Targeting the Multidrug Transporter Ptch1 Potentiates Chemotherapy Efficiency



Abstract

1. Introduction
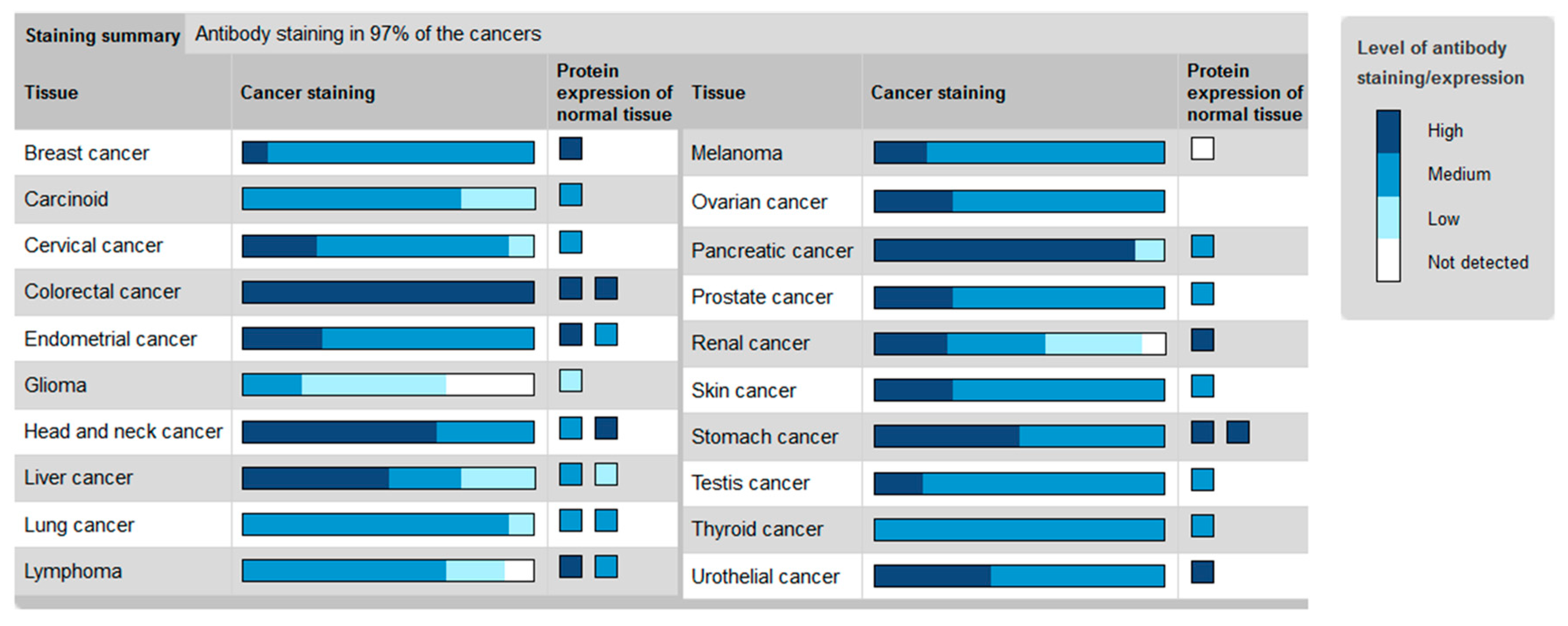
2. The Hh Receptor Ptch1 Is Overexpressed in Many Aggressive Cancers
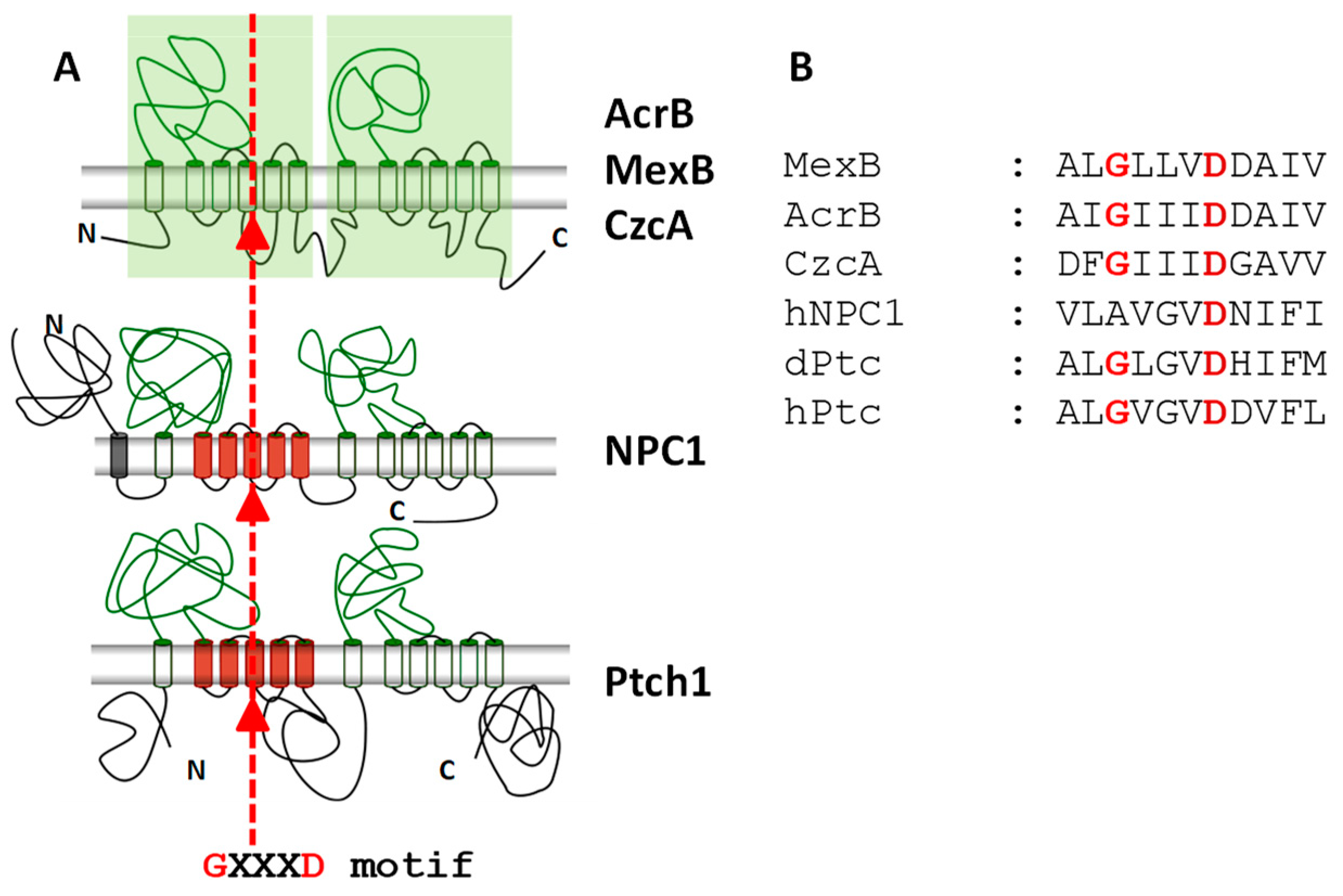
3. Ptch1 Is a Multidrug Transporter Involved in Chemotherapy Resistance
4. Patched Drug Efflux Activity and Cancer Cell Metabolism
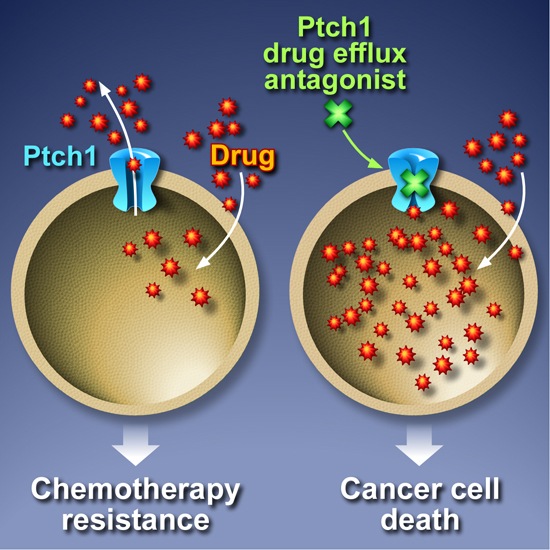
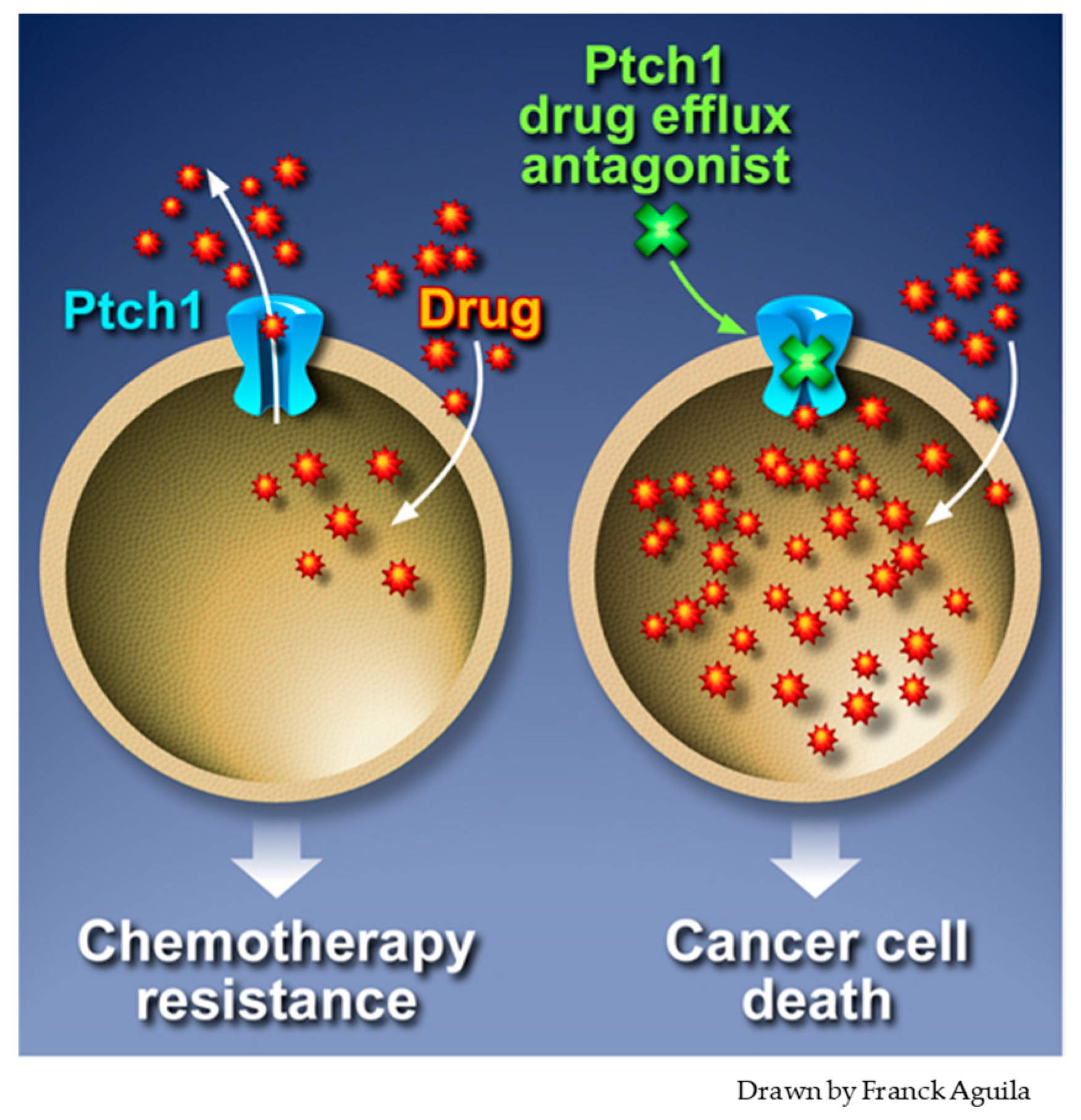
5. Inhibition of Ptch1 Drug Efflux Activity Increases Chemotherapy Efficacy
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Fidler, I.J. Regulation of neoplastic angiogenesis. J. Natl. Cancer Inst. Monogr. 2001, 28, 10–14. [Google Scholar] [CrossRef]
- Jacobson, L.O.; Spurr, C.L.; Guzman, E.S.; Barron, E.S.; Smith, T.; Lushbaugh, C.; Dick, G.F. Nitrogen mustard therapy. Studies on the Effect of Methyl-Bis (Beta-Chloroethyl) Amine Hydrochloride on Neoplastic Diseases and Allied Disorders of the Hemopoietic System. JAMA 1946, 132, 263–271. [Google Scholar] [CrossRef]
- Goodman, L.S.; Wintrobe, M.M. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J. Am. Med. Assoc. 1946, 132, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Amin, L. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef] [PubMed]
- Ozben, T. Mechanisms and strategies to overcome multiple drug resistance in cancer. FEBS Lett. 2006, 580, 2903–2909. [Google Scholar] [CrossRef] [PubMed]
- Stavrovskaya, A.A.; Stromskaya, T.P. Transport proteins of the ABC family and multidrug resistance of tumor cells. Biochemistry 2008, 73, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Couture, L.; Nash, J.A.; Turgeon, J. The ATP-binding cassette transporters and their implication in drug disposition: A special look at the heart. Pharmacol. Rev. 2006, 58, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, A.C.; Al Saig, F.; Cloos, J.; Jansen, G.; Peters, J.G. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance? Cancer Drug Resist. 2018, 1, 6–29. [Google Scholar] [CrossRef]
- Sims-Mourtada, J.; Izzo, J.G.; Ajani, J.; Chao, K.S. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007, 26, 5674–5679. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, K.C.; Ruela-de-Sousa, R.R.; Fuhler, G.M.; Aberson, H.L.; Ferreira, C.V.; Peppelenbosch, M.P.; Spek, C.A. