Mitochondrial Quality Control in COPD and IPF


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
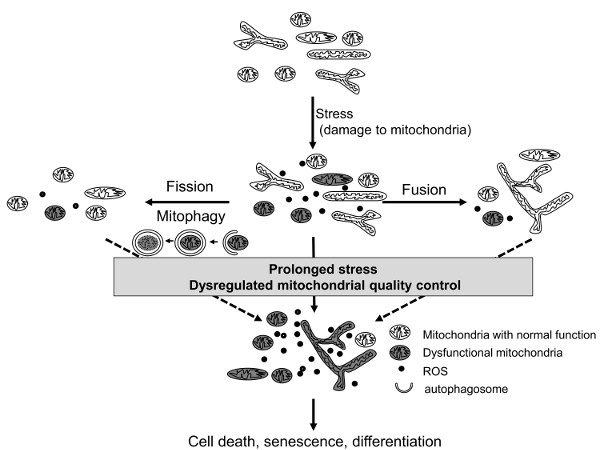
2. Mitochondrial Quality Control Systems
2.1. Mitochondria Dynamics
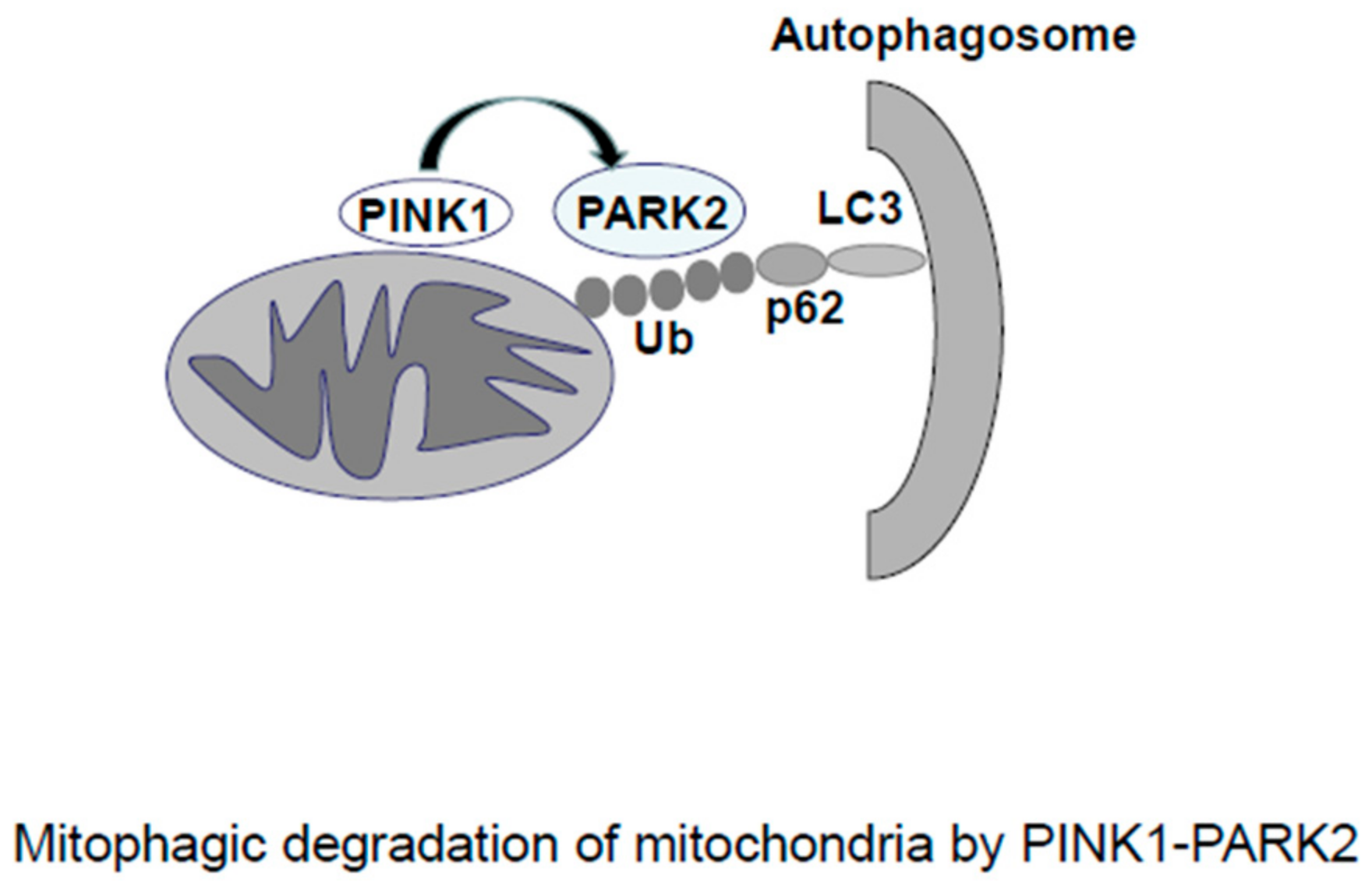
2.2. Mitophagy
2.3. Mitochondrial Biogenesis
2.4. Other Mitochondrial Quality Control Pathways
3. COPD
3.1. Mitochondrial Quality Control in COPD
3.2. Mitochondrial Dynamics in COPD
3.3. Mitophagy in COPD
3.4. Mitochondria Biogenesis in COPD
3.5. Mitochondria Function in COPD
4. IPF
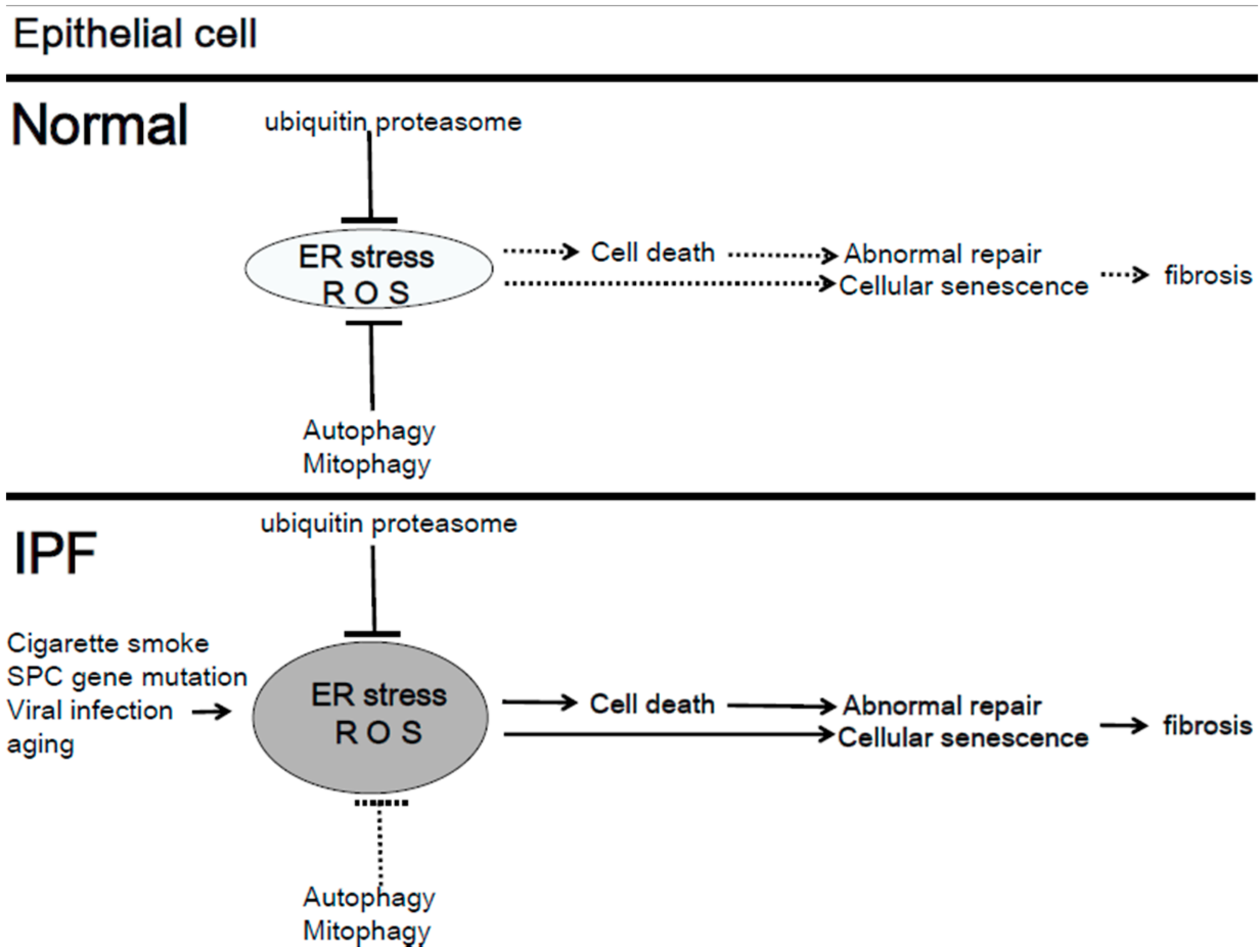
4.1. Mitochondria Quality Control in IPF
4.2. Mitochondrial Dynamics in IPF
4.3. Mitophagy in IPF
4.3.1. Mitophagy in Lung Epithelial Cells
4.3.2. Mitophagy in Lung Fibroblasts
4.4. Mitochondria Biogenesis in IPF
4.5. Other Mitochondrial Quality Control Systems in IPF
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell 2015, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zemirli, N.; Morel, E.; Molino, D. Mitochondrial Dynamics in Basal and Stressful Conditions. Int. J. Mol. Sci. 2018, 19, 564. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. Intra- and Intercellular Quality Control Mechanisms of Mitochondria. Cells 2017, 7, 1. [Google Scholar]
- Shi, R.; Guberman, M.; Kirshenbaum, L.A. Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc. Med. 2018, 28, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.B.; Hanss, Z.; Kruger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta 2015, 1847, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Hamalainen, R.H.; Yatsuga, S.; Uutela, M.; Terzioglu, M.; Gotz, A.; Forsström, S.; Salven, P.; Angers-Loustau, A.; Kopra, O.H.; et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012, 15, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Araya, J.; Ito, S.; Kobayashi, K.; Takasaka, N.; Yoshii, Y.; Wakui, H.; Kojima, J.; Shimizu, K.; Numata, T.; et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Bhavsar, P.K.; Mumby, S.; Chung, K.F.; Adcock, I.M. Dealing with Stress: Defective Metabolic Adaptation in Chronic Obstructive Pulmonary Disease Pathogenesis. Ann. Am. Thorac. Soc. 2017, 14 (Suppl. S5), S374–S382. