Lysosomal Sequestration Impairs the Activity of the Preclinical FGFR Inhibitor PD173074

 , , ,
, , ,  and
and 
Abstract
:
1. Introduction
2. Material and methods
2.1. Drugs and Chemicals
2.2. Cell Culture
2.3. Fluorescence Spectroscopy
2.4. RNA Isolation and Quantitative Real-Time PCR (qPCR)
2.5. Flow Cytometry
2.6. Live Cell Microscopy
2.7. Confocal Fluorescence Microscopy
2.8. Western Blot Analysis
2.9. Cell Viability Assay
2.10. Statistical Analysis
3. Results
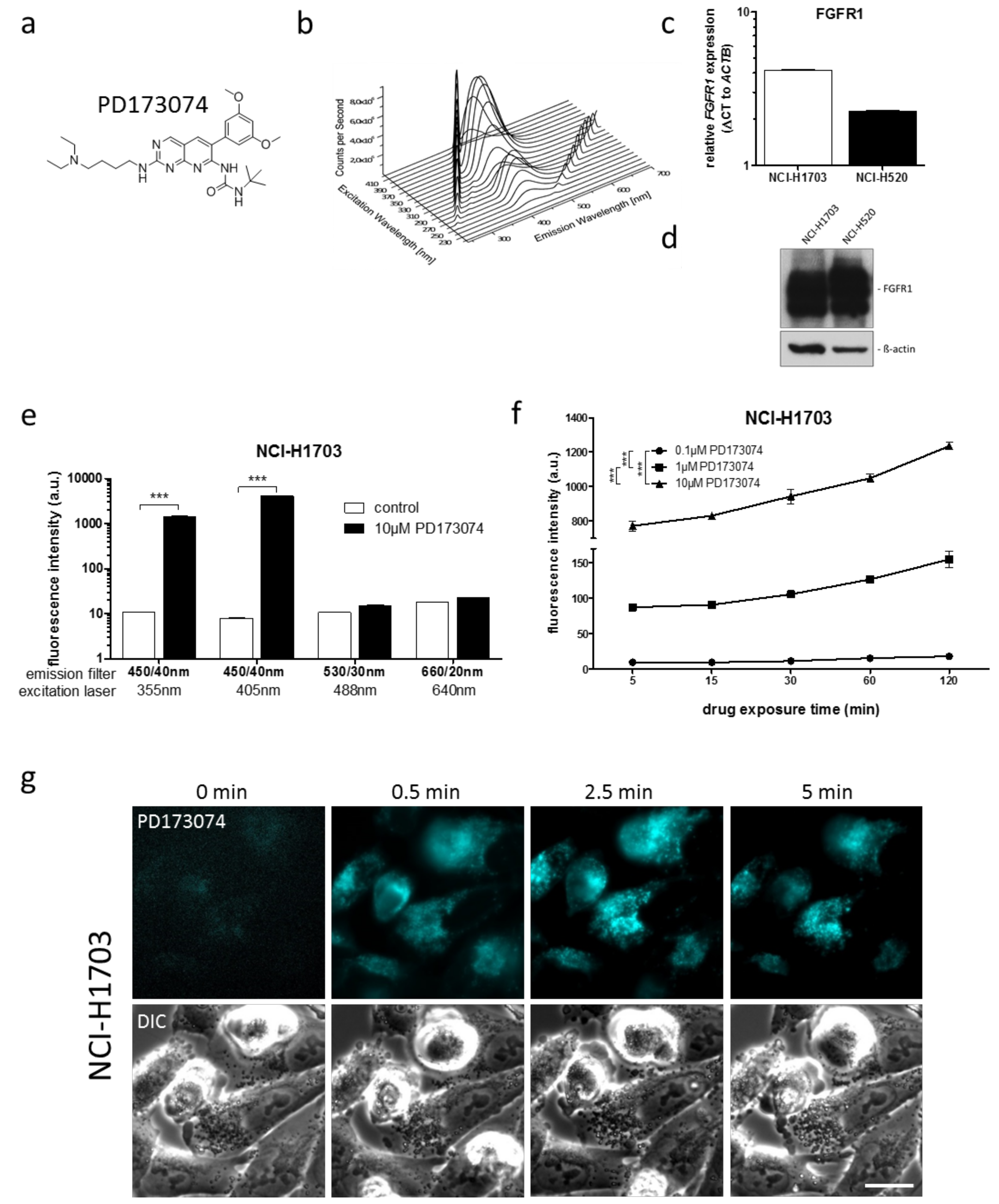
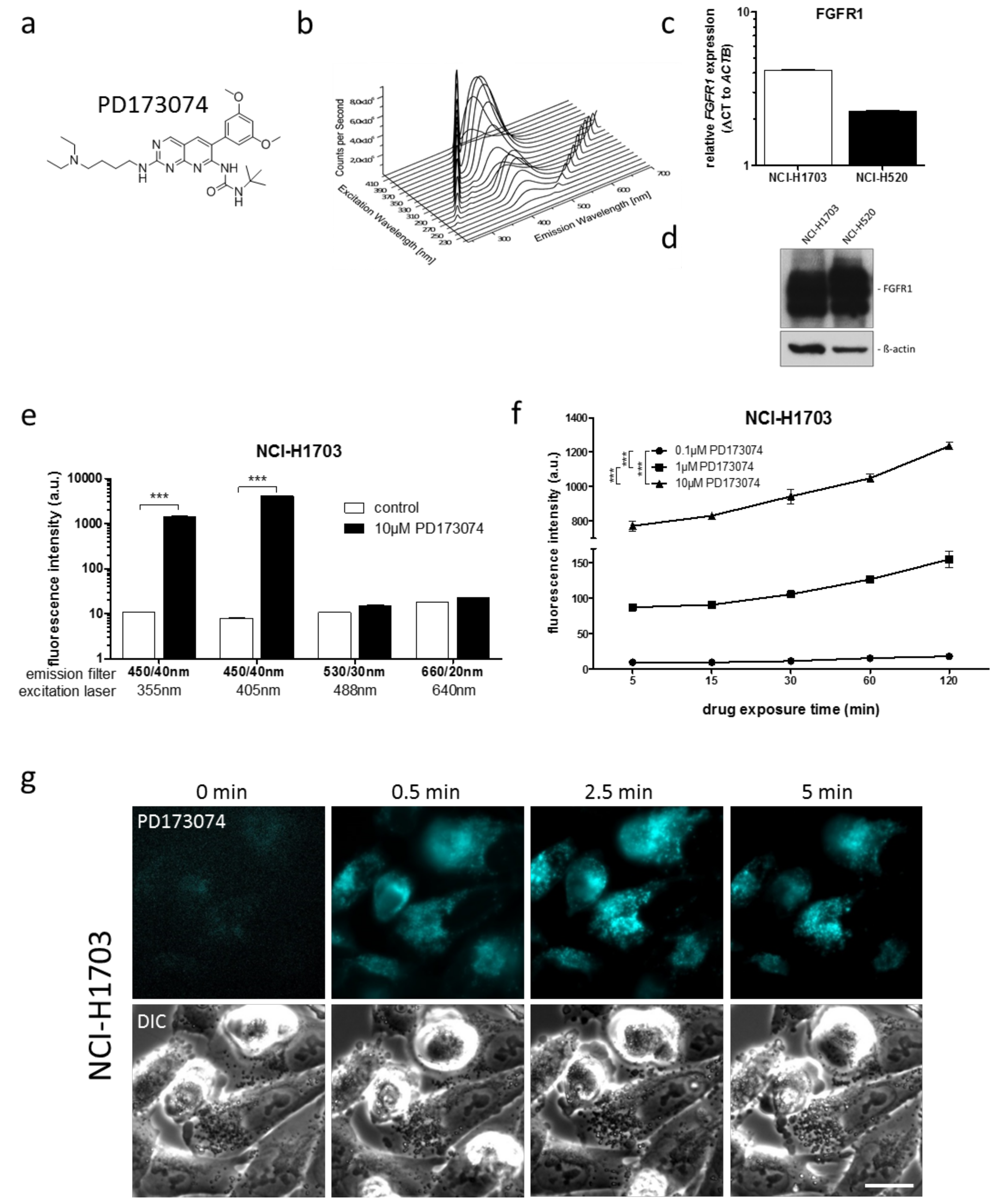
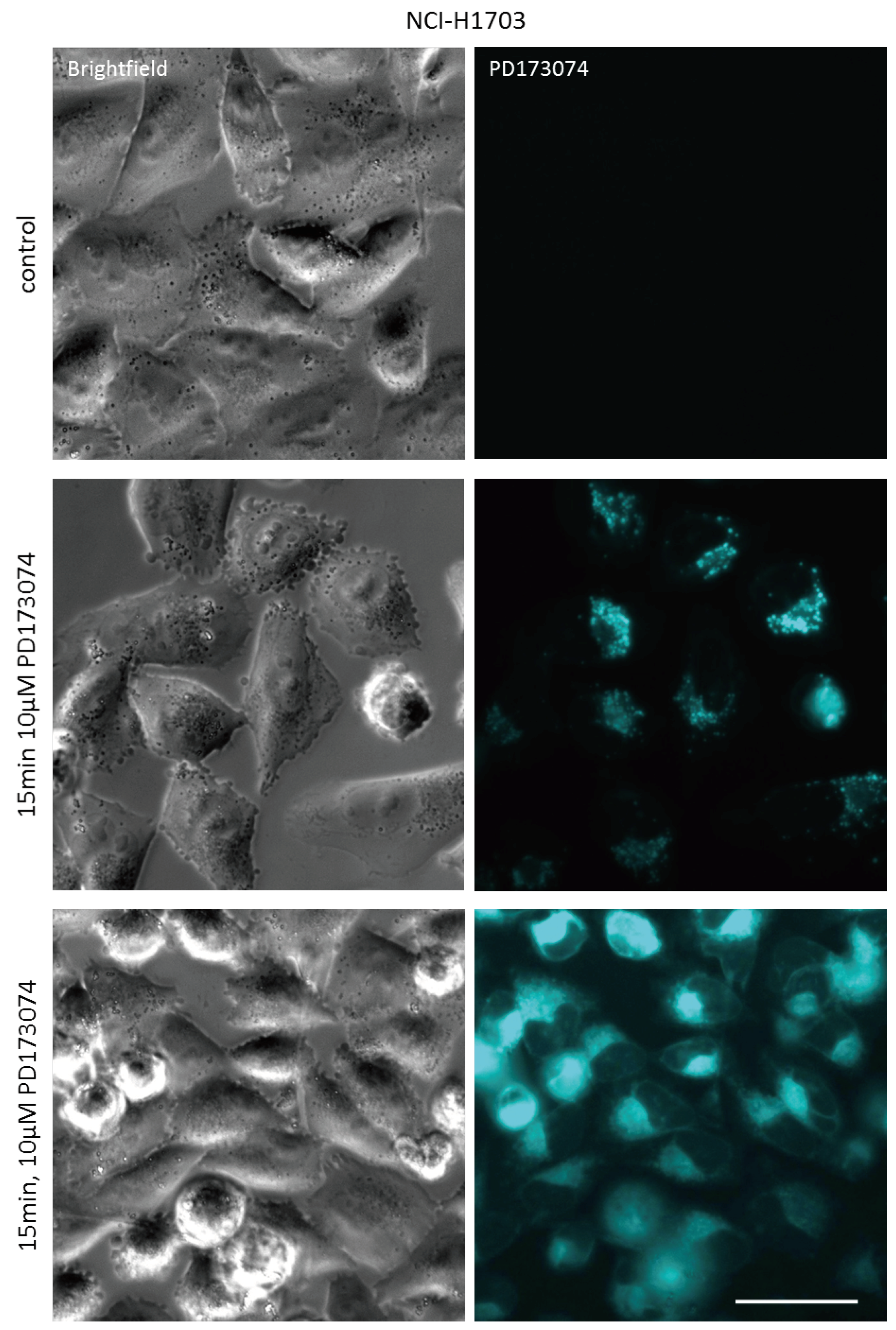
3.1. PD173074 Emits Fluorescence Under Cell-Free Conditions and in Treated Lung Cancer Cells
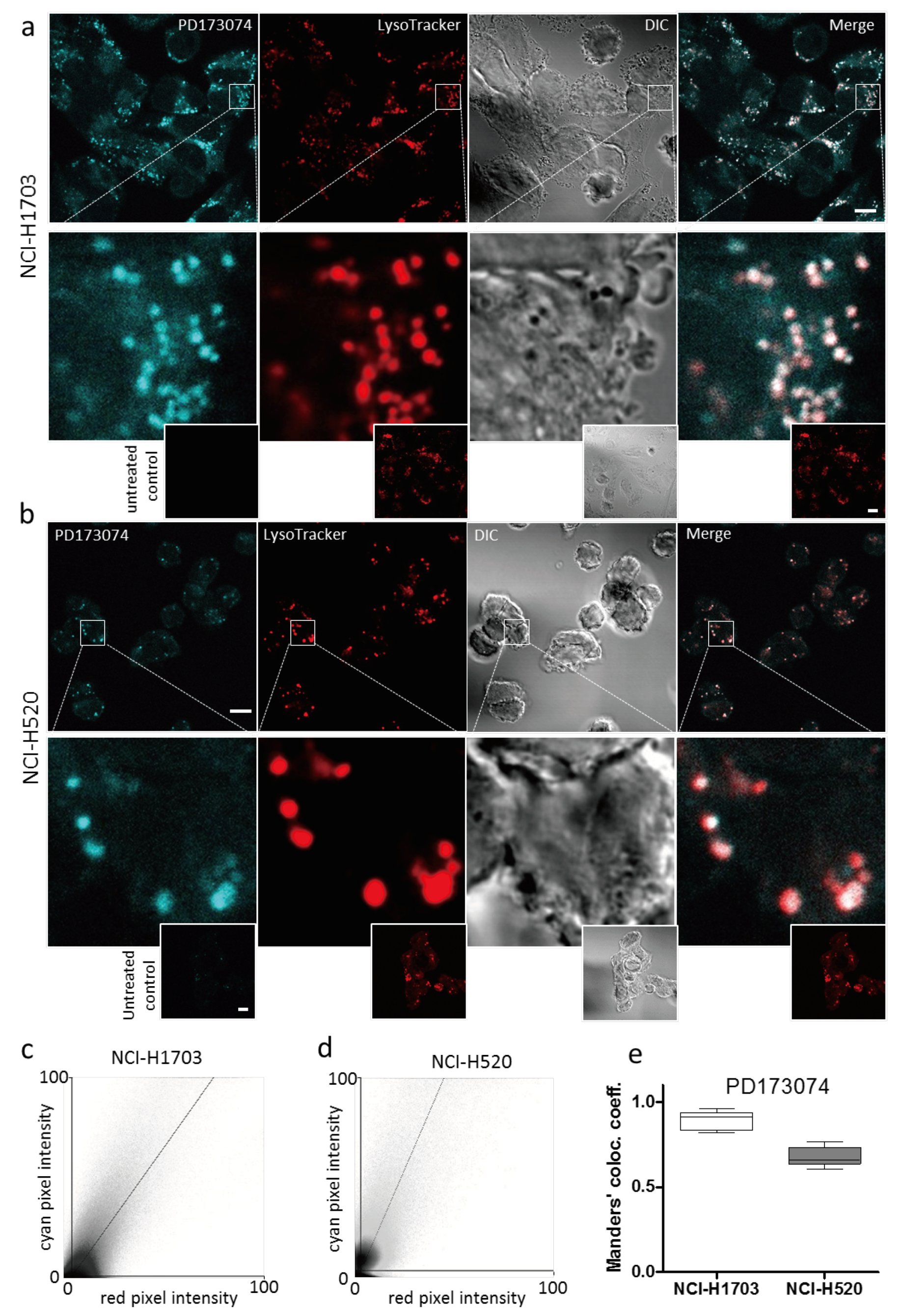
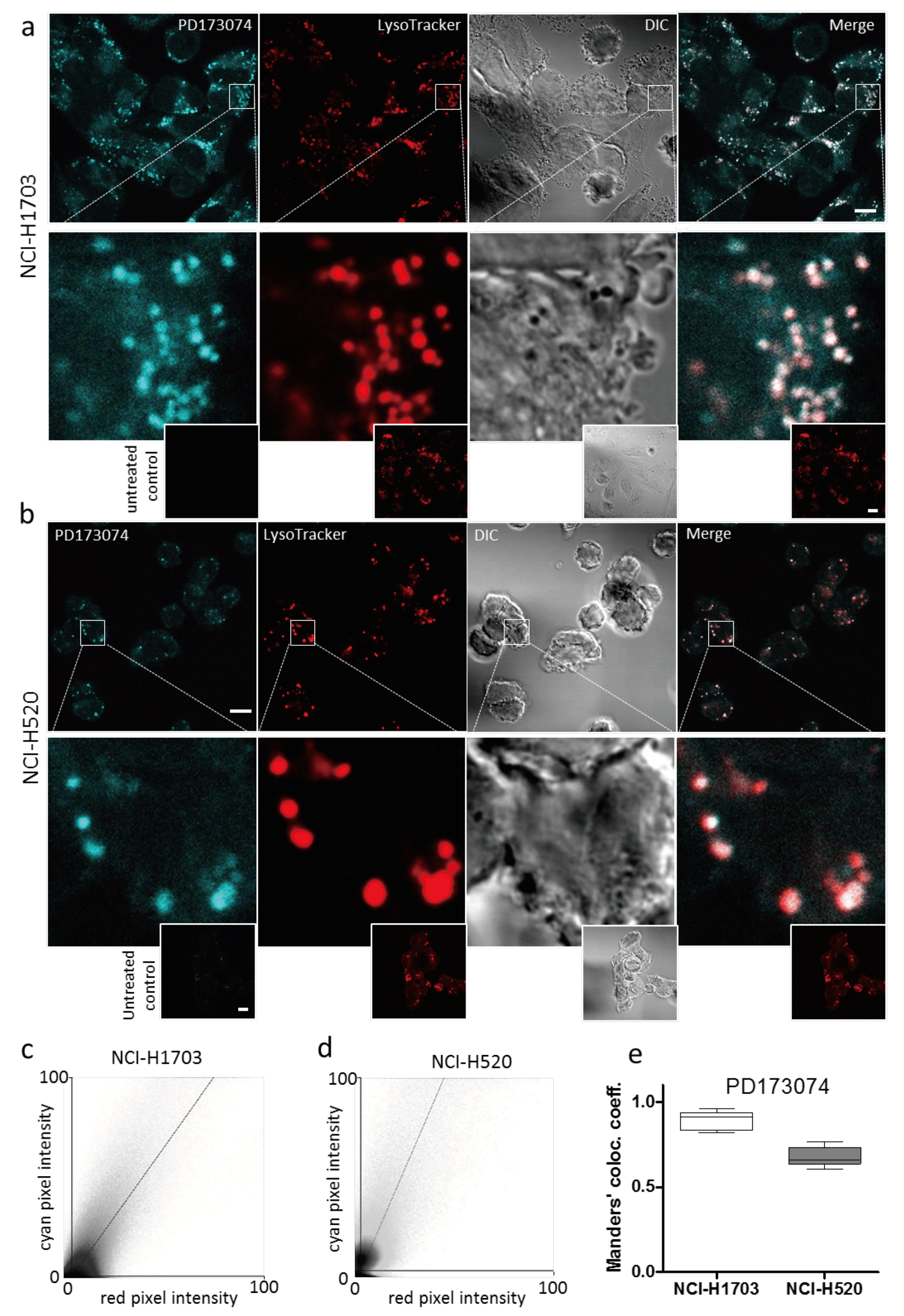
3.2. PD173074 Localizes Selectively to Lysosomes of FGFR1-Driven Lung Cancer Cells
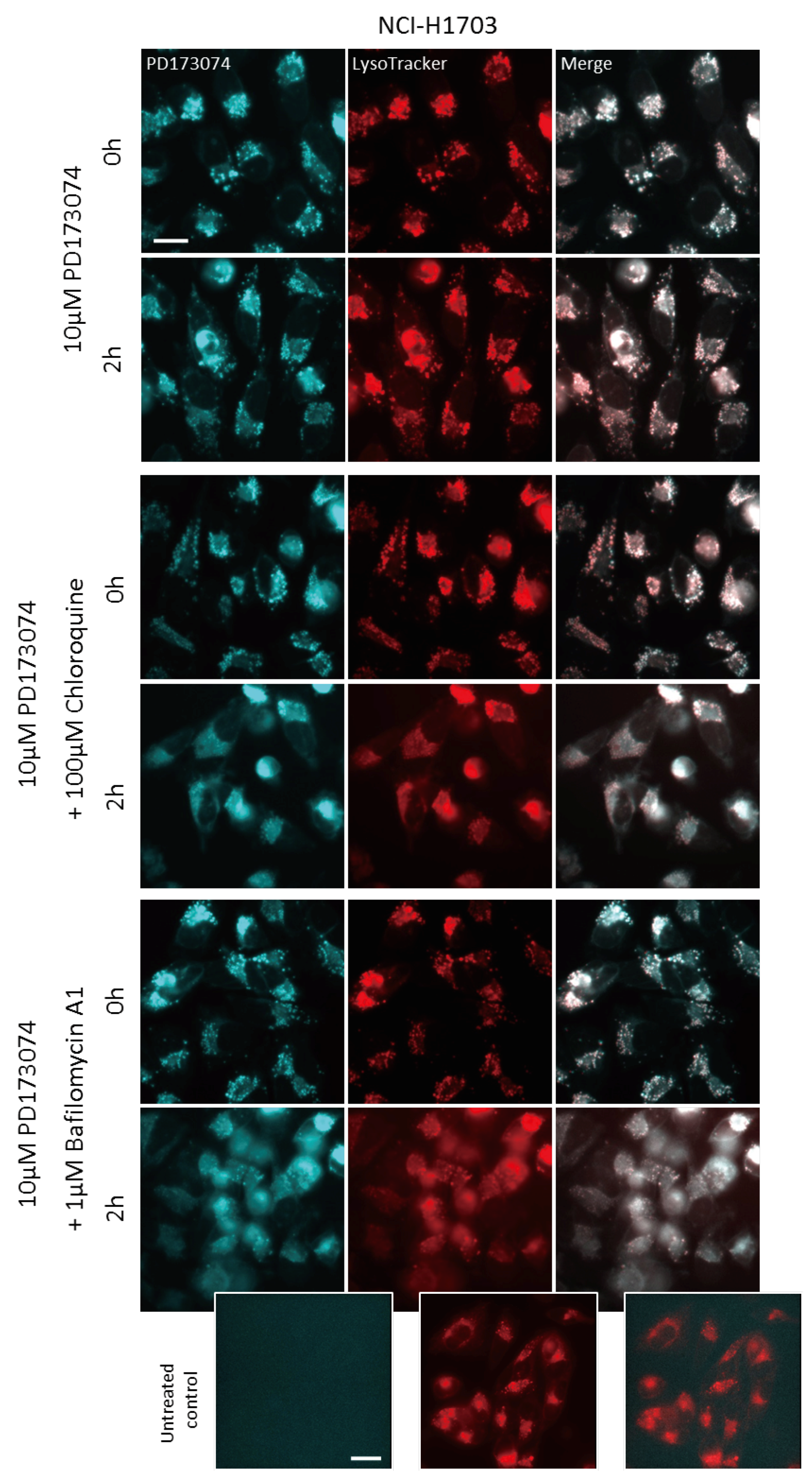