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, C.; Jacinto, A.; Ingham, P.W. Transcriptional activation of hedgehog target genes in Drosophila is mediated directly by the cubitus interruptus protein, a member of the GLI family of zinc finger DNA-binding proteins. Genes Dev. 1996, 10, 2003–2013. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A. Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development 1998, 125, 2203–2212. [Google Scholar] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef] [PubMed]
- Scales, S.; de Sauvage, F. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Shevde, L.A. Hedgehog signaling: Modulation of cancer properies and tumor mircroenvironment. Mol. Cancer 2016, 18, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J. Nevoid basal-cell carcinoma syndrome. Medicine 1987, 66, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Svärd, J.; Rozell, B.; Toftgård, R.; Teglund, S. Tumor suppressor gene co-operativity in compound Patched1 and suppressor of fused heterozygous mutant mice. Mol. Carcinog. 2009, 48, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, J.W.; De Sauvage, F.J. Paracrine Hedgehog Signaling in Cancer. Cancer Res. 2009, 69, 6007–6010. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Callahan, C.A.; Dupree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, R.K. Stroma-initiated Hedgehog signaling takes center stage in B-cell lymphoma. Cancer Res. 2008, 68, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, Y.H.; Chen, J.L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Xie, J. Non-Canonical Hh Signaling in Cancer-Current Understanding and Future Directions. Cancers 2015, 7, 1684–1698. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical Hedgehog signaling. Vitam. Horm. 2012, 88, 55–72. [Google Scholar] [PubMed]
- Blotta, S.; Jakubikova, J.; Calimeri, T.; Roccaro, A.M.; Amodio, N.; Azab, A.K.; Foresta, U.; Mitsiades, C.S.; Rossi, M.; Todoerti, K.; et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood 2012, 120, 5002–5013. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Sheen, I.S.; Jeng, W.J.; Yu, M.C.; Hsiau, H.; Chang, F.Y. High expression of Sonic Hedgehog signaling pathway genes indicates a risk of recurrence of breast carcinoma. Onco-Targets Ther. 2013, 7, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779, Erratum in Nature 2009, 460, 652. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Björling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [PubMed]
