Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency


Abstract
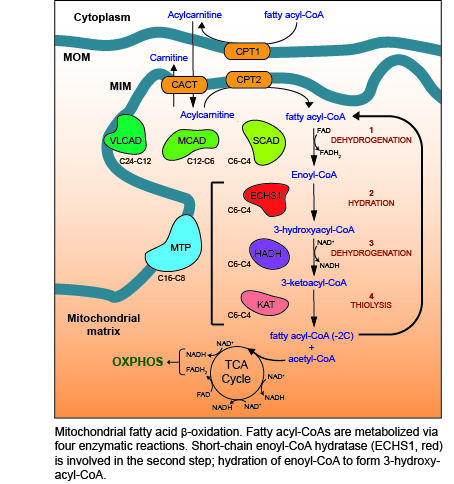
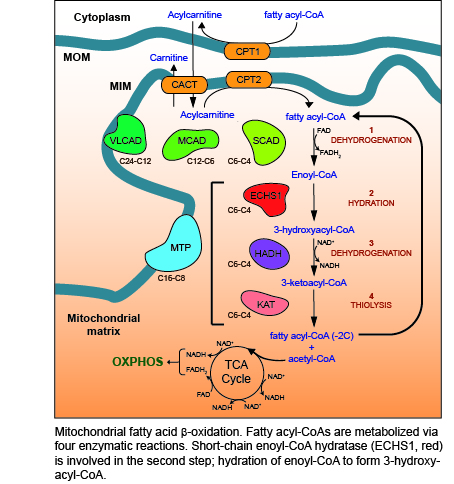
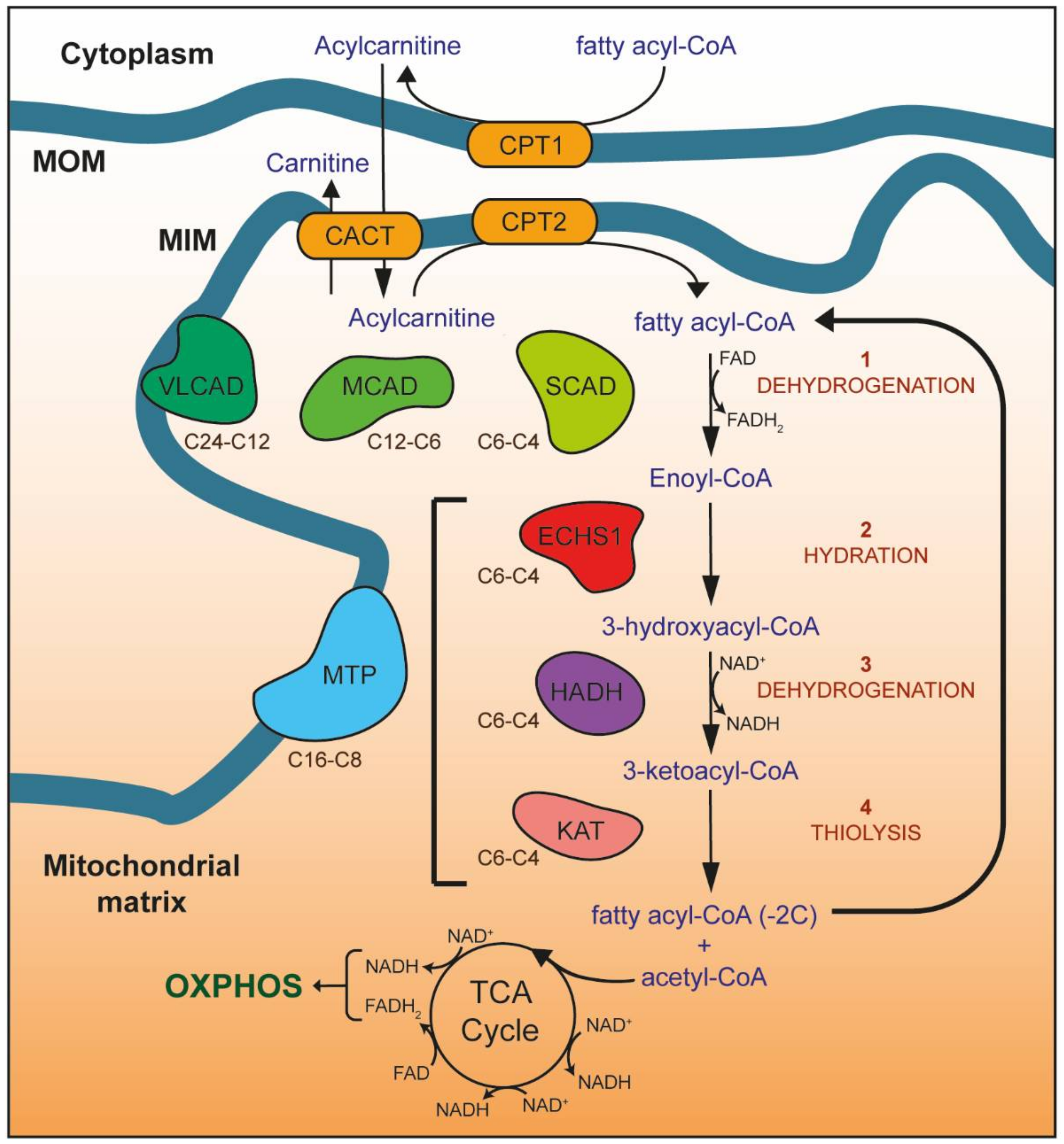
1. Mitochondrial Metabolism
2. Fatty Acid β-Oxidation (FAO)
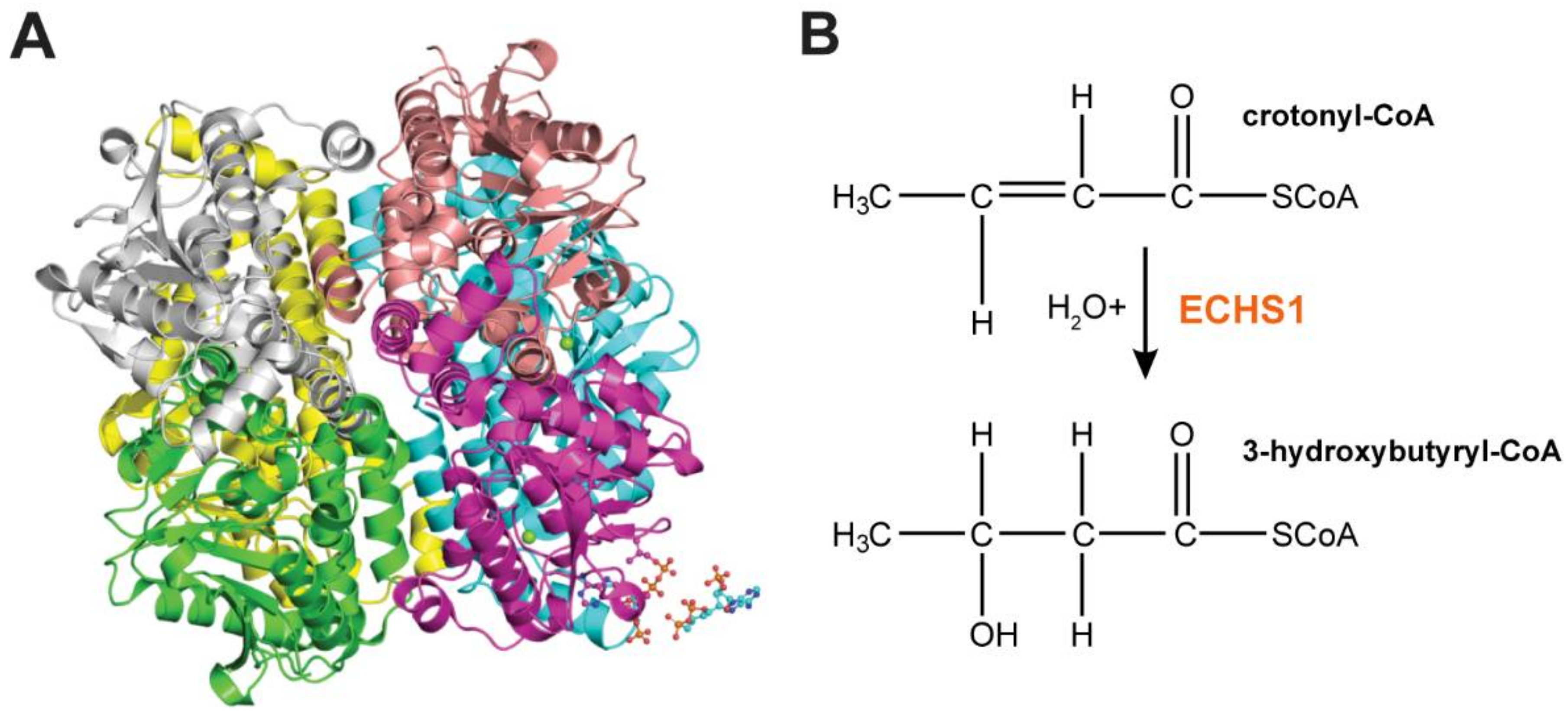
3. ECHS1 is a Multifunctional Enzyme
4. FAO Disease
5. ECHS1 Deficiency (ECHS1D)
6. Pathogenic Mutations in ECHS1
7. Biochemical and Metabolic Characterization of ECHS1D
8. Secondary OXPHOS Defects in ECHS1D
9. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Nsiah-Sefaa, A.; McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid beta-oxidation in mitochondrial disease. Biosci. Rep. 2016, 36, e00313. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.H.; Distelmaier, F.; Smeitink, J.A.M.; Willems, P.H.G.M. OXPHOS mutations and neurodegeneration. EMBO J. 2013, 32, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001, 2, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Wajner, M.; Amaral, A.U. Mitochondrial dysfunction in fatty acid oxidation disorders: Insights from human and animal studies. Biosci. Rep. 2016, 36, e00281. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, K.; Eaton, S. Mitochondrial beta-oxidation. Eur. J. Biochem. 2004, 271, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Van Eunen, K.; Volker-Touw, C.M.L.; Gerding, A.; Bleeker, A.; Wolters, J.C.; van Rijt, W.J.; Martines, A.-C.M.F.; Niezen-Koning, K.E.; Heiner, R.M.; Permentier, H.; et al. Living on the edge: Substrate competition explains loss of robustness in mitochondrial fatty-acid oxidation disorders. BMC Biol. 2016, 14, 107. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.; Pollitt, R.J.; Middleton, B. Human liver long-chain 3-hydroxyacyl-coenzyme a dehydrogenase is a multifunctional membrane-bound beta-oxidation enzyme of mitochondria. Biochem. Biophys. Res. Commun. 1992, 183, 443–448. [Google Scholar] [CrossRef]
- Kanazawa, M.; Ohtake, A.; Abe, H.; Yamamoto, S.; Satoh, Y.; Takayanagi, M.; Niimi, H.; Mori, M.; Hashimoto, T. Molecular cloning and sequence analysis of the cDNA for human mitochondrial short-chain enoyl-CoA hydratase. Enzyme Protein 1993, 47, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Janßen, U.; Davis, E.M.; Le Beau, M.M.; Stoffel, W. Human mitochondrial enoyl-CoA hydratase gene (ECHS1): Structural organization and assignment to chromosome 10q26. 2–q26. 3. Genomics 1997, 40, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Vaca Jacome, A.S.; Rabilloud, T.; Schaeffer-Reiss, C.; Rompais, M.; Ayoub, D.; Lane, L.; Bairoch, A.; Van Dorsselaer, A.; Carapito, C. N-terminome analysis of the human mitochondrial proteome. Proteomics 2015, 15, 2519–2524. [Google Scholar] [CrossRef] [PubMed]
