Oxidative Stress Induces Telomere Dysfunction and Senescence by Replication Fork Arrest


Abstract
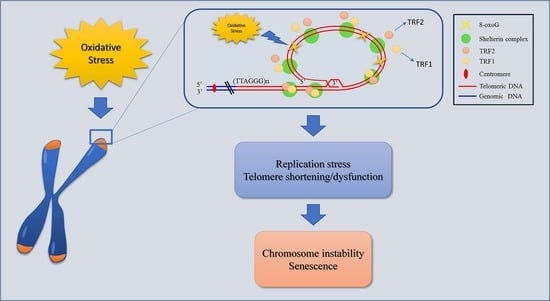
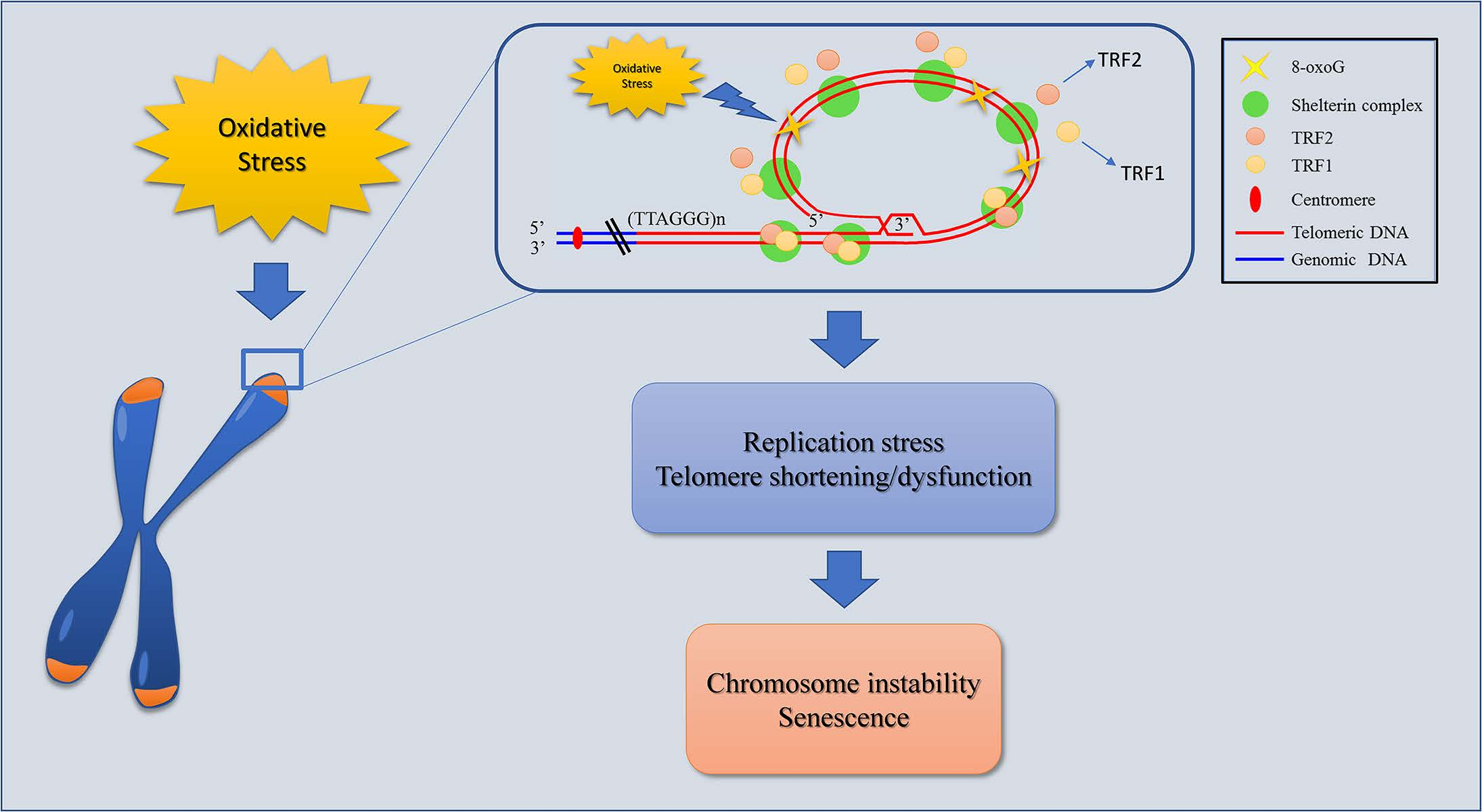{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell and Culture Conditions
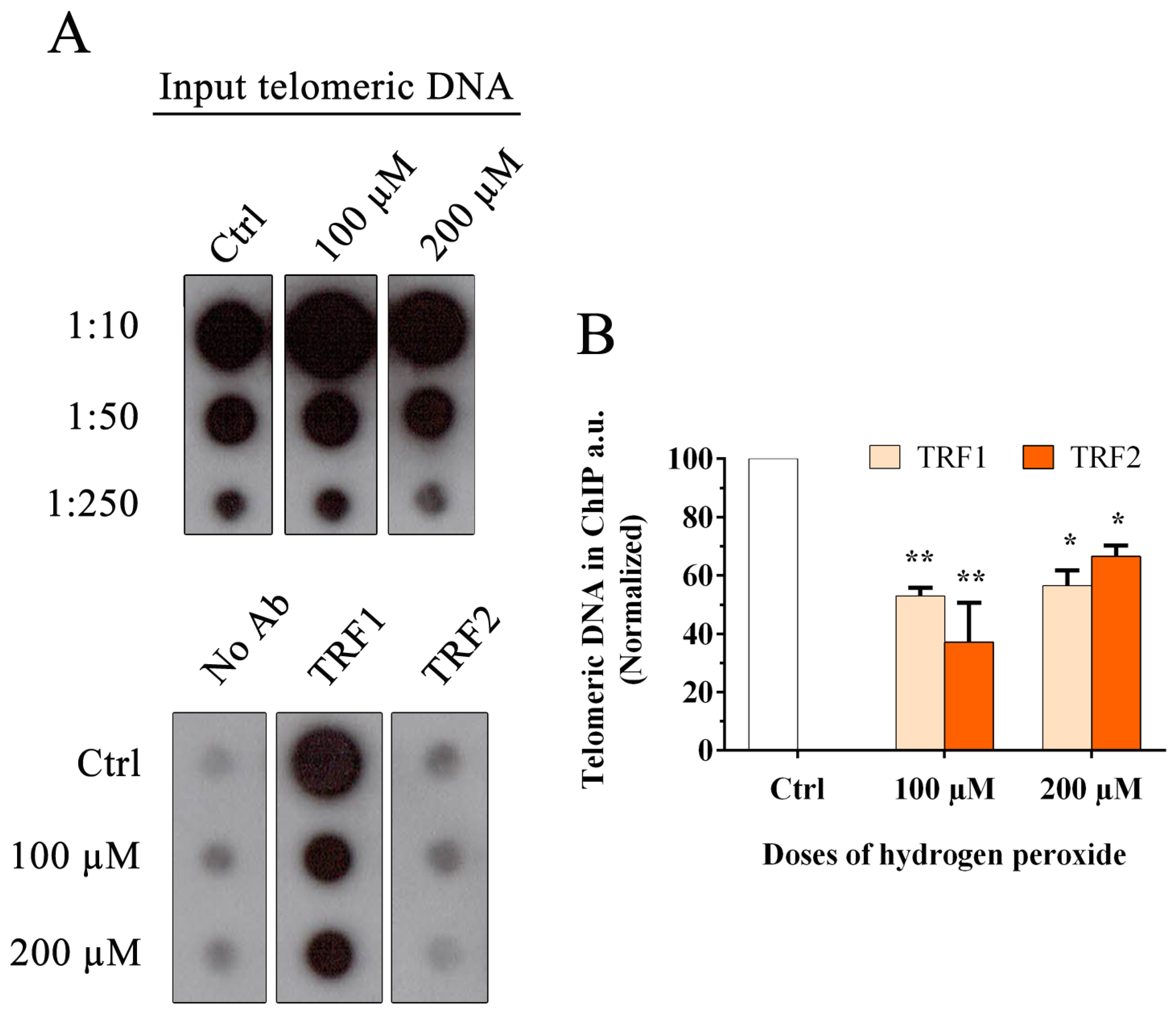
2.2. Chromatin Immunoprecipitation (ChIP)
2.3. γH2AX and 53BP1 Immunofluorescence Staining
2.4. TIF Immunofish Staining
2.5. Senescence Associated β–Galactosidase Assay
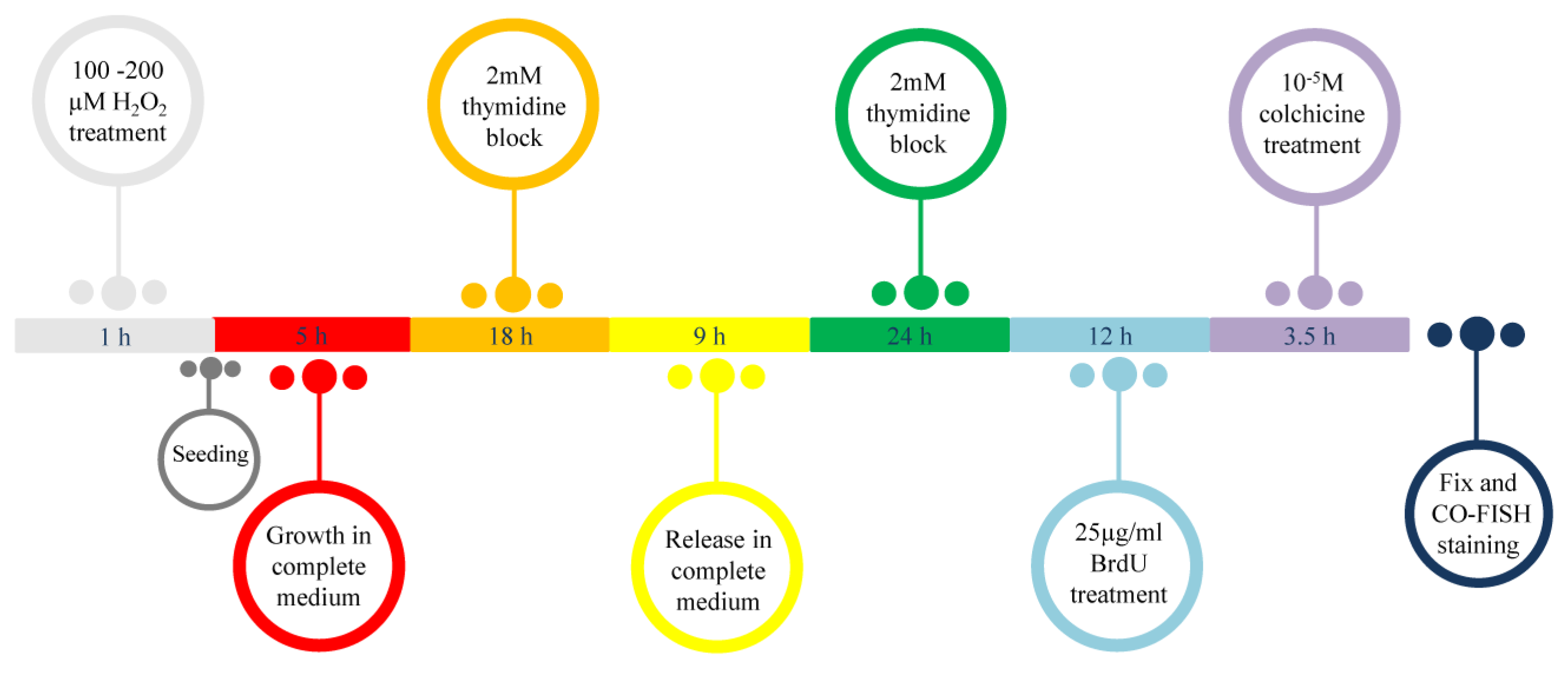
2.6. Cell Synchronization Protocol
2.7. Cell Cycle
2.8. BrdU Incorporation
2.9. Metaphase Preparation and CO-FISH (Chromosome Orientation-FISH) Analysis
2.10. Data Analysis
3. Results
3.1. Oxidative Stress Induces a Reduction in Telomeric Binding Proteins TRF1 and TRF2
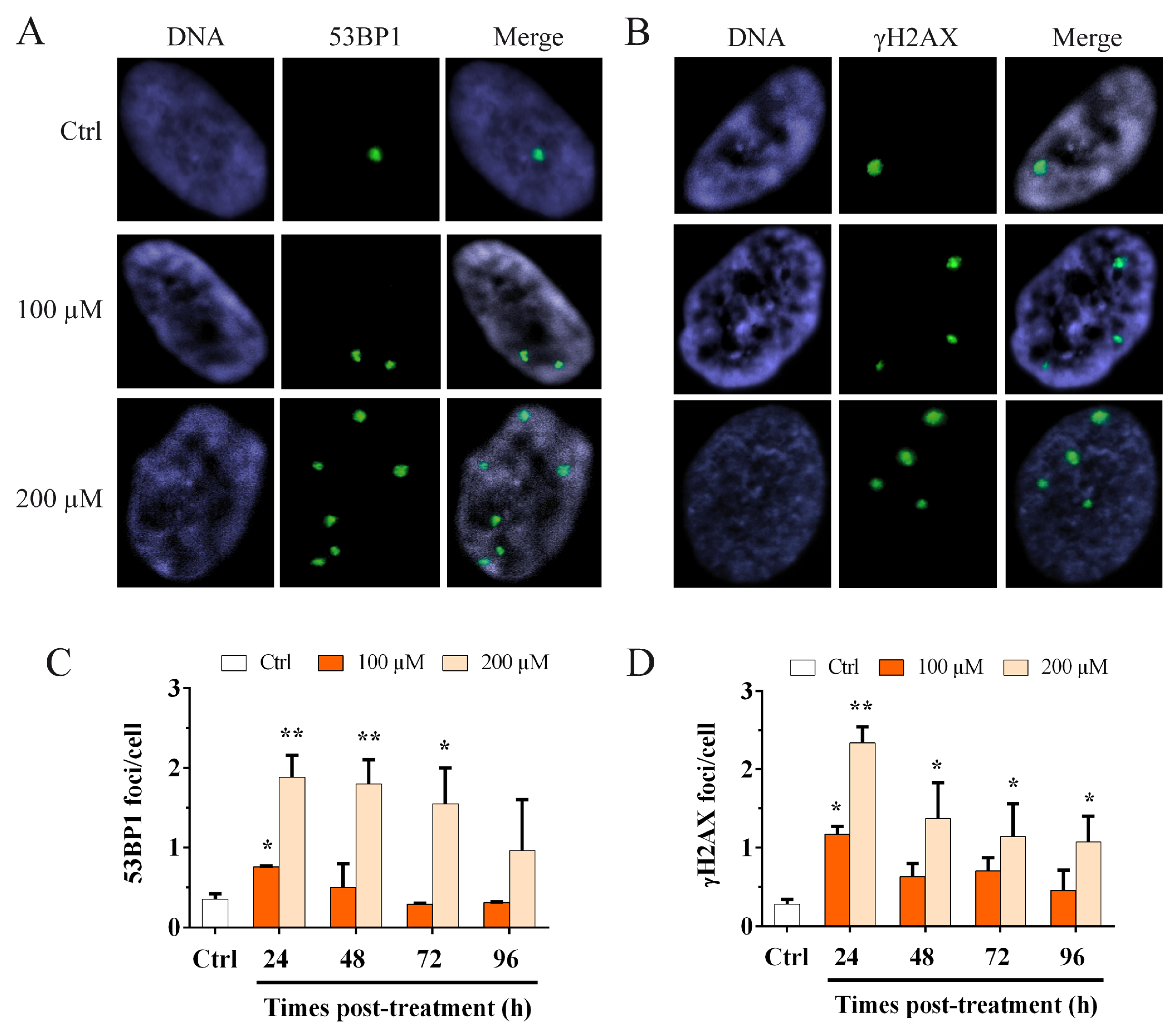
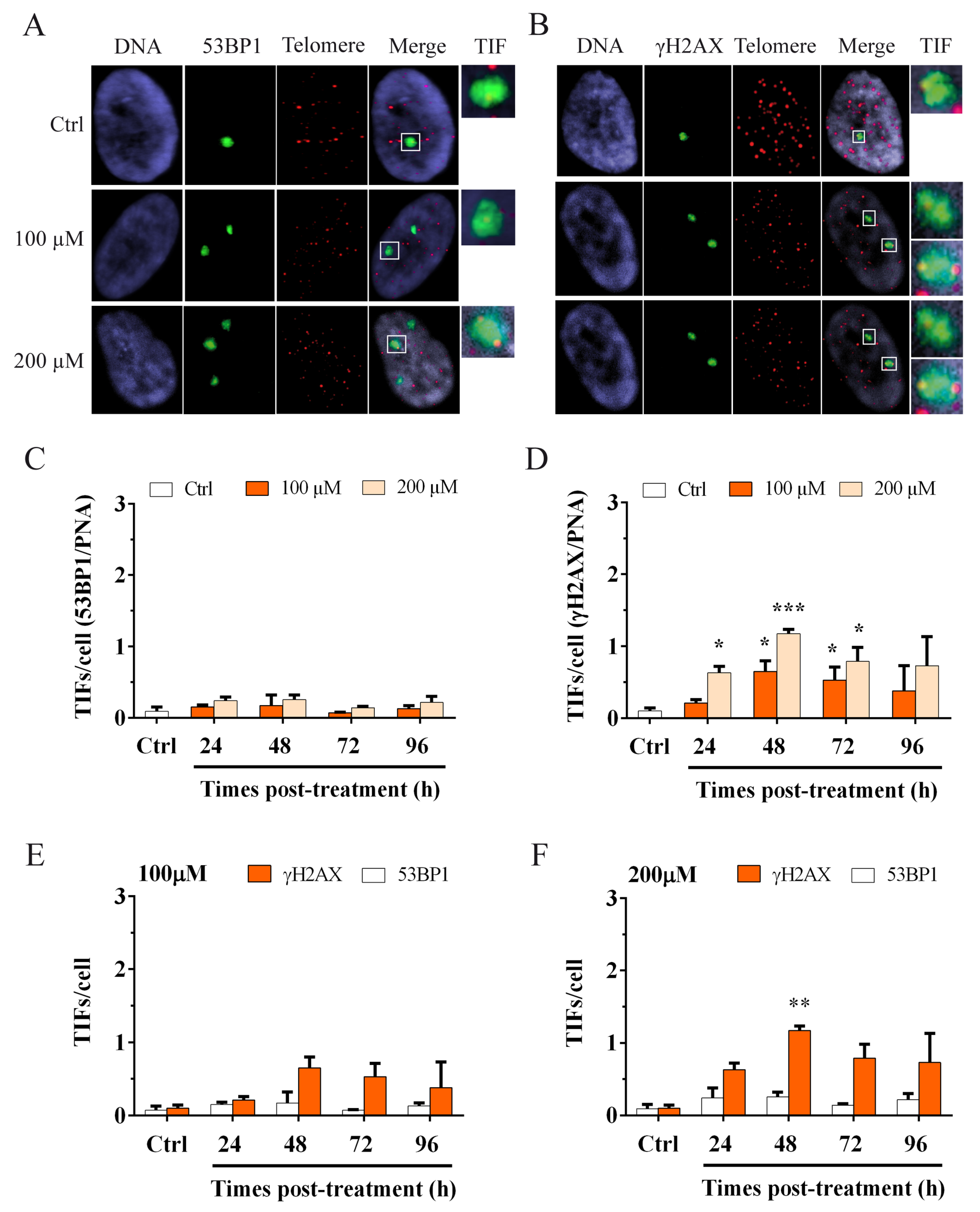
3.2. Oxidative Stress Induces an Increase in the Genomic Damage and an Increase in γH2AX Telomere Dysfunction-Induced Foci (TIFs)
3.3. Hydrogen Peroxide Treatment Induces a Telomeric Replication Fork Block Rather Than a DSB
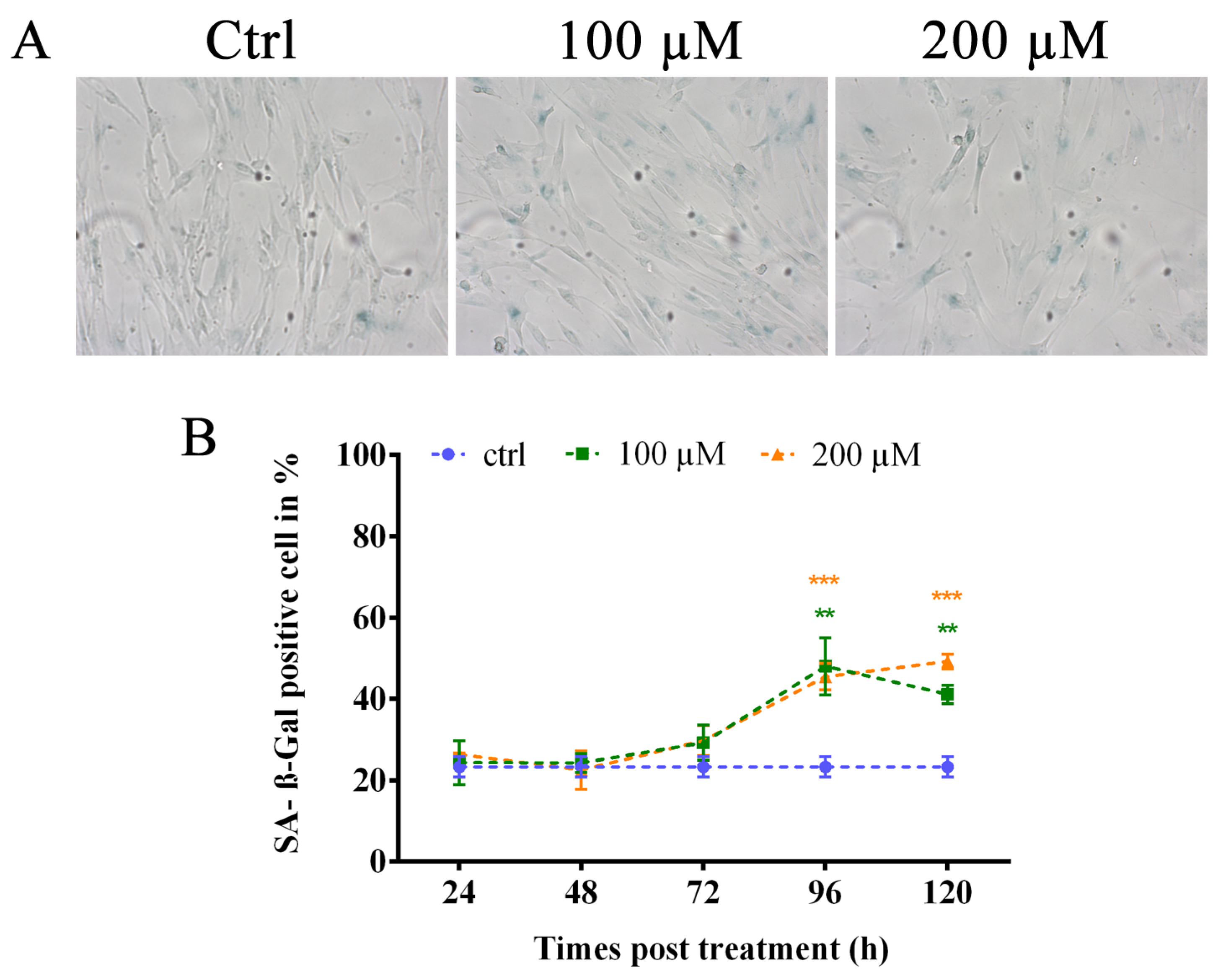
3.4. Oxidative Stress Induces Premature Senescence
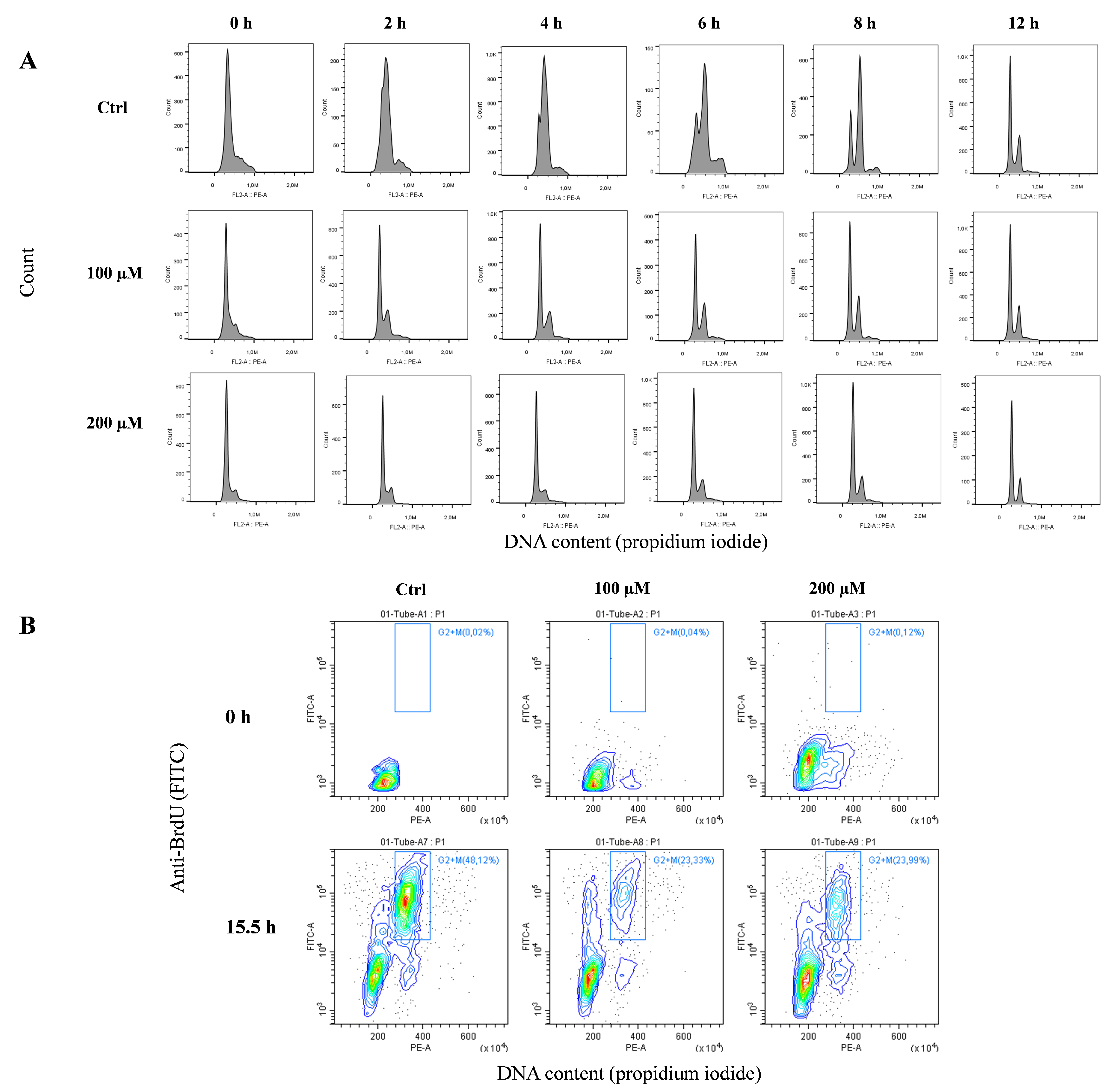
3.5. Oxidative Stress Induces a Genomic Reduction of Replication Rate
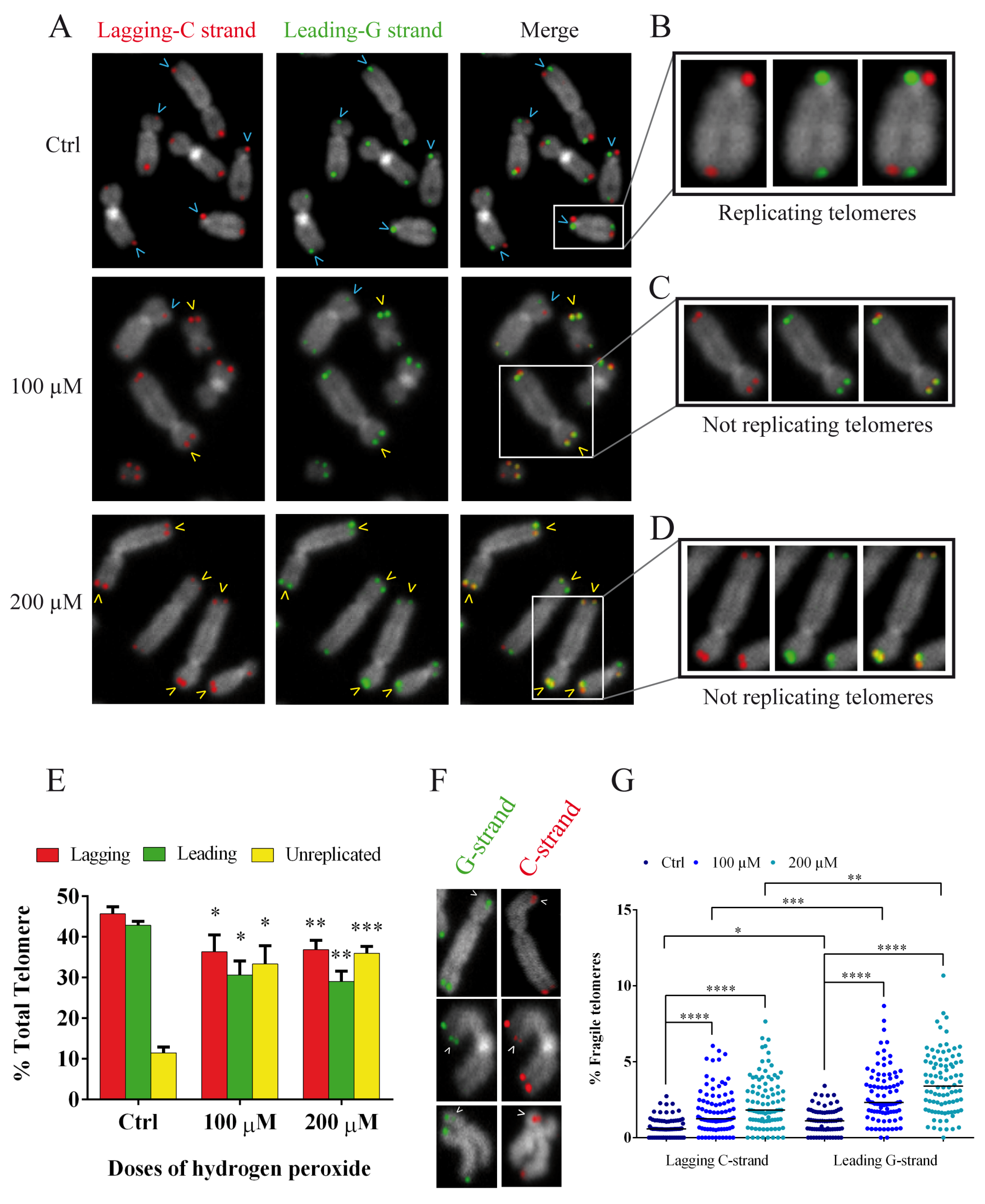
3.6. Oxidative Stress Induces Replication Fork Arrest at Telomerse
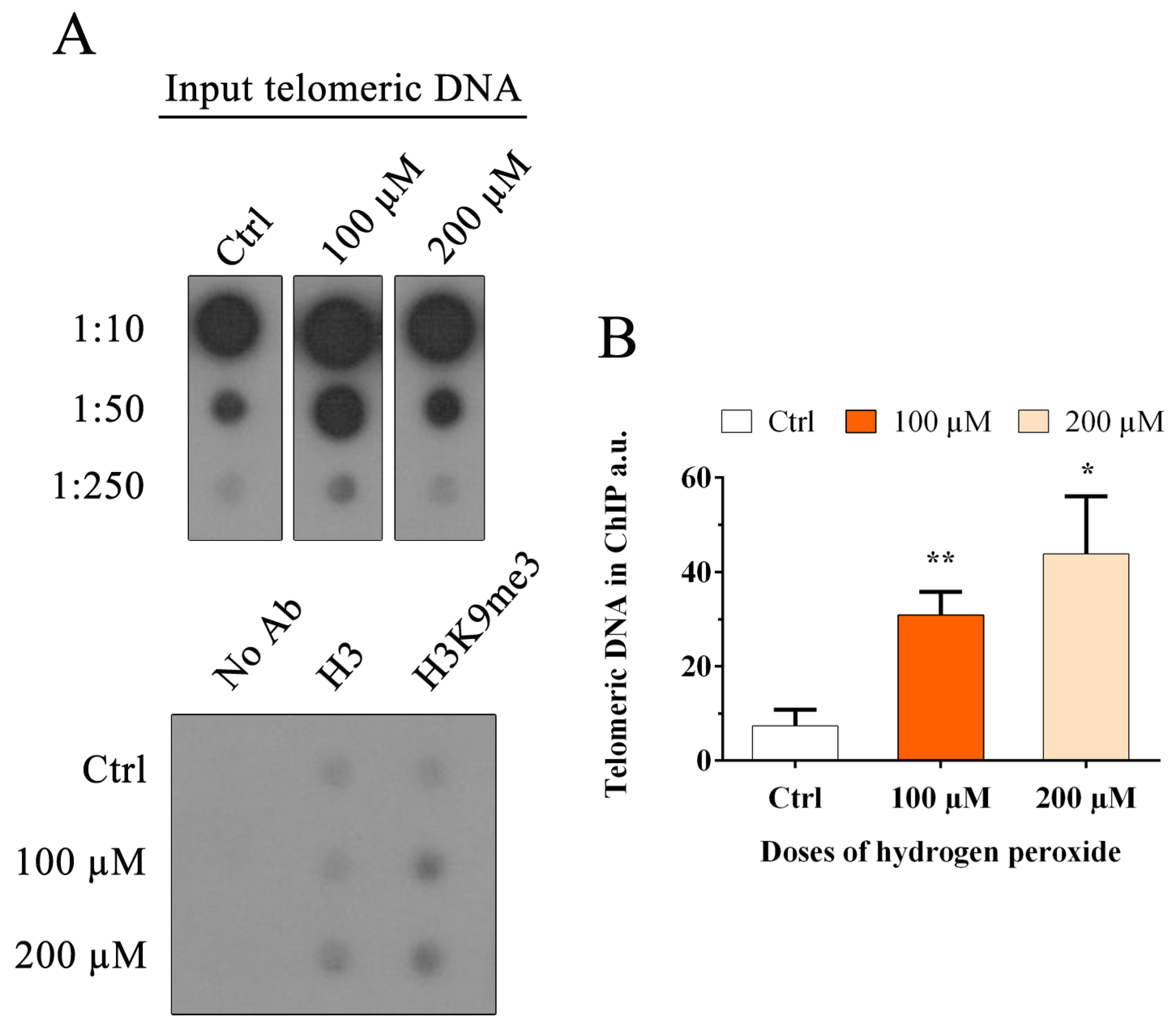
3.7. Oxidative Stress Induces an Increase in the Heterochromatin Mark H3K9me3