- Im, S.; Choi, H.J.; Yoo, C.; Jung, J.H.; Jeon, Y.W.; Suh, Y.J.; Kang, C.S. Hedgehog related protein expression in breast cancer: Gli-2 is associated with poor overall survival. Korean J. Pathol. 2013, 47, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Idress, R.; Habib, S.; Azam, I.; Lalani, E.M. Expression of Androgen Receptor and Cancer Stem Cell Markers (CD44(+)/CD24(−) and ALDH1(+)): Prognostic Implications in Invasive Breast Cancer. Transl. Oncol. 2018, 11, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Tsapakidis, K.; Riobo Del Galdo, N.A.; Papandreou, C.N.; Del Galdo, F.; Anthoney, A.; Sakellaridis, N.; Dimas, K.; Kamposioras, K. The Prognostic Significance of the Hedgehog Signaling Pathway in Colorectal Cancer. Clin. Colorectal Cancer 2016, 15, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Hasanovic, A.; Ruggiero, C.; Jung, S.; Rapa, I.; Signetti, L.; Ben Hadj, M.; Terzolo, M.; Beuschlein, F.; Volante, M.; Hantel, C.; et al. Targeting the multidrug transporter Patched potentiates chemotherapy efficiency on adrenocortical carcinoma in vitro and in vivo. Int. J. Cancer 2018, 143, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Saze, Z.; Terashima, M.; Kogure, M.; Ohsuka, F.; Suzuki, H.; Gotoh, M. Activation of the sonic hedgehog pathway and its prognostic impact in patients with gastric cancer. Dig. Surg. 2012, 29, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ding, H.; Rao, G.; Arora, S.; Saclarides, C.P.; Esparaz, J.; Gattuso, P.; Solorzano, C.C.; Prinz, R.A. Activation of the Sonic Hedgehog pathway in thyroid neoplasms and its potential role in tumor cell proliferation. Endocr. Relat. Cancer 2012, 19, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; You, Z.; Li, T.; Yu, C.; Tao, G.; Hu, M.; Chen, X. Correlation of hedgehog signal activation with chemoradiotherapy sensitivity and survival in esophageal squamous cell carcinomas. Jpn. J. Clin. Oncol. 2011, 41, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Perneel, C.; McKee, C.M.; Van Utterbeeck, F.; Lerut, E.; Verrill, C.; Bryant, R.J.; Joniau, S.; Muschel, R.J.; et al. Patched 1 Expression Correlates with Biochemical Relapse in High-Risk Prostate Cancer Patients. Am. J. Pathol. 2018, 188, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Cooper, M.; Maiti, T.; Beachy, P. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002, 418, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Low, J.A.; de Sauvage, F.J. Clinical experience with Hedgehog pathway inhibitors. J. Clin. Oncol. 2010, 28, 5321–5326. