dis. 2005, 40, 1244–1249. [Google Scholar] [CrossRef]
- Paolino, K.; Sande, J.; Perez, E.; Loechelt, B.; Jantausch, B.; Painter, W.; Anderson, M.; Tippin, T.; Lanier, E.R.; Fry, T.; et al. Eradication of disseminated adenovirus infection in a pediatric hematopoietic stem cell transplantation recipient using the novel antiviral agent CMX001. J. Clin. Virol. 2011, 50, 167–170. [Google Scholar] [CrossRef]
- Hiwarkar, P.; Amrolia, P.; Sivaprakasam, P. Brincidofovir is highly efficacious in controlling adenoviremia in pediatric recipients of hematopoietic cell transplant. Blood 2017, 129, 2033–2037. [Google Scholar] [CrossRef]
- George, B.; Pati, N.; Gilroy, N.; Ratnamohan, M.; Huang, G.; Kerridge, I.; Hertzberg, M.; Gottlieb, D.; Bradstock, K. Pre-transplant cytomegalovirus (CMV) serostatus remains the most important determinant of CMV reactivation after allogeneic hematopoietic stem cell transplantation in the era of surveillance and preemptive therapy: CMV reactivation in HSCT. Transpl. Infect. Dis. 2010, 12, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Boeckh, M.; Murphy, W.J.; Peggs, K.S. Recent advances in Cytomegalovirus: An update on pharmacologic and cellular therapies. Biol. Blood Marrow Transplant. 2015, 21, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Atay, D.; Erbey, F.; Akcay, A.; Dag, A.; Ozturk, G. Oral valganciclovir as preemptive therapy for cytomegalovirus reactivation in pediatric hematopoietic stem cell transplant patients. J. Pediatr. Hematol. Oncol. 2015, 37, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Barkam, C.; Kamal, H.; Dammann, E.; Diedrich, H.; Buchholz, S.; Eder, M.; Krauter, J.; Ganser, A.; Stadler, M. Improving safety of preemptive therapy with oral valganciclovir for cytomegalovirus infection after allogeneic hematopoietic stem cell transplantation. Bone Marrow Res. 2012, 2012, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sellar, R.S.; Peggs, K.S. Therapeutic strategies for cytomegalovirus infection in haematopoietic transplant recipients: A focused update. Expert Opin. Biol. Ther. 2014, 14, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Styczynski, J.; van der Velden, W.; Fox, C.P.; Engelhard, D.; de la Camara, R.; Cordonnier, C.; Ljungman, P. Management of Epstein-Barr virus infections and post-transplant lymphoproliferative disorders in patients after allogeneic hematopoietic stem cell transplantation: Sixth European Conference on Infections in Leukemia (ECIL-6) guidelines. Haematologica 2016, 101, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Kapp, M.; Einsele, H.; Mielke, S. EBV-induced post transplant lymphoproliferative disorders: A persisting challenge in allogeneic hematopoetic SCT. Bone Marrow Transplant. 2014, 49, 163–167. [Google Scholar] [CrossRef]
- Quintela, A.; Escuret, V.; Roux, S.; Bonnafous, P.; Gilis, L.; Barraco, F.; Labussière-Wallet, H.; Duscastelle-Leprêtre, S.; Nicolini, F.-E.; Thomas, X.; et al. HHV-6 infection after allogeneic hematopoietic stem cell transplantation: From chromosomal integration to viral co-infections and T-cell reconstitution patterns. J. Infect. 2016, 72, 214–222. [Google Scholar] [CrossRef]
- Janeczko, M.; Mielcarek, M.; Rybka, B.; Ryczan-Krawczyk, R.; Noworolska-Sauren, D.; Kałwak, K. Immune recovery and the risk of CMV/ EBV reactivation in children post allogeneic haematopoietic stem cell transplantation. Cent. Eur. J. Immunol. 2016, 3, 287–296. [Google Scholar] [CrossRef]