- Hass, G.M.; Hill, R.L. The subunit structure of crotonase. J. Biol. Chem. 1969, 244, 6080–6086. [Google Scholar] [PubMed]
- Fong, J.C.; Schulz, H. Purification and properties of pig heart crotonase and the presence of short chain and long chain enoyl coenzyme A hydratases in pig and guinea pig tissues. J. Biol. Chem. 1977, 252, 542–547. [Google Scholar] [PubMed]
- Stern, J.R.; Del Campillo, A. Enzymatic reaction of crotonyl coenzyme A. J. Am. Chem. Soc. 1953, 75, 2277–2278. [Google Scholar] [CrossRef]
- Yamada, K.; Aiba, K.; Kitaura, Y.; Kondo, Y.; Nomura, N.; Nakamura, Y.; Fukushi, D.; Murayama, K.; Shimomura, Y.; Pitt, J.; et al. Clinical, biochemical and metabolic characterisation of a mild form of human short-chain enoyl-CoA hydratase deficiency: Significance of increased N-acetyl-S-(2-carboxypropyl)cysteine excretion. J. Med. Genet. 2015, 52, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Friederich, M.W.; Burlina, A.; Ruiter, J.P.N.; Coughlin, C.R.; Dishop, M.K.; Gallagher, R.C.; Bedoyan, J.K.; Vaz, F.M.; Waterham, H.R.; et al. Clinical and biochemical characterization of four patients with mutations in ECHS1. Orphanet J. Rare Dis. 2015, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; DiMauro, P.M. Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science 1973, 182, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Karpati, G.; Carpenter, S.; Engel, A.G.; Watters, G.; Allen, J.; Rothman, S.; Klassen, G.; Mamer, O.A. The syndrome of systemic carnitine deficiency. Clinical, morphologic, biochemical, and pathophysiologic features. Neurology 1975, 25, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Lauritzen, R.; Rasmussen, K. Suberylglycine excretion in the urine from a patient with dicarboxylic aciduria. Clin. Chim. Acta 1976, 70, 417–425. [Google Scholar] [CrossRef]
- Kelly, D.P.; Whelan, A.J.; Ogden, M.L.; Alpers, R.; Zhang, Z.F.; Bellus, G.; Gregersen, N.; Dorland, L.; Strauss, A.W. Molecular characterization of inherited medium-chain acyl-CoA dehydrogenase deficiency. Proc. Natl. Acad. Sci. USA 1990, 87, 9236–9240. [Google Scholar] [CrossRef] [PubMed]
- Yokota, I.; Indo, Y.; Coates, P.M.; Tanaka, K. Molecular basis of medium chain acyl-coenzyme A dehydrogenase deficiency. An A to G transition at position 985 that causes a lysine-304 to glutamate substitution in the mature protein is the single prevalent mutation. J. Clin. Investig. 1990, 86, 1000–1003. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, Y.; Narisawa, K.; Miyabayashi, S.; Tada, K.; Coates, P.M. Molecular lesion in patients with medium-chain acyl-CoA dehydrogenase deficiency. Lancet 1990, 335, 1589. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Kompare, M.; Rizzo, W.B. Mitochondrial fatty-acid oxidation disorders. Semin. Pediatr. Neurol. 2008, 15, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Tyni, T.; Pihko, H. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatr. 1999, 88, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [PubMed]