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Spek, A.; Zivkovic, D.; van de Water, S.; Rezaee, F.; Peppelenbosch, M.P. Repression of smoothened by patched-dependent (pro-)vitamin D3 secretion. PLoS Biol. 2006, 4, e232. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.; Scott, M. Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8408–8413. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.R.; Sever, N.; Carlson, M.; Nelson, S.F.; Beachy, P.A.; Parhami, F. Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. J. Biol. Chem. 2007, 282, 8959–8968. [Google Scholar] [CrossRef] [PubMed]
- Yavari, A.; Nagaraj, R.; Owusu-Ansah, E.; Folick, A.; Ngo, K.; Hillman, T.; Call, G.; Rohatgi, R.; Scott, M.P.; Banerjee, U. Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev. Cell 2010, 19, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.K.; Wassif, C.A.; Krakowiak, P.A.; Taipale, J.; Gong, R.; Kelley, R.I.; Porter, F.D.; Beachy, P.A. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet. 2003, 33, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.W.; Davis, H.R., Jr.; Zhu, L.J.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.; Maguire, M.; Golovko, A.; Zeng, M.; et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.R., Jr.; Zhu, L.J.; Hoos, L.M.; Tetzloff, G.; Maguire, M.; Liu, J.; Yao, X.; Iyer, S.P.; Lam, M.H.; Lund, E.G.; et al. Niemann-Pick C1 Like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. J. Biol. Chem. 2004, 279, 33586–33592. [Google Scholar] [CrossRef] [PubMed]
- Infante, R.E.; Abi-Mosleh, L.; Radhakrishnan, A.; Dale, J.D.; Brown, M.S.; Goldstein, J.L. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J. Biol. Chem. 2008, 283, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lu, P.; Chu, J.; Sharom, F. Characterization of fluorescent sterol binding to purified human NPC1. J. Biol. Chem. 2009, 284, 1840–1852. [Google Scholar] [CrossRef] [PubMed]
- Bidet, M.; Joubert, O.; Lacombe, B.; Ciantar, M.; Nehmé, R.; Mollat, P.; Brétillon, L.; Faure, H.; Bittman, R.; Ruat, M.; Mus-Veteau, I. The hedgehog receptor patched is involved in cholesterol transport. PLoS ONE 2011, 6, e23834. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, P.; Labouesse, M. The sterol-sensing domain: Multiple families, a unique role? Trends Genet. 2002, 18, 193–201. [Google Scholar] [CrossRef]
- Martin, V.; Carrillo, G.; Torroja, C.; Guerrero, I. The sterol-sensing domain of Patched protein seems to control Smoothened activity through Patched vesicular trafficking. Curr. Biol. 2001, 11, 601–607. [Google Scholar] [CrossRef]