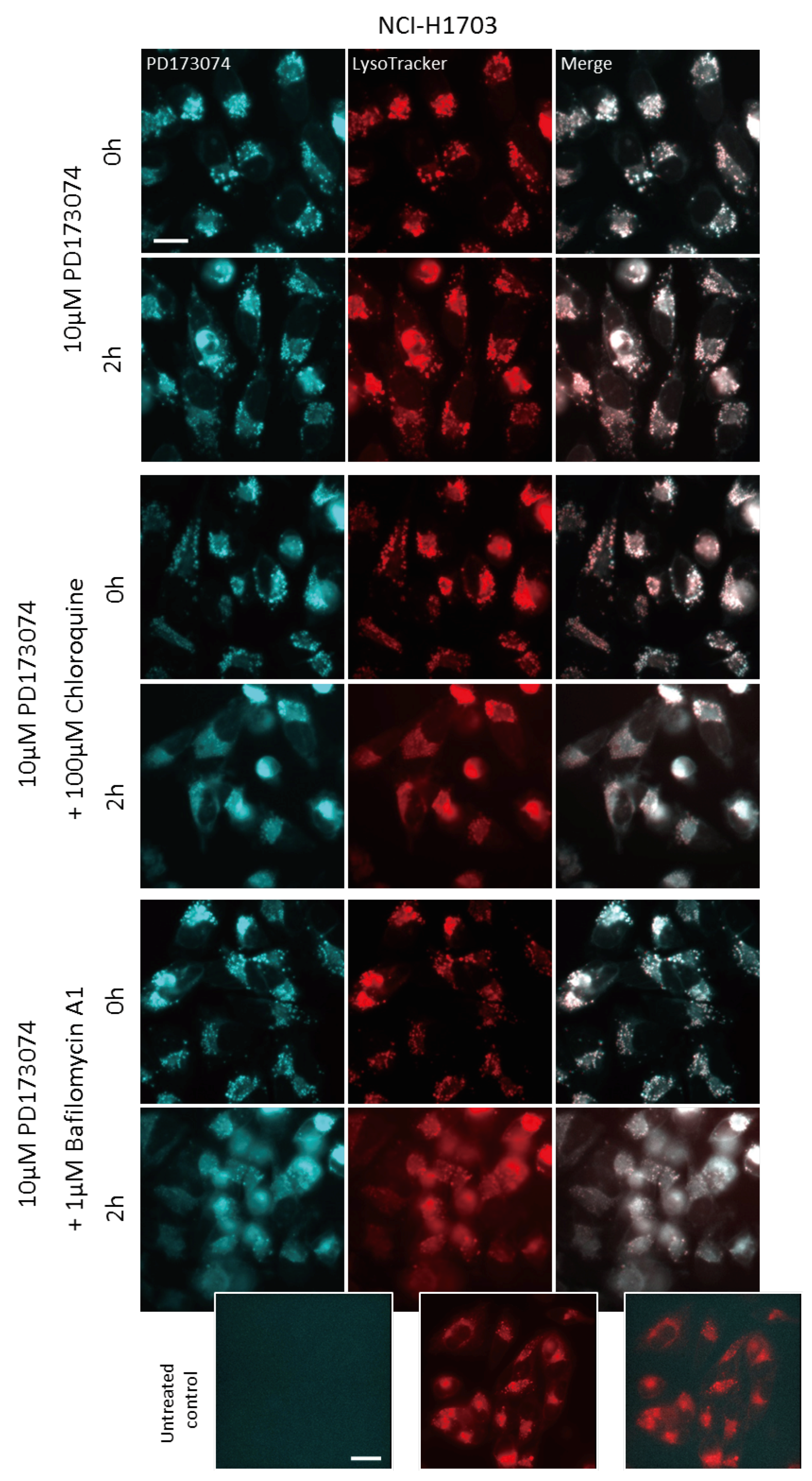
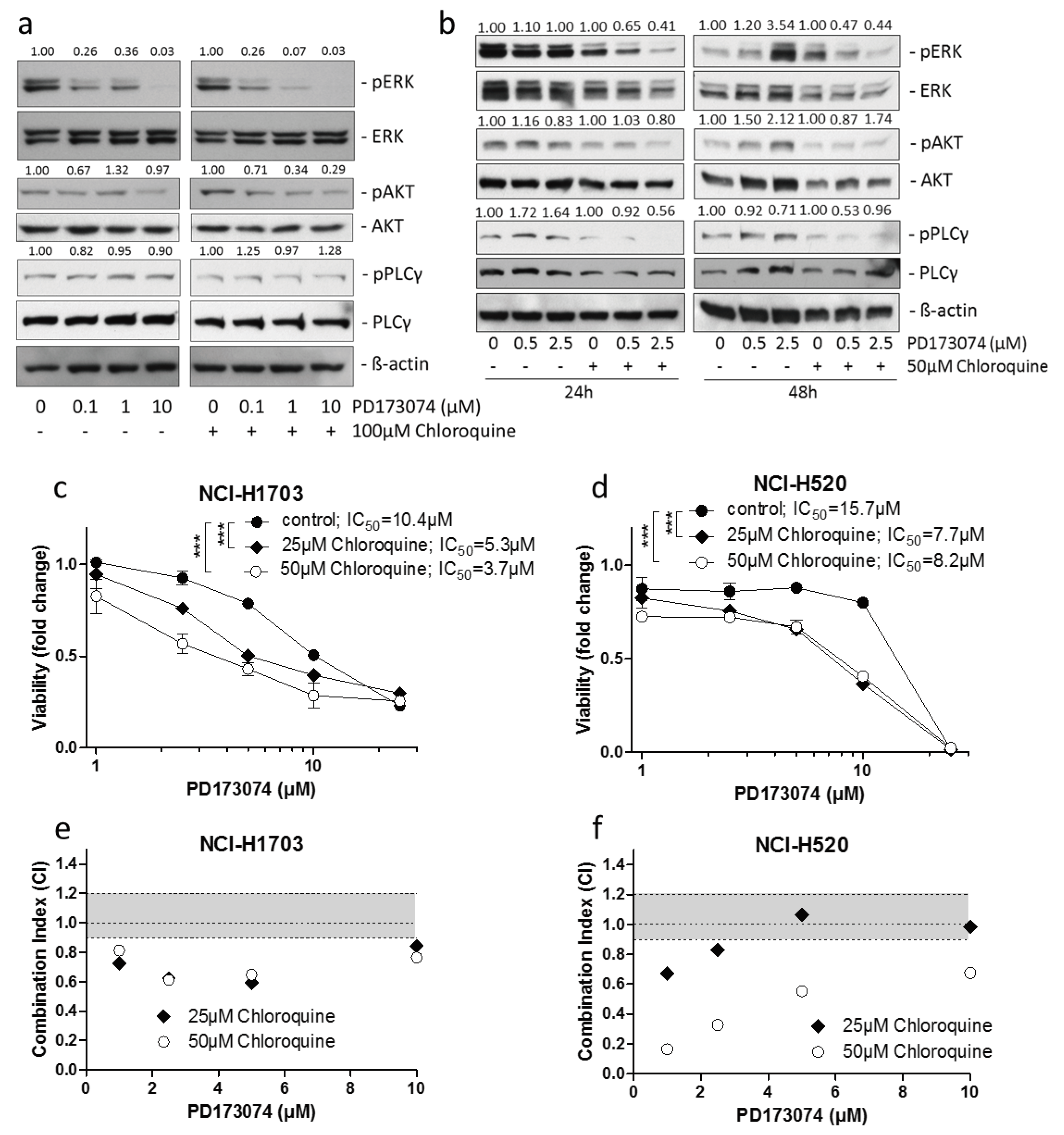
3.3. Luminal alkalinization Abrogates Lysosomal PD173074 Sequestration
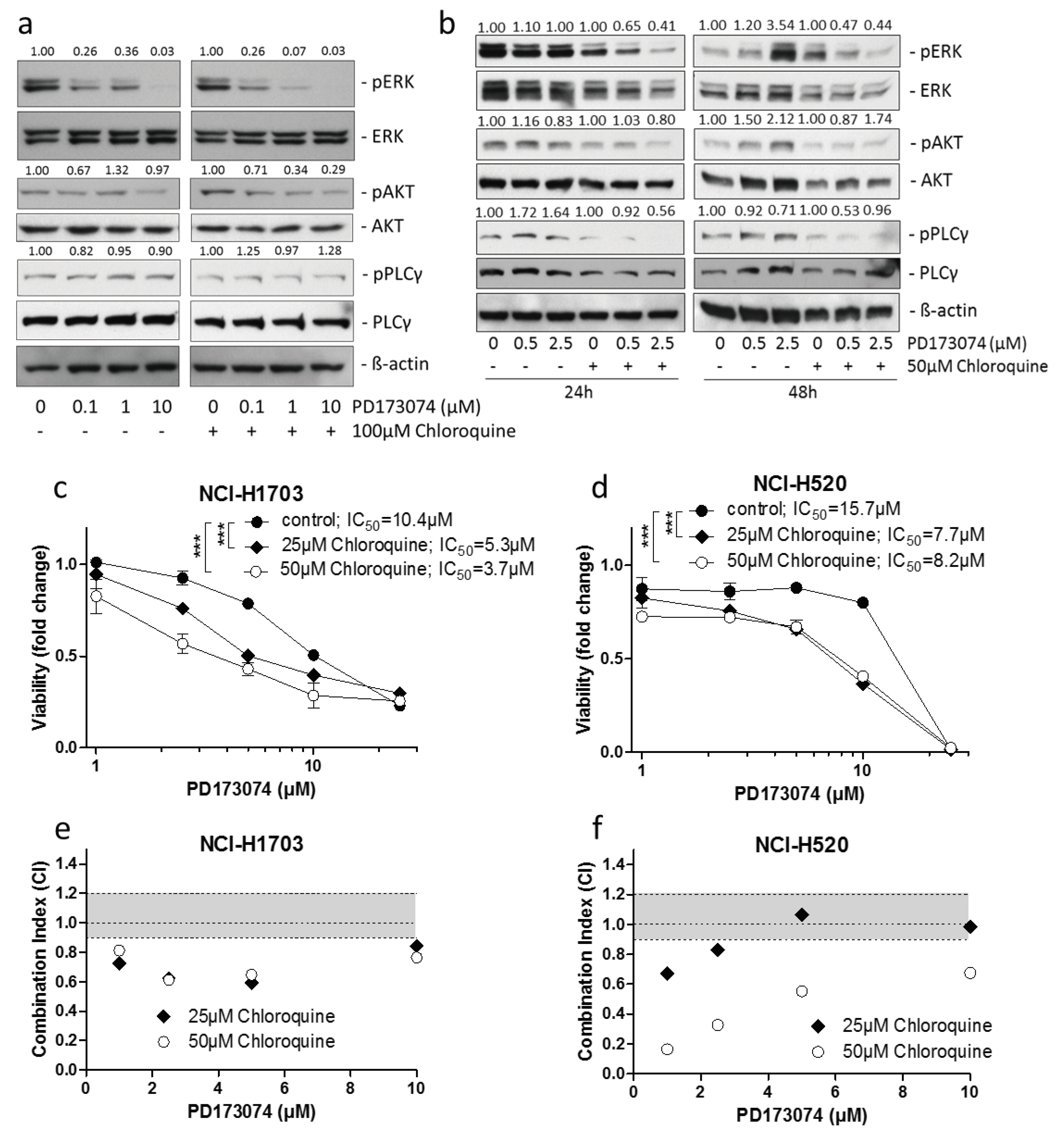
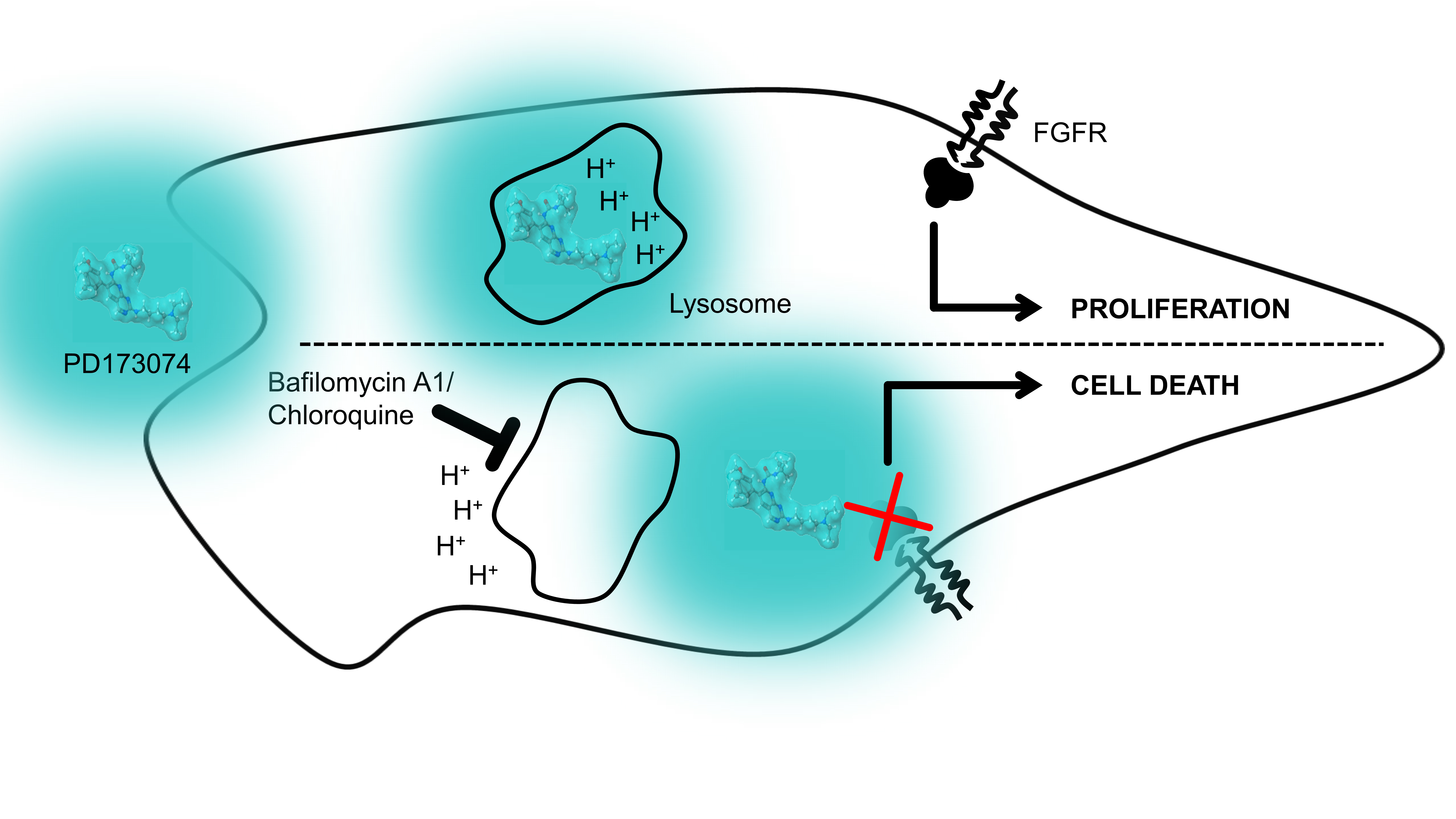
3.4. Lysosomal alkalinization Enhances the Cytotoxic Potential of PD173074
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Heinzle, C.; Sutterluty, H.; Grusch, M.; Grasl-Kraupp, B.; Berger, W.; Marian, B. Targeting