- De Lonlay, P.; Giurgea, I.; Touati, G.; Saudubray, J.M. Neonatal hypoglycaemia: Aetiologies. Semin. Neonatol. 2004, 9, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Kottlors, M.; Jaksch, M.; Ketelsen, U.P.; Weiner, S.; Glocker, F.X.; Lucking, C.H. Valproic acid triggers acute rhabdomyolysis in a patient with carnitine palmitoyltransferase type II deficiency. Neuromuscul. Disord. 2001, 11, 757–759. [Google Scholar] [CrossRef]
- Moczulski, D.; Majak, I.; Mamczur, D. An overview of beta-oxidation disorders. Postepy Hig. Med. Dosw. 2009, 63, 266–277. [Google Scholar]
- Emery, J.L.; Howat, A.J.; Variend, S.; Vawter, G.F. Investigation of inborn errors of metabolism in unexpected infant deaths. Lancet 1988, 2, 29–31. [Google Scholar] [CrossRef]
- Wanders, R.J.; Duran, M.; Ijlst, L.; de Jager, J.P.; van Gennip, A.H.; Jakobs, C.; Dorland, L.; van Sprang, F.J. Sudden infant death and long-chain 3-hydroxyacyl-CoA dehydrogenase. Lancet 1989, 2, 52–53. [Google Scholar] [CrossRef]
- Sim, K.G.; Hammond, J.; Wilcken, B. Strategies for the diagnosis of mitochondrial fatty acid beta-oxidation disorders. Clin. Chim. Acta 2002, 323, 37–58. [Google Scholar] [CrossRef]
- Mansouri, A.; Fromenty, B.; Durand, F.; Degott, C.; Bernuau, J.; Pessayre, D. Assessment of the prevalence of genetic metabolic defects in acute fatty liver of pregnancy. J. Hepatol. 1996, 25, 781. [Google Scholar] [CrossRef]
- Al Mutairi, F.; Shamseldin, H.E.; Alfadhel, M.; Rodenburg, R.J.; Alkuraya, F.S. A lethal neonatal phenotype of mitochondrial short-chain enoyl-CoA hydratase-1 deficiency. Clin. Genet. 2017, 91, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Aulbert, W.; Weigt-Usinger, K.; Thiels, C.; Köhler, C.; Vorgerd, M.; Schreiner, A.; Hoffjan, S.; Rothoeft, T.; Wortmann, S.B.; Heyer, C.M.; et al. Long survival in Leigh syndrome: New cases and review of literature. Neuropediatrics 2014, 45, 346–353. [Google Scholar] [PubMed]
- Saudubray, J.M.; Martin, D.; de Lonlay, P.; Touati, G.; Poggi-Travert, F.; Bonnet, D.; Jouvet, P.; Boutron, M.; Slama, A.; Vianey-Saban, C.; et al. Recognition and management of fatty acid oxidation defects: A series of 107 patients. J. Inherit. Metab. Dis. 1999, 22, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Blok, R.B.; Dahl, H.H.M.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Jackson, C.B.; Murayama, K.; Kremer, L.S.; Schaller, A.; Kotzaeridou, U.; de Vries, M.C.; Schottmann, G.; Santra, S.; Buchner, B.; et al. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann. Clin. Transl. Neurol. 2015, 2, 492–509. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Constantinou, J.; Sidiropoulos, C. ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: Case presentation and initial benefit of intervention. J. Neurol. 2017, 264, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Olgiati, S.; Skorvanek, M.; Quadri, M.; Minneboo, M.; Graafland, J.; Breedveld, G.J.; Bonte, R.; Ozgur, Z.; van den Hout, M.C.G.N.; Schoonderwoerd, K.; et al. Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov. Disord. 2016, 31, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Riley, L.G.; Bratkovic, D.; Ketteridge, D.; Manton, N.; Cowley, M.J.; Gayevskiy, V.; Roscioli, T.; Mohamed, M.; Gardeitchik, T.; et al. Unique presentation of cutis laxa with Leigh-like syndrome due to ECHS1 deficiency. J. Inherit. Metab. Dis. 2017, 40, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, K.P. Paroxysmal dyskinesias. Mov. Disord. 2011, 26, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Buck, N.; Wanders, R.; Ruiter, J.; Waterham, H.; Koster, J.; Yaplito-Lee, J.; Ferdinandusse, S.; Pitt, J. ECHS1 mutations in Leigh disease: A new inborn error of metabolism affecting valine metabolism. Brain 2014, 137, 2903–2908. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Hamzeh, A.R.; Mohamed, M.; Malik, E.M.; Al-Ali, M.T.; Bastaki, F. Novel ECHS1 mutation in an Emirati neonate with severe metabolic acidosis. Metab. Brain Dis. 2016, 31, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Tetreault, M.; Fahiminiya, S.; Antonicka, H.; Mitchell, G.A.; Geraghty, M.T.; Lines, M.; Boycott, K.M.; Shoubridge, E.A.; Mitchell, J.J.; Michaud, J.L.; et al. Whole-exome sequencing identifies novel ECHS1 mutations in Leigh syndrome. Hum. Genet. 2015, 134, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimons, P.E.; Alston, C.L.; Bonnen, P.E.; Hughes, J.; Crushell, E.; Geraghty, M.T.; Tetreault, M.; O’Reilly, P.; Twomey, E.; Sheikh, Y.; et al. Clinical, biochemical, and genetic features of four patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency. Am. J. Med. Genet. A 2018, 176, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Sakai, C.; Yamaguchi, S.; Sasaki, M.; Miyamoto, Y.; Matsushima, Y.; Goto, Y.I. ECHS1 mutations cause combined respiratory chain deficiency resulting in leigh syndrome. Hum. Mutat. 2015, 36, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Ganetzky, R.D.; Bloom, K.; Ahrens-Nicklas, R.; Edmondson, A.; Deardorff, M.A.; Bennett, M.J.; Ficicioglu, C. ECHS1 Deficiency as a Cause of Severe Neonatal Lactic Acidosis. JIMD Rep. 2016, 30, 33–37. [Google Scholar] [PubMed]