- Strutt, H.; Thomas, C.; Nakano, Y.; Stark, D.; Neave, B.; Taylor, A.M.; Ingham, P.W. Mutations in the sterol-sensing domain of Patched suggest a role for vesicular trafficking in Smoothened regulation. Curr. Biol. 2001, 11, 608–613. [Google Scholar] [CrossRef]
- Khaliullina, H.; Panáková, D.; Eugster, C.; Riedel, F.; Carvalho, M.; Eaton, S. Patched regulates Smoothened trafficking using lipoprotein-derived lipids. Development 2009, 136, 4111–4121. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.; Muenke, M. Abnormal sterol metabolism in holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Minogue, S.; Chu, K.M.; Westover, E.J.; Covey, D.F.; Hsuan, J.J.; Waugh, M.G. Relationship between phosphatidylinositol 4-phosphate synthesis, membrane organization, and lateral diffusion of PI4KIIalpha at the trans-Golgi network. J. Lipid Res. 2010, 51, 2314–2324. [Google Scholar] [CrossRef] [PubMed]
- Lindström, E.; Shimokawa, T.; Toftgård, R.; Zaphiropoulos, P.G. PTCH mutations: Distribution and analyses. Hum. Mutat. 2006, 27, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Soloviev, A.; Gallagher, J.; Marnef, A.; Kuwabara, P.E. C. elegans patched-3 is an essential gene implicated in osmoregulation and requiring an intact permease transporter domain. Dev. Biol. 2011, 351, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.P.; Levy, B.; Ioannou, Y.A. Evidence for a Niemann-pick C (NPC) gene family: Identification and characterization of NPC1L1. Genomics 2000, 65, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Nakae, T. Identification of essential charged residues in transmembrane segments of the multidrug transporter MexB of Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Bidet, M.; Tomico, A.; Martin, P.; Guizouarn, H.; Mollat, P.; Mus-Veteau, I. The Hedgehog receptor patched functions in multidrug transport and chemotherapy resistance. Mol. Cancer Res. 2012, 10, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Detmer, K.; Walker, A.N.; Jenkins, T.M.; Steele, T.A.; Dannawi, H. Erythroid differentiation in vitro is blocked by cyclopamine, an inhibitor of hedgehog signaling. Blood Cells Mol. Dis. 2000, 26, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Hammer, G.D.; Fassnacht, M.; Lalli, E. Adrenal cancer: Scientific advances. Mol. Cell. Endocrinol. 2011, 351, 1. [Google Scholar] [CrossRef] [PubMed]