- Willemsen, L.; Jol-van der Zijde, C.M.; Admiraal, R.; Putter, H.; Jansen-Hoogendijk, A.M.; Ostaijen-ten Dam, M.M.; Wijnen, J.T.; van Kesteren, C.; Waaijer, J.L.M.; Lankester, A.C.; et al. Impact of serotherapy on immune reconstitution and survival outcomes after stem cell transplantations in children: Thymoglobulin versus alemtuzumab. Biol. Blood Marrow Transplant. 2015, 21, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Park, B.G.; Park, C.-J.; Jang, S.; Chi, H.-S.; Kim, D.-Y.; Lee, J.-H.; Lee, J.-H.; Lee, K.-H. Reconstitution of lymphocyte subpopulations after hematopoietic stem cell transplantation: Comparison of hematologic malignancies and donor types in event-free patients. Leuk. Res. 2015, 39, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Lindemans, C.A.; Chiesa, R.; Amrolia, P.J.; Rao, K.; Nikolajeva, O.; de Wildt, A.; Gerhardt, C.E.; Gilmour, K.C.; B. Bierings, M.; Veys, P.; et al. Impact of thymoglobulin prior to pediatric unrelated umbilical cord blood transplantation on immune reconstitution and clinical outcome. Blood 2014, 123, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Solano, C.; Talaya, A.; Giménez, E.; Albert, E.; Piñana, J.L.; Hernández-Boluda, J.C.; Pérez, A.; Navarro, D. Refractory cytomegalovirus DNAemia after allogeneic hematopoietic stem cell transplantation: When should genotypic drug resistance testing be requested? Bone Marrow Transplant. 2018, 53, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, E.; Ladanyi, M.; Emanuel, D.; Mackninnon, S.; Boulad, F.; Caraasi, M. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogenic bone marrow transplantation. N. Engl. J. Med. 1994, 330, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Hromas, R.; Cornetta, K.; Srour, E. Donor leukocyte infusion as therapy of life-threatening adenoviral infections after T-cell-depleted bone marrow transplantation. Blood 1994, 84, 1689–1690. [Google Scholar] [PubMed]
- Witt, V.; Fritsch, G.; Peters, C.; Matthes-Martin, S.; Ladenstein, R.; Gadner, H. Resolution of early cytomegalovirus (CMV) infection after leukocyte transfusion therapy from a CMV seropositive donor. Bone Marrow Transplant. 1998, 22, 289–292. [Google Scholar] [CrossRef]
- Fehse, B.; Goldmann, M.; Frerk, O.; Bulduk, M.; Zander, A. Depletion of alloreactive donor T cells using immunomagnetic cell selection. Bone Marrow Transplant. 2000, 25, S39–S42. [Google Scholar] [CrossRef]
- Walter, E.A.; Finch, R.J.; Watanabe, K.S.; Thomas, E.D.; Riddell, S.R. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-Cell clones from the donor. N. Engl. J. Med. 1995, 7, 1038–1044. [Google Scholar] [CrossRef]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.C.; Loftin, S.K.; Sixbey, J.W.; Gan, Y.; Srivastava, D.-K.; Bowman, L.C.; Krance, R.A.; Brenner, M.K.; et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus–Induced Lymphoma in allogeneic transplant recipients. Blood 1998, 8, 1549–1555. [Google Scholar]
- Hanley, P.J.; Shaffer, D.R.; Cruz, C.R.Y.; Ku, S.; Tzou, B.; Liu, H.; Demmler-Harrison, G.; Heslop, H.E.; Rooney, C.M.; Gottschalk, S.; et al. Expansion of T cells targeting multiple antigens of cytomegalovirus, Epstein–Barr virus and adenovirus to provide broad antiviral specificity after stem cell transplantation. Cytotherapy 2011, 13, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Leen, A.M.; Myers, G.D.; Sili, U.; Huls, M.H.; Weiss, H.; Leung, K.S.; Carrum, G.; Krance, R.A.; Chang, C.-C.; Molldrem, J.J.; et al. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat. Med. 2006, 12, 1160–1166. [Google Scholar] [CrossRef]