- Bedoyan, J.K.; Yang, S.P.; Ferdinandusse, S.; Jack, R.M.; Miron, A.; Grahame, G.; DeBrosse, S.D.; Hoppel, C.L.; Kerr, D.S.; Wanders, R.J.A. Lethal neonatal case and review of primary short-chain enoyl-CoA hydratase (SCEH) deficiency associated with secondary lymphocyte pyruvate dehydrogenase complex (PDC) deficiency. Mol. Genet. Metab. 2017, 120, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Huffnagel, I.C.; Redeker, E.J.W.; Reneman, L.; Vaz, F.M.; Ferdinandusse, S.; Poll-The, B.T. Mitochondrial Encephalopathy and Transient 3-Methylglutaconic Aciduria in ECHS1 Deficiency: Long-Term Follow-Up. JIMD Rep. 2017, 39, 83–87. [Google Scholar] [PubMed]
- Ogawa, E.; Shimura, M.; Fushimi, T.; Tajika, M.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; Ishige, M.; Fuchigami, T.; Yamazaki, T.; et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. J. Inherit. Metab. Dis. 2017, 40, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Ferdinandusse, S.; Ruiter, J.P.; Wanders, R.J.A.; Boneh, A.; Pitt, J. Metabolite studies in HIBCH and ECHS1 defects: Implications for screening. Mol. Genet. Metab. 2015, 115, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.K.; Hunt, S.M.; Scholem, R.; Fowler, K.; Grimes, A.; Mercer, J.F.; Truscott, R.M.; Cotton, R.G.; Rogers, J.G.; Danks, D.M. β-Hydroxyisobutyryl coenzyme A deacylase deficiency: A defect in valine metabolism associated with physical malformations. Pediatrics 1982, 70, 532–538. [Google Scholar] [PubMed]
- Sumegi, B.; Srere, P.A. Complex I binds several mitochondrial NAD-coupled dehydrogenases. J. Biol. Chem. 1984, 259, 15040–15045. [Google Scholar] [PubMed]
- Parker, A.; Engel, P.C. Preliminary evidence for the existence of specific functional assemblies between enzymes of the beta-oxidation pathway and the respiratory chain. Biochem. J. 2000, 345 Pt 3, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mohsen, A.W.; Mihalik, S.J.; Goetzman, E.S.; Vockley, J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J. Biol. Chem. 2010, 285, 29834–29841. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; Fingerhut, R.; Wanders, R.J.; Ullrich, K. Secondary respiratory chain defect in a boy with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Possible diagnostic pitfalls. Eur. J. Pediatr. 2000, 159, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Tajika, M.; Shimura, M.; Carey, K.T.; Stroud, D.A.; Murayama, K.; Ohtake, A.; McKenzie, M. Loss of the Mitochondrial Fatty Acid β-Oxidation Protein Medium-Chain Acyl-Coenzyme A Dehydrogenase Disrupts Oxidative Phosphorylation Protein Complex Stability and Function. Sci. Rep. 2018, 8, 153. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Reference | Peters et al. 2014 [45] | Sakai et al. 2015 [49] | Haack et al. 2015 [40] | |||||
| Patient ID | Patient 1 | Patient 2 | Patient 1 | FI, II:2 | F2: II:1 | F3. II:6 | F4; II:1 | |
| Age at presentation | Birth | 3 months | 2 months | Birth | Birth | Birth | Birth | |