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scandalios, J.G. Oxidative stress: Molecular perception and transduction of signals triggering antioxidant gene defenses. Braz. J. Med. Biol. Res. 2005, 38, 995–1014. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Delatour, T.; Douki, T.; Gasparutto, D.; Pouget, J.P.; Ravanat, J.L.; Sauvaigo, S. Hydroxyl radicals and DNA base damage. Mutat. Res. 1999, 424, 9–21. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxy radicals, lipid peroxidation and DNA damage. Toxicology 2002, 181–182, 219–222. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Wallace, S.S. Biological consequences of free radical-damaged DNA bases. Free Radic. Biol. Med. 2002, 33, 1–14. [Google Scholar] [CrossRef]
- Daroui, P.; Desai, S.D.; Li, T.K.; Liu, A.A.; Liu, L.F. Hydrogen peroxide induces topoisomerase I-mediated DNA damage and cell death. J. Biol. Chem. 2004, 279, 14587–14594. [Google Scholar] [CrossRef]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef]
- Magdalou, I.; Lopez, B.S.; Pasero, P.; Lambert, S.A. The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol. 2014, 30, 154–164. [Google Scholar] [CrossRef]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef]
- Blackburn, E.H. Structure and function of telomeres. Nature 1991, 350, 569–573. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Nishimura, Y.; Kurumizaka, H.; Shimizu, M. Nucleosome organization and chromatin dynamics in telomeres. Biomol. Concepts 2015, 6, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Frippiat, C.; Chen, Q.M.; Zdanov, S.; Magalhaes, J.P.; Remacle, J.; Toussaint, O. Subcytotoxic H2O2 stress triggers a release of transforming growth factor-beta 1, which induces biomarkers of cellular senescence of human diploid fibroblasts. J. Biol. Chem. 2001, 276, 2531–2537. [Google Scholar] [CrossRef]
- Cristofalo, V.J.; Lorenzini, A.; Allen, R.G.; Torres, C.; Tresini, M. Replicative senescence: A critical review. Mech. Ageing Dev. 2004, 125, 827–848. [Google Scholar] [CrossRef]