- Cobbold, M.; Khan, N.; Pourgheysari, B.; Tauro, S.; McDonald, D.; Osman, H.; Assenmacher, M.; Billingham, L.; Steward, C.; Crawley, C.; et al. Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA–peptide tetramers. J. Exp. Med. 2005, 202, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Thomson, K.; Samuel, E.; Dyer, G.; Armoogum, J.; Chakraverty, R.; Pang, K.; Mackinnon, S.; Lowdell, M.W. Directly selected cytomegalovirus-reactive donor T cells confer rapid and safe systemic reconstitution of virus-specific immunity following stem cell transplantation. Clin. Infect. Dis. 2011, 52, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Heslop, H.E.; Slobod, K.S.; Pule, M.A.; Hale, G.A.; Rousseau, A.; Smith, C.A.; Bollard, C.M.; Liu, H.; Wu, M.-F.; Rochester, R.J.; et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010, 115, 12. [Google Scholar] [CrossRef]
- Blyth, E.; Clancy, L.; Simms, R.; Ma, C.K.K.; Burgess, J.; Deo, S.; Byth, K.; Dubosq, M.-C.; Shaw, P.J.; Micklethwaite, K.P.; et al. Donor-derived CMV-specific T cells reduce the requirement for CMV-directed pharmacotherapy after allogeneic stem cell transplantation. Blood 2013, 121, 3745–3758. [Google Scholar] [CrossRef]
- Ip, W.; Silva, J.M.F.; Gaspar, H.; Mitra, A.; Patel, S.; Rao, K.; Chiesa, R.; Amrolia, P.; Gilmour, K.; Ahsan, G.; et al. Multicenter phase 1/2 application of adenovirus-specific T cells in high-risk pediatric patients after allogeneic stem cell transplantation. Cytotherapy 2018, 20, 830–838. [Google Scholar] [CrossRef]
- Icheva, V.; Kayser, S.; Wolff, D.; Tuve, S.; Kyzirakos, C.; Bethge, W.; Greil, J.; Albert, M.H.; Schwinger, W.; Nathrath, M.; et al. Adoptive transfer of Epstein-Barr Virus (EBV) nuclear antigen 1–specific T cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J. Clin. Oncol. 2013, 31, 39–48. [Google Scholar] [CrossRef]
- Feucht, J.; Opherk, K.; Lang, P.; Kayser, S.; Hartl, L.; Bethge, W.; Matthes-Martin, S.; Bader, P.; Albert, M.H.; Maecker-Kolhoff, B.; et al. Adoptive T-cell therapy with hexon-specific Th1 cells as a treatment of refractory adenovirus infection after HSCT. Blood 2015, 125, 1986–1994. [Google Scholar] [CrossRef]
- Blyth, E.; Clancy, L.; Simms, R.; Gaundar, S.; O′Connell, P.; Micklethwaite, K.; Gottlieb, D.J. BK Virus-specific T cells for use in cellular therapy show specificity to multiple antigens and polyfunctional cytokine responses. Transplantation 2011, 92, 1077–1084. [Google Scholar] [CrossRef]
- Mani, J.; Jin, N.; Schmitt, M. Cellular immunotherapy for patients with reactivation of JC and BK polyomaviruses after transplantation. Cytotherapy 2014, 16, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Balduzzi, A.; Lucchini, G.; Hirsch, H.H.; Basso, S.; Cioni, M.; Rovelli, A.; Zincone, A.; Grimaldi, M.; Corti, P.; Bonanomi, S.; et al. Polyomavirus JC-targeted T-cell therapy for progressive multiple leukoencephalopathy in a hematopoietic cell transplantation recipient. Bone Marrow Transplant. 2011, 46, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, A.; Gerdemann, U.; Katari, U.L.; Tzannou, I.; Liu, H.; Martinez, C.; Leung, K.; Carrum, G.; Gee, A.P.; Vera, J.F.; et al. Activity of broad-spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci. Transl. Med. 2014, 6, 242ra83. [Google Scholar] [CrossRef] [PubMed]
- Sili, U.; Leen, A.M.; Vera, J.F.; Gee, A.P.; Huls, H.; Heslop, H.E.; Bollard, C.M.; Rooney, C.M. Production of good manufacturing practice-grade cytotoxic T lymphocytes specific for Epstein–Barr virus, cytomegalovirus and adenovirus to prevent or treat viral infections post-allogeneic hematopoietic stem cell transplant. Cytotherapy 2012, 14, 7–11. [Google Scholar] [CrossRef]
- Leen, A.M.; Christin, A.; Myers, G.D.; Liu, H.; Cruz, C.R.; Hanley, P.J.; Kennedy-Nasser, A.A.; Leung, K.S.; Gee, A.P.; Krance, R.A.; et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood 2009, 114, 4283–4292. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.K.K.; Blyth, E.; Clancy, L.; Simms, R.; Burgess, J.; Brown, R.; Deo, S.; Micklethwaite, K.P.; Gottlieb, D.J. Addition of varicella zoster virus–specific T cells to cytomegalovirus, Epstein-Barr virus and adenovirus tri-specific T cells as adoptive immunotherapy in patients undergoing allogeneic hematopoietic stem cell transplantation. Cytotherapy 2015, 17, 1406–1420. [Google Scholar] [CrossRef] [PubMed]