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. pH sensing and regulation in cancer. Front. Physiol. 2013, 17, 370. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Gerweck, L.E.; Seetharaman, K. Cellular pH gradient in tumor versus normal tissue: Potential exploitation for the treatment of cancer. Cancer Res. 1996, 56, 1194–1198. [Google Scholar] [PubMed]
- Fiorini, L.; Mus-Veteau, I. Method to Screen Multidrug Transport Inhibitors Using Yeast Overexpressing a Human MDR Transporter. Methods Mol. Biol. 2016, 1432, 303–318. [Google Scholar] [PubMed]
- Fiorini, L.; Tribalat, M.A.; Sauvard, L.; Cazareth, J.; Lalli, E.; Broutin, I.; Thomas, O.P.; Mus-Veteau, I. Natural paniceins from mediterranean sponge inhibit the multidrug resistance activity of Patched and increase chemotherapy efficiency on melanoma cells. Oncotarget 2015, 6, 22282–22297. [Google Scholar] [CrossRef] [PubMed]
- Monachon, M.A.; Burkard, W.P.; Jalfre, M.; Haefely, W. Blockade of central 5-hydroxytryptamine receptors by methiothepin. Naunyn. Schmiedeberg Arch. Pharmacol. 1972, 274, 192–197. [Google Scholar] [CrossRef]
- Dall’Olio, R.; Vaccheri, A.; Montanaro, N. Reduced head-twitch response to quipazine of rats previously treated with methiothepin: Possible involvement of dopaminergic system. Pharmacol. Biochem. Behav. 1985, 23, 43–48. [Google Scholar] [CrossRef]
- Sridhar, R.; Dwivedi, C.; Anderson, J.; Baker, P.B.; Sharma, H.M.; Desai, P.; Engineer, F.N. Effects of verapamil on the acute toxicity of doxorubicin in vivo. J. Natl. Cancer Inst. 1992, 84, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Colombo, T.; Zucchetti, M.; D’Incalci, M. Cyclosporin A markedly changes the distribution of doxorubicin in mice and rats. J. Pharmacol. Exp. Ther. 1994, 269, 22–27. [Google Scholar] [PubMed]
- Bellamy, W.T.; Peng, Y.M.; Odeleye, A.; Ellsworth, L.; Xu, M.J.; Grogan, T.M.; Weinstein, R.S. Cardiotoxicity in the SCID mouse following administration of doxorubicin and cyclosporin A. Anticancer Drugs 1995, 6, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Magdy, T.; Burmeister, B.T.; Burridge, P.W. Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: What is missing? Pharmacol. Ther. 2016, 168, 113–125. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Exogenous Chemotherapy Substance |
|---|---|