- Dimri, G.P. What has senescence got to do with cancer? Cancer Cell 2005, 7, 505–512. [Google Scholar] [CrossRef]
- Duan, J.; Duan, J.; Zhang, Z.; Tong, T. Irreversible cellular senescence induced by prolonged exposure to H2O2 involves DNA-damage-and-repair genes and telomere shortening. Int. J. Biochem. Cell Biol. 2005, 37, 1407–1420. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Coluzzi, E.; Colamartino, M.; Cozzi, R.; Leone, S.; Meneghini, C.; O’Callaghan, N.; Sgura, A. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS ONE 2014, 9, e110963. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Kawanishi, S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999, 453, 365–368. [Google Scholar] [CrossRef]
- Opresko, P.L.; Fan, J.; Danzy, S.; Wilson, D.M., 3rd; Bohr, V.A. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005, 33, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Shiue, L.; Kaplan, S.; de Lange, T. A mammalian factor that binds telomeric TTAGGG repeats in vitro. Mol. Cell. Biol. 1992, 12, 4834–4843. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Martinez, P.; Thanasoula, M.; Munoz, P.; Liao, C.; Tejera, A.; McNees, C.; Flores, J.M.; Fernandez-Capetillo, O.; Tarsounas, M.; Blasco, M.A. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009, 23, 2060–2075. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef]
- Badie, S.; Escandell, J.M.; Bouwman, P.; Carlos, A.R.; Thanasoula, M.; Gallardo, M.M.; Suram, A.; Jaco, I.; Benitez, J.; Herbig, U.; et al. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat. Struct. Mol. Biol. 2010, 17, 1461–1469. [Google Scholar] [CrossRef]
- Chan, S.W.; Blackburn, E.H. New ways not to make ends meet: Telomerase, DNA damage proteins and heterochromatin. Oncogene 2002, 21, 553–563. [Google Scholar] [CrossRef]