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Mannam, P.; Zhang, J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L433–L452. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Araya, J.; Minagawa, S.; Hara, H.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Tsubouchi, K.; Kurita, Y.; et al. Involvement of PARK2-Mediated Mitophagy in Idiopathic Pulmonary Fibrosis Pathogenesis. J. Immunol. 2016, 197, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jeong, S.Y.; Lim, W.C.; Kim, S.; Park, Y.Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, K.; Araya, J.; Hara, H.; Minagawa, S.; Takasaka, N.; Ito, S.; Kobayashi, K.; Nakayama, K. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir. Investig. 2016, 54, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, X.; Gao, J.; Zhang, Q.; Sun, S.; Zhu, H.; Yang, X.; Yan, H. PINK1/Parkin-mediated mitophagy was activated against 1,4-Benzoquinone-induced apoptosis in HL-60 cells. Toxicol. In Vitro 2018, 50, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; De Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.M.; Ding, W.X. The reciprocal roles of PARK2 and mitofusins in mitophagy and mitochondrial spheroid formation. Autophagy 2013, 9, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar cell senescence in patients with pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Demedts, I.K.; Demoor, T.; Bracke, K.R.; Joos, G.F.; Brusselle, G.G. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir. Res. 2006, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Yao, H. Mitochondrial dysfunction in inflammatory responses and cellular senescence: Pathogenesis and pharmacological targets for chronic lung diseases. Br. J. Pharmacol. 2016, 173, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Ballweg, K.; Mutze, K.; Konigshoff, M.; Eickelberg, O.; Meiners, S. Cigarette smoke extract affects mitochondrial function in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L895–L907. [Google Scholar] [CrossRef] [PubMed]