| MDR1, ABCB1, P-GP | Anthracyclines (doxorubucin, daunorubicin, epirubicin), actinomycin D, colchicine, podophyllotoxin (etoposide, teniposide), methotrexate (only in carrier-deficient cells), mitomycin C, mitoxantrone, taxenes (paclitaxel, docetaxel), vinca alkaloids (vincristine, vinblastine) |
| MRP1, ABCC1 | Anthracyclines, cochicine, etoposide, heavy metals (arsenite, arsenate, antimonials), vincristine, vinblastine, paclitaxel |
| MRP2, ABCC2, cMOAT | Cisplatin, CPT-11, doxorubicin, etoposide, methotrexate, SN-38, vincristine, vinblastine |
| MRP3, ABCC3 | Cisplatin, doxorubicin, etoposide, methotrexate, teniopside, vincristine |
| MRP4, ABCC4 | Methotrexate, nucleotide analogs, PMEA * |
| MRP5, ABCC5 | Doxorubicin, methotrexate, nucleotide analogs, topotecan |
| MRP6, ABCC6 | Doxorubicin, etoposide, teniposide |
| MRP8, ABCC11 | 5′-Fluorouracil, 5′-fluoro-2′-deoxyuridine, 5′-fluoro-5′-deoxyuridine, PMEA* |
| BCRP, ABCG2, MXR1, ABCP | Anthracyclines, bisantrene, camptothecin, epirubicin, flavopiridol, mitoxantrone, S-38, topotecan |
| Hh Signaling | Example of Cancers | |
|---|---|---|
| Type I | Mutations on Ptch1, Smo, or suppressor of Fused (SUFU). Ligand independent cancers with autonomous Hh signaling | Nevoid basal cell carcinoma syndrome (NBCCS), medulloblastomas, basal cell carcinomas (BCCs), rhabdomyosarcoma |
| Type II | Ligand dependent with autocrine activation | Small-cell lung cancer, prostate, pancreatic, breast cancers |
| Type IIIa | Ligand-dependent, paracrine activation | Pancreatic, ovarian, prostate and colorectal cancers |
| Type IIIb | Ligand-dependent, reverse paracrine activation | B-cell lymphoma, multiple-myeloma and leukemia |
| Type IV | Regulation of stemness-determining genes | Cancer stem cells present in hematological malignancies and in solid tumors |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasanovic, A.; Mus-Veteau, I. Targeting the Multidrug Transporter Ptch1 Potentiates Chemotherapy Efficiency. Cells 2018, 7, 107. https://doi.org/10.3390/cells7080107
Hasanovic A, Mus-Veteau I. Targeting the Multidrug Transporter Ptch1 Potentiates Chemotherapy Efficiency. Cells. 2018; 7(8):107. https://doi.org/10.3390/cells7080107
Chicago/Turabian StyleHasanovic, Anida, and Isabelle Mus-Veteau. 2018. "Targeting the Multidrug Transporter Ptch1 Potentiates Chemotherapy Efficiency" Cells 7, no. 8: 107. https://doi.org/10.3390/cells7080107
APA StyleHasanovic, A., & Mus-Veteau, I. (2018). Targeting the Multidrug Transporter Ptch1 Potentiates Chemotherapy Efficiency. Cells, 7(8), 107. https://doi.org/10.3390/cells7080107