- Blasco, M.A. The epigenetic regulation of mammalian telomeres. Nat. Rev. Genet. 2007, 8, 299–309. [Google Scholar] [CrossRef]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009, 10, 192–206. [Google Scholar] [CrossRef]
- Garcia-Cao, M.; O’Sullivan, R.; Peters, A.H.; Jenuwein, T.; Blasco, M.A. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 2004, 36, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Jaco, I.; Fraga, M.F.; Chen, T.; Li, E.; Esteller, M.; Blasco, M.A. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat. Cell Biol. 2006, 8, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Blasco, M.A. Replicating through telomeres: A means to an end. Trends Biochem. Sci. 2015, 40, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M.; Nagasaka, T.; Ikai, I.; Goel, A. Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma. Hepatology 2012, 56, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Arizumi, T.; Takita, M.; Kitai, S.; Yada, N.; Hagiwara, S.; Inoue, T.; Minami, Y.; Ueshima, K.; Sakurai, T.; et al. Reactive oxygen species induce epigenetic instability through the formation of 8-hydroxydeoxyguanosine in human hepatocarcinogenesis. Dig. Dis. 2013, 31, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free Radic. Biol. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef]
- Rivera, C.; Gurard-Levin, Z.A.; Almouzni, G.; Loyola, A. Histone lysine methylation and chromatin replication. Biochim. Biophys. Acta 2014, 1839, 1433–1439. [Google Scholar] [CrossRef]
- Nikolov, I.; Taddei, A. Linking replication stress with heterochromatin formation. Chromosoma 2016, 125, 523–533. [Google Scholar] [CrossRef]
- Benetti, R.; Garcia-Cao, M.; Blasco, M.A. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat. Genet. 2007, 39, 243–250. [Google Scholar] [CrossRef]