- Einsele, H. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 2002, 99, 3916–3922. [Google Scholar] [CrossRef] [PubMed]
- Rooney, C.; Ng, C.Y.; Loftin, S.; Smith, C.; Li, C.; Krance, R.; Brenner, M.; Heslop, H.; Rooney, C.; Brenner, M.; et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar] [CrossRef]
- Perruccio, K. Transferring functional immune responses to pathogens after haploidentical hematopoietic transplantation. Blood 2005, 106, 4397–4406. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Verfuerth, S.; Pizzey, A.; Khan, N.; Guiver, M.; Moss, P.A.; Mackinnon, S. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet 2003, 362, 1375–1377. [Google Scholar] [CrossRef]
- Koehne, G.; Hasan, A.; Doubrovina, E.; Prockop, S.; Tyler, E.; Wasilewski, G.; O’Reilly, R.J. Immunotherapy with Donor T Cells Sensitized with Overlapping Pentadecapeptides for Treatment of Persistent Cytomegalovirus Infection or Viremia. Biol. Blood Marrow Transplant. 2015, 21, 1663–1678. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Wang, Y.; Reppel, L.; D’aveni, M.; Campidelli, A.; Decot, V.; Bensoussan, D. Viral-specific T-cell transfer from HSCT donor for the treatment of viral infections or diseases after HSCT. Bone Marrow Transplant. 2018, 53, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Doubrovina, E.; Oflaz-Sozmen, B.; Prockop, S.E.; Kernan, N.A.; Abramson, S.; Teruya-Feldstein, J.; Hedvat, C.; Chou, J.F.; Heller, G.; Barker, J.N.; et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012, 119, 2644–2656. [Google Scholar] [CrossRef] [PubMed]
- Qesari, M.; Richter, A.; Ogonek, J.; Mischak-Weissinger, E.; Wang, X.; Dickinson, A.M. Cytomegalovirus-specific T cells isolated by IFN-γ secretion assay do not induce significant graft-versus-host reactions in vitro. Transplantation 2016, 100, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Waruiru, C.; Slatter, M.A.; Taylor, C.; Ramesh, V.; Flood, T.J.; Abinun, M.; Cant, A.J.; Gennery, A.R. Outcome of hematopoietic stem cell transplantation in severe combined immune deficiency with central nervous system viral infection. Pediatr. Infect. Dis. J. 2007, 26, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Scheinberg, P.; Melenhorst, J.J.; Brenchley, J.M.; Hill, B.J.; Hensel, N.F.; Chattopadhyay, P.K.; Roederer, M.; Picker, L.J.; Price, D.A.; Barrett, A.J.; et al. The transfer of adaptive immunity to CMV during hematopoietic stem cell transplantation is dependent on the specificity and phenotype of CMV-specific T cells in the donor. Blood 2009, 114, 5071–5080. [Google Scholar] [CrossRef] [PubMed]
- Feuchtinger, T.; Opherk, K.; Bethge, W.A.; Topp, M.S.; Schuster, F.R.; Weissinger, E.M.; Mohty, M.; Or, R.; Maschan, M.; Schumm, M.; et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010, 116, 4360–4367. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, S. An Epstein-Barr virus deletion mutant associated with fatal lymphoproliferative disease unresponsive to therapy with virus-specific CTLs. Blood 2001, 97, 835–843. [Google Scholar] [CrossRef]
- Haque, T.; Wilkie, G.M.; Jones, M.M.; Higgins, C.D.; Urquhart, G.; Wingate, P.; Burns, D.; McAulay, K.; Turner, M.; Bellamy, C.; et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: Results of a phase 2 multicenter clinical trial. Blood 2007, 110, 1123–1131. [Google Scholar] [CrossRef]
- Uhlin, M.; Okas, M.; Gertow, J.; Uzunel, M.; Brismar, T.B.; Mattsson, J. A novel haplo-identical adoptive CTL therapy as a treatment for EBV-associated lymphoma after stem cell transplantation. Cancer Immunol. Immunother. 2010, 59, 473–477. [Google Scholar] [CrossRef]