| Death | 4 months | 8 months | NL | 4 months | 11 months | 3 years | 7.5 years | |
| Parental consanguinity | No | No | No | No | No | Yes | No | |
| Mutation 1 (genetic level; protein effect) | c.473C > A; p.Ala158Asp | c.473C > A; p.Ala158Asp | c.2T > G; p.Met1Arg | c.176A > G; p.Asn59Ser | c.197T > C; p.Ile66Thr | c.476A > G; p.Gln159Arg | c.161G > A; p.Arg54His | |
| Mutation 2 (genetic level; protein effect) | c.414 + 3G > C; splicing | c.414 + 3G > C; splicing | c.5C > T; p.Ala2Val | c.476A > G; p.Gln159Arg | c.449A > G; p.Asp150Gly | c.476A > G; p.Gln159Arg | c.817A > G; p.Lys273Glu | |
| T2 hyperintensity | Yes | NL | Yes | Yes | Yes | Yes | NL | |
| Acylcarnitine profile | ND | ND | Normal | Normal | Normal | Normal | NL | |
| PDC activity | Reduced | Reduced | ND | ND | Reduced | ND | ND | |
| OXPHOS activity | ND | ND | Reduced CI, CIII and CIV (patient cells), reduced CI, CIV and CV (immortalized myoblasts) | Reduced CI in liver, normal in heart and muscle | Normal | ND | Normal (but reduced overall ATP production) | |
| OXPHOS complex steady-state levels | ND | ND | Normal | ND | ND | ND | ND | |
| Reference | Haack et al. 2015 [40] (continued) | |||||||
| Patient ID | F5; II:3 | F6, II:1 | F7, II:2 | F8, II:1 | F9, II:2 | F10, II:1 | ||
| Age at presentation | Birth | Birth | 2 years | 1 year | Birth | 11 months | ||
| Death | Alive at 2.3 years | Alive at 3 years | Alive at 5 years | Alive at 8 years | Alive at 16 years | Alive at 31 years | ||
| Parental consanguinity | Yes | No | No | No | No | No | ||
| Mutation 1 (genetic level; protein effect) | c.673T > C; p.Cys225Arg | c.98T > C; p.Phe33Ser | c.268G > A, p.Gly90Arg | c.161G > A; p.Arg54His | c.161G > A; p.Arg54His | c.229G > C; p.Glu77Gln | ||
| Mutation 2 (genetic level; protein effect) | c.673T > C; p.Cys225Arg | c.176A > G; p.Asn59Ser | c.583G > A; p.Gly195Ser | c.394G > A; p.Ala132Thr | c.431dup; p.Leu145Alafs*6 | c.476A > G; p.Gln159Arg | ||
| T2 hyperintensity | Yes | Yes | Yes | ND | Yes | Yes | ||
| Acylcarnitine profile | Normal | Normal | NL | NL | NL | NL | ||
| PDC activity | ND | ND | ND | ND | Normal | ND | ||
| OXPHOS activity | Normal | Reduced CIV in muscle | Normal | ND | Normal | Normal | ||
| OXPHOS complex steady-state levels | ND | ND | ND | ND | ND | ND | ||
| Reference | Ferdinandusse et al. 2015 [17] | Tetreault et al. 2015 [47] | ||||||
| Patient ID | Patient 1 | Patient 2 | Patient 3 | Patient 4 | P1 | P2 | P3 | P4 |
| Age at presentation | Birth | Birth | Early infancy | 1 year | 2.5 months | 2.9 years | 10 months | 6 months |
| Death | 24 h | 2 days | Alive at 7 years | 3 years | 10 months | Alive at 18 years | Alive at 13 years | Alive at 12 years |
| Parental consanguinity | Yes | Yes | No | No | No | No | No | No |