- Berardinelli, F.; Antoccia, A.; Cherubini, R.; De Nadal, V.; Gerardi, S.; Cirrone, G.A.; Tanzarella, C.; Sgura, A. Transient activation of the ALT pathway in human primary fibroblasts exposed to high-LET radiation. Radiat. Res. 2010, 174, 539–549. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. How telomeres solve the end-protection problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, D.; Xiao, H. Methods of cellular senescence induction using oxidative stress. Methods Mol. Biol. 2013, 1048, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Mondello, C.; d’Adda di Fagagna, F. Stable cellular senescence is associated with persistent DDR activation. PLoS ONE 2014, 9, e110969. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.; Meyne, J. Strand-specific FISH reveals orientation of chromosome 18 alphoid DNA. Cytogenet. Cell Genet. 1993, 63, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Cornforth, M.N.; Kurimasa, A.; Chen, D.J.; Goodwin, E.H. Strand-specific postreplicative processing of mammalian telomeres. Science 2001, 293, 2462–2465. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Min, J.N.; Wang, Y.; Huang, C.; Peng, T.; Chai, W.; Chang, S. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J. 2012, 31, 2309–2321. [Google Scholar] [CrossRef]
- Chai, W.; Du, Q.; Shay, J.W.; Wright, W.E. Human telomeres have different overhang sizes at leading versus lagging strands. Mol. Cell 2006, 21, 427–435. [Google Scholar] [CrossRef]
- Pedram, M.; Sprung, C.N.; Gao, Q.; Lo, A.W.; Reynolds, G.E.; Murnane, J.P. Telomere position effect and silencing of transgenes near telomeres in the mouse. Mol. Cell. Biol. 2006, 26, 1865–1878. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann. N. Y. Acad. Sci. 2000, 908, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Karlseder, J.; Broccoli, D.; Dai, Y.; Hardy, S.; de Lange, T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 1999, 283, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef]