fibroblast-growth-factor-receptor-dependent signaling for cancer therapy. Expert Opin. Ther. Targets 2011, 15, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Tanner, Y.; Grose, R.P. Dysregulated fgf signalling in neoplastic disorders. Semin. Cell Dev. Biol. 2016, 53, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.P.; Fearon, A.E.; Grose, R.P. Careless talk costs lives: Fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015, 25, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Hallinan, N.; Finn, S.; Cuffe, S.; Rafee, S.; O’Byrne, K.; Gately, K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat. Rev. 2016, 46, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M. Therapeutics targeting fgf signaling network in human diseases. Trends Pharmacol. Sci. 2016, 37, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Paur, J.; Nika, L.; Maier, C.; Moscu-Gregor, A.; Kostka, J.; Huber, D.; Mohr, T.; Heffeter, P.; Schrottmaier, W.C.; Kappel, S.; et al. Fibroblast growth factor receptor 3 isoforms: Novel therapeutic targets for hepatocellular carcinoma? Hepatology 2015, 62, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Schelch, K.; Hoda, M.A.; Klikovits, T.; Munzker, J.; Ghanim, B.; Wagner, C.; Garay, T.; Laszlo, V.; Setinek, U.; Dome, B.; et al. Fibroblast growth factor receptor inhibition is active against mesothelioma and synergizes with radio- and chemotherapy. Am. J. Respir. Crit. Care Med. 2014, 190, 763–772. [Google Scholar] [CrossRef]
- Roth, G.J.; Binder, R.; Colbatzky, F.; Dallinger, C.; Schlenker-Herceg, R.; Hilberg, F.; Wollin, S.L.; Kaiser, R. Nintedanib: From discovery to the clinic. J. Med. Chem. 2015, 58, 1053–1063. [Google Scholar] [CrossRef]
- Dey, N.; Williams, C.; Leyland-Jones, B.; De, P. Mutation matters in precision medicine: A future to believe in. Cancer Treat. Rev. 2017, 55, 136–149. [Google Scholar] [CrossRef]
- Chell, V.; Balmanno, K.; Little, A.S.; Wilson, M.; Andrews, S.; Blockley, L.; Hampson, M.; Gavine, P.R.; Cook, S.J. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in fgfr3 as a mechanism of acquired resistance. Oncogene 2013, 32, 3059–3070. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Abreu, M.T.; Pearson, A.; Campbell, J.; Shnyder, S.D.; Knowles, M.A.; Ashworth, A.; Turner, N.C