- Van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koeter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [PubMed]
- Kanaji, N.; Basma, H.; Nelson, A.; Farid, M.; Sato, T.; Nakanishi, M.; Wang, X.; Michalski, J.; Li, Y.; Gunji, Y.; et al. Fibroblasts that resist cigarette smoke-induced senescence acquire profibrotic phenotypes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L364–L373. [Google Scholar] [CrossRef] [PubMed]
- Soulitzis, N.; Neofytou, E.; Psarrou, M.; Anagnostis, A.; Tavernarakis, N.; Siafakas, N.; Tzortzaki, E.G. Downregulation of lung mitochondrial prohibitin in COPD. Respir. Med. 2012, 106, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cheng, X.; Yue, L.; Cui, W.; Zhou, W.; Gao, J.; Yao, H. Molecular pathogenesis in chronic obstructive pulmonary disease and therapeutic potential by targeting AMP-activated protein kinase. J. Cell. Physiol. 2018, 233, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.J.; Wang, H.Y.; Wang, J.Y.; Lee, C.C.; Tseng, H.W.; Wu, Y.L.; Shyue, S.K.; Lee, T.S.; Kou, Y.R. Novel role of AMP-activated protein kinase signaling in cigarette smoke induction of IL-8 in human lung epithelial cells and lung inflammation in mice. Free. Radic. Biol. Med. 2011, 50, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, X.; Lopez de Silanes, I.; Carling, D.; Gorospe, M. Increased AMP:ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J. Biol. Chem. 2003, 278, 27016–27023. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.Y.; Li, Y.Y.; Huang, C.; Li, J.; Yao, H.W. AMP-activated protein kinase reduces inflammatory responses and cellular senescence in pulmonary emphysema. Oncotarget 2017, 8, 22513–22523. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dai, A.; Hu, R.; Zhu, L.; Tan, S. Positive correlation between PPARgamma/PGC-1alpha and gamma-GCS in lungs of rats and patients with chronic obstructive pulmonary disease. Acta Biochim. Biophys. Sin. 2010, 42, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Zank, D.C.; Bueno, M.; Mora, A.L.; Rojas, M. Idiopathic Pulmonary Fibrosis: Aging, Mitochondrial Dysfunction, and Cellular Bioenergetics. Front. Med. 2018, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Burman, A.; Tanjore, H.; Blackwell, T.S. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 2018, 68–69, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.M.; Seeger, W.; et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K. Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Int. J. Clin. Exp. Med. 2010, 3, 33–40. [Google Scholar] [PubMed]
- Fujimoto, M.; Hayashi, T. New insights into the role of mitochondria-associated endoplasmic reticulum membrane. Int. Rev. Cell Mol. Biol. 2011, 292, 73–117. [Google Scholar] [PubMed]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; St Croix, C.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17, e12720. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef] [PubMed]
- Sosulski, M.L.; Gongora, R.; Danchuk, S.; Dong, C.; Luo, F.; Sanchez, C.G. Deregulation of selective autophagy during aging and pulmonary fibrosis: The role of TGFbeta1. Aging Cell 2015, 14, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, R.P.; Kim, S.J.; Cheresh, P.; Williams, D.B.; Morales-Nebreda, L.; Cheng, Y.; Yeldandi, A.; Bhorade, S.; Pardo, A.; Selman, M.; et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J. 2017, 31, 2520–2532. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hara, H.; Kuwano, K.; Araya, J. Mitochondrial Quality Control in COPD and IPF. Cells 2018, 7, 86. https://doi.org/10.3390/cells7080086
Hara H, Kuwano K, Araya J. Mitochondrial Quality Control in COPD and IPF. Cells. 2018; 7(8):86. https://doi.org/10.3390/cells7080086
Chicago/Turabian StyleHara, Hiromichi, Kazuyoshi Kuwano, and Jun Araya. 2018. "Mitochondrial Quality Control in COPD and IPF" Cells 7, no. 8: 86. https://doi.org/10.3390/cells7080086
APA StyleHara, H., Kuwano, K., & Araya, J. (2018). Mitochondrial Quality Control in COPD and IPF. Cells, 7(8), 86. https://doi.org/10.3390/cells7080086