- Dimitrova, N.; Chen, Y.C.; Spector, D.L.; de Lange, T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008, 456, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Bartocci, C.; Ouzounov, I.; Diedrich, J.K.; Yates, J.R., 3rd; Denchi, E.L. A two-step mechanism for TRF2-mediated chromosome-end protection. Nature 2013, 494, 502–505. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T.; Pilger, R.; Sitte, N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 2000, 28, 64–74. [Google Scholar] [CrossRef]
- Ten Hagen, K.G.; Cohen, S.N. Timing of replication of beta satellite repeats of human chromosomes. Nucleic Acids Res. 1993, 21, 2139–2142. [Google Scholar] [CrossRef]
- Wright, W.E.; Tesmer, V.M.; Liao, M.L.; Shay, J.W. Normal human telomeres are not late replicating. Exp. Cell Res. 1999, 251, 492–499. [Google Scholar] [CrossRef]
- Arnoult, N.; Shin-Ya, K.; Londono-Vallejo, J.A. Studying telomere replication by Q-CO-FISH: The effect of telomestatin, a potent G-quadruplex ligand. Cytogenet. Genome Res. 2008, 122, 229–236. [Google Scholar] [CrossRef]
- Arnoult, N.; Schluth-Bolard, C.; Letessier, A.; Drascovic, I.; Bouarich-Bourimi, R.; Campisi, J.; Kim, S.H.; Boussouar, A.; Ottaviani, A.; Magdinier, F.; et al. Replication timing of human telomeres is chromosome arm-specific, influenced by subtelomeric structures and connected to nuclear localization. PLoS Genet. 2010, 6, e1000920. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Gryaznov, S.M.; Shay, J.W.; Wright, W.E.; Cornforth, M.N. Asynchronous replication timing of telomeres at opposite arms of mammalian chromosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 12928–12933. [Google Scholar] [CrossRef]