- Qasim, W.; Derniame, S.; Gilmour, K.; Chiesa, R.; Weber, M.; Adams, S.; Rao, K.; Amrolia, P.; Goulden, N.; Veys, P.; et al. Third-party virus-specific T cells eradicate adenoviraemia but trigger bystander graft-versus-host disease: Correspondence. Br. J. Haematol. 2011, 154, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Leen, A.M.; Bollard, C.M.; Mendizabal, A.M.; Shpall, E.J.; Szabolcs, P.; Antin, J.H.; Kapoor, N.; Pai, S.-Y.; Rowley, S.D.; Kebriaei, P.; et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood 2013, 121, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- Withers, B.; Blyth, E.; Clancy, L.E.; Yong, A.; Fraser, C.; Burgess, J.; Simms, R.; Brown, R.; Kliman, D.; Dubosq, M.-C.; et al. Long-term control of recurrent or refractory viral infections after allogeneic HSCT with third-party virus-specific T cells. Blood Adv. 2017, 1, 13. [Google Scholar]


{kind=link}
| Patients | Viremia | Viral Disease | Treatment | Response Rate |
|---|---|---|---|---|
| AdV | ||||
| Children | 15–30% | 6–11% | Cidofovir, brincidofovir | 60–80% |
| Adults | 6–15% | 2% | ||
| CMV | ||||
| Children | 15–20% | 4% | Gancyclovir, foscarnet, valgancyclovir | 70–80% |
| Adults | 39% | 13% | ||
| EBV | ||||
| Children | 11% | 1–7% | Rituximab | 60–70% |
| Adults | 22% | 1–3% | ||
| Patients (n) | Population | Viral Infection | VST Stimulated or Isolated by | Citation |
|---|---|---|---|---|
| Ex-Vivo Expanded | ||||
| 113 | Adults/Children | EBV | LBC | Heslop, 2010 [35] |
| 50 | Adults/Children | CMV | Mo-DC pp65-restricted | Blyth, 2013 [36] |
| 8 | Children | ADV | Multi-peptides AdV5 | Ip, 2018 [37] |
| Direct Selection | ||||
| 10 | Adults/Children | EBV | IFN-γ capture | Icheva, 2013 [38] |
| 18 | Unknown | CMV | IFN-γ capture | Peggs, 2011 [34] |
| 30 | Adults/Children | ADV | IFN-γ capture | Feucht, 2015 [39] |
| Patients (n) | Population | Viral Specificity | VST Stimulated by | Citation |
|---|---|---|---|---|
| 13 | Children | EBV/ADV | LBCs transduced with ADV vector | Leen, 2009 [45] |
| 15 | Adults/Children | ADV/CMV/EBV | LBCs transduced with ADV vector encoding CMVpp65 | Leen, 2006 [32] |
| 10 | Adults | ADV/CMV/EBV/VZV | Mo-DC transduced with ADV vector encoding CMVpp65, EBNA1, VZV vaccine | Ma, 2015 [46] |
| 11 | Adults/Children | ADV/CMV/EBV/BKV/HHV6 | Pepmixes of immunodominant antigens | Papadopulou, 2014 [43] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottaviano, G.; Chiesa, R.; Feuchtinger, T.; Vickers, M.A.; Dickinson, A.; Gennery, A.R.; Veys, P.; Todryk, S. Adoptive T Cell Therapy Strategies for Viral Infections in Patients Receiving Haematopoietic Stem Cell Transplantation. Cells 2019, 8, 47. https://doi.org/10.3390/cells8010047
Ottaviano G, Chiesa R, Feuchtinger T, Vickers MA, Dickinson A, Gennery AR, Veys P, Todryk S. Adoptive T Cell Therapy Strategies for Viral Infections in Patients Receiving Haematopoietic Stem Cell Transplantation. Cells. 2019; 8(1):47. https://doi.org/10.3390/cells8010047
Chicago/Turabian StyleOttaviano, Giorgio, Robert Chiesa, Tobias Feuchtinger, Mark A. Vickers, Anne Dickinson, Andrew R. Gennery, Paul Veys, and Stephen Todryk. 2019. "Adoptive T Cell Therapy Strategies for Viral Infections in Patients Receiving Haematopoietic Stem Cell Transplantation" Cells 8, no. 1: 47. https://doi.org/10.3390/cells8010047
APA StyleOttaviano, G., Chiesa, R., Feuchtinger, T., Vickers, M. A., Dickinson, A., Gennery, A. R., Veys, P., & Todryk, S. (2019). Adoptive T Cell Therapy Strategies for Viral Infections in Patients Receiving Haematopoietic Stem Cell Transplantation. Cells, 8(1), 47. https://doi.org/10.3390/cells8010047