| Mutation 1 (genetic level; protein effect) | c.817A > G; p.Lys273Glu | c.817 > G; p.Lys273Glu | c.433C > T; p.Leu145Phe | c.673T > C; p.Cys225Arg | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala |
| Mutation 2 (genetic level; protein effect) | c.817A > G; p.Lys273Glu | c.817A > G; p.Lys273Glu | c.476A > G; p.Gln159Arg | c.674G > C; p.Cys225Ser | c.583G > A; p.Gly195Ser | c.713C > T; p.Ala238Val | c.713C > T; p.Ala238Val | c.476A > G; p.Gln159Arg |
| T2 hyperintensity | ND | Yes | NL | Yes | Yes | Yes | Yes | Yes |
| Acylcarnitine profile | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| PDC activity | Reduced | Reduced | ND | ND | Reduced | Normal | ND | Normal |
| OXPHOS activity | Normal | Normal | ND | ND | Mild reduction of CI and CIII in muscle | Normal | Normal | Normal |
| OXPHOS complex steady-state levels | Normal | Normal | ND | ND | ND | ND | ND | Reduced CIV in fibroblasts |
| Reference | Yamada et al. 2015 [16] | Ganetzky et al. 2016 [50] | Olgiati et al. 2016 [42] | Nair et al. 2016 [46] | ||||
| Patient ID | III-2 | III-3 | Patient 1 | Patient 2 | II-1 | II-2 | Patient 1 | |
| Age at presentation | 10 months | 7 months | Prenatal | Prenatal | 3.5 years | 4.5 years | Birth | |
| Death | Alive at 7 years | 5 years | 16 h | 24 h | Alive at 17 years | Alive at 15 years | 24 h | |
| Parental consanguinity | No | No | No | No | No | No | Yes | |
| Mutation 1 (genetic level; protein effect) | c.176A > G; p.Asn59Ser | c.176A > G; p.Asn59Ser | c.8C > A; p.Ala3Asp | c.8C > A; p.Ala3Asp | c.232G > T; p.Glu78Ter | c.232G > T; p.Glu78Ter | c.842A > G; p.Glu281Gly | |
| Mutation 2 (genetic level; protein effect) | c.413C > T; p.Ala138Val | c.413C > T; p.Ala138Val | c.389T > A; p.Val130Asp | c.389T > A; p.Val130Asp | c.518C > T; p.Ala173Val | c.518C > T; p.Ala173Val | c.842A > G; p.Glu281Gly | |
| T2 hyperintensity | Yes | Yes | ND | ND | Yes | Yes | ND | |
| Acylcarnitine profile | Normal | Normal | Mild C4 elevation | Mild C4 elevation | ND | ND | Elevated C4 and C6 | |
| PDC activity | ND | ND | ND | ND | ND | ND | ND | |
| OXPHOS activity | Normal | ND | ND | ND | ND | ND | ND | |
| OXPHOS complex steady-state levels | ND | ND | ND | ND | ND | ND | ND | |
| Reference | Mahajan et al. 2017 [41] | Al Mutairi et al. 2017 [35] | Balasubramaniam et al. 2017 [43] | Bedoyan et al. 2017 [51] | Huffnagel et al. 2017 [52] | |||
| Patient ID | Patient 1 | Patient 1 | Patient 2 | Patient 1 | Patient 1 | Patient 1 | ||
| Age at presentation | 8 years | Birth | Birth | 17 months | Birth | 6 weeks | ||
| Death | Alive at 8 years | 2 days | 8 ho | Alive at 4.5 years | 39 days | Alive at 26 years | ||
| Parental consanguinity | No | Yes | Yes | No | No | No | ||