- Verdun, R.E.; Karlseder, J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 2006, 127, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, H.M.; Wang, W.; Sen, S.; Destefano Shields, C.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Zou, Y.; Shay, J.W.; Wright, W.E. Telomere position effect in human cells. Science 2001, 292, 2075–2077. [Google Scholar] [CrossRef] [PubMed]
- Koering, C.E.; Pollice, A.; Zibella, M.P.; Bauwens, S.; Puisieux, A.; Brunori, M.; Brun, C.; Martins, L.; Sabatier, L.; Pulitzer, J.F.; et al. Human telomeric position effect is determined by chromosomal context and telomeric chromatin integrity. EMBO Rep. 2002, 3, 1055–1061. [Google Scholar] [CrossRef]
- Ottaviani, A.; Gilson, E.; Magdinier, F. Telomeric position effect: From the yeast paradigm to human pathologies? Biochimie 2008, 90, 93–107. [Google Scholar] [CrossRef]
- Surace, C.; Berardinelli, F.; Masotti, A.; Roberti, M.C.; Da Sacco, L.; D’Elia, G.; Sirleto, P.; Digilio, M.C.; Cusmai, R.; Grotta, S.; et al. Telomere shortening and telomere position effect in mild ring 17 syndrome. Epigenetics Chromatin 2014, 7, 1. [Google Scholar] [CrossRef]
- Surace, C.; Piazzolla, S.; Sirleto, P.; Digilio, M.C.; Roberti, M.C.; Lombardo, A.; D’Elia, G.; Tomaiuolo, A.C.; Petrocchi, S.; Capolino, R.; et al. Mild ring 17 syndrome shares common phenotypic features irrespective of the chromosomal breakpoints location. Clin. Genet. 2009, 76, 256–262. [Google Scholar] [CrossRef]
- Kloc, A.; Zaratiegui, M.; Nora, E.; Martienssen, R. RNA interference guides histone modification during the S phase of chromosomal replication. Curr. Biol. 2008, 18, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Zaratiegui, M.; Castel, S.E.; Irvine, D.V.; Kloc, A.; Ren, J.; Li, F.; de Castro, E.; Marin, L.; Chang, A.Y.; Goto, D.; et al. RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature 2011, 479, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Ayrapetov, M.K.; Gursoy-Yuzugullu, O.; Xu, C.; Xu, Y.; Price, B.D. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. USA 2014, 111, 9169–9174. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, C.; Soutoglou, E. Double strand break (DSB) repair in heterochromatin and heterochromatin proteins in DSB repair. DNA Repair 2014, 19, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Bartocci, C.; Diedrich, J.K.; Ouzounov, I.; Li, J.; Piunti, A.; Pasini, D.; Yates, J.R., 3rd; Lazzerini Denchi, E. Isolation of chromatin from dysfunctional telomeres reveals an important role for Ring1b in NHEJ-mediated chromosome fusions. Cell Rep. 2014, 7, 1320–1332. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coluzzi, E.; Leone, S.; Sgura, A. Oxidative Stress Induces Telomere Dysfunction and Senescence by Replication Fork Arrest. Cells 2019, 8, 19. https://doi.org/10.3390/cells8010019
Coluzzi E, Leone S, Sgura A. Oxidative Stress Induces Telomere Dysfunction and Senescence by Replication Fork Arrest. Cells. 2019; 8(1):19. https://doi.org/10.3390/cells8010019
Chicago/Turabian StyleColuzzi, Elisa, Stefano Leone, and Antonella Sgura. 2019. "Oxidative Stress Induces Telomere Dysfunction and Senescence by Replication Fork Arrest" Cells 8, no. 1: 19. https://doi.org/10.3390/cells8010019
APA StyleColuzzi, E., Leone, S., & Sgura, A. (2019). Oxidative Stress Induces Telomere Dysfunction and Senescence by Replication Fork Arrest. Cells, 8(1), 19. https://doi.org/10.3390/cells8010019