| Mutation 1 (genetic level; protein effect) | c.518C > T; p.Ala173Val | c.88 + 5G > A; p.Ala31Glufs*23 | c.88 + 5G > A; p.Ala31Glufs*23 | c.476A > G; p.Gln159Arg | c.836T > C; p.Phe279Ser | c.229G > C p.Glu77Gln | ||
| Mutation 2 (genetic level; protein effect) | c.817A > G; p.Lys273Glu | c.88 + 5G > A; p.Ala31Glufs*23 | c.88 + 5G > A; p.Ala31Glufs*23 | c.538A > G; p.Thr180Ala | c.8C > A; p.Ala3Asp | c.563C > T p.Ala188Val | ||
| T2 hyperintensity | Yes | ND | ND | Yes | Yes | Yes | ||
| Acylcarnitine profile | ND | Mild C3, C4, C5 and C10 elevation | Normal | ND | ND | Normal | ||
| PDC activity | ND | Reduced | Normal | ND | Reduced | ND | ||
| OXPHOS activity | ND | ND | Normal | ND | Reduced | Normal | ||
| OXPHOS complex steady-state levels | ND | ND | ND | ND | ND | ND | ||
| Reference | Ogawa et al. 2017 [53] | Fitzsimons et al. 2018 [48] | ||||||
| Patient ID | Pt376 | Pt536 | Pt1038 | Pt1135 | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
| Age at presentation | NL | NL | NL | NL | 5 months | 3 months | 5 months | 2 weeks |
| Death | NL | NL | NL | NL | 3 years | 21 months | 28 months | 13 months |
| Parental consanguinity | No | No | No | No | Yes | Yes | Yes | Yes |
| Mutation 1 (genetic level; protein effect) | c.98T > C; p.Phe33Ser | c.5C > T; p.Ala2Val | c.5C > T; p.Ala2Val | c.5C > T; p.Ala2Val | c.476A > G; p.Gln159Arg | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala |
| Mutation 2 (genetic level; protein effect) | c.176A > G; p.Asn59Ser | c.1A > G; p.Met1Val | c.176A > G; p.Asn59Ser | c.176A > G; p.Asn59Ser | c.476A > G; p.Gln159Arg | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala |
| T2 hyperintensity | ND | ND | ND | ND | Yes | Yes | Yes | Yes |
| Acylcarnitine profile | ND | ND | ND | ND | Normal | ND | Mild reduction of free carnitine and long-chain acylcarnitines | Normal |
| PDC activity | ND | ND | ND | ND | Reduced | ND | Normal | ND |
| OXPHOS activity | Reduced CIV | Normal | Normal (but reduced oxygen consumption rate) | Reduced CI | Normal | ND | Reduced CIII in muscle | ND |
| OXPHOS complex steady-state levels | ND | ND | ND | ND | ND | ND | ND | ND |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharpe, A.J.; McKenzie, M. Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency. Cells 2018, 7, 46. https://doi.org/10.3390/cells7060046
Sharpe AJ, McKenzie M. Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency. Cells. 2018; 7(6):46. https://doi.org/10.3390/cells7060046
Chicago/Turabian StyleSharpe, Alice J., and Matthew McKenzie. 2018. "Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency" Cells 7, no. 6: 46. https://doi.org/10.3390/cells7060046
APA StyleSharpe, A. J., & McKenzie, M. (2018). Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency. Cells, 7(6), 46. https://doi.org/10.3390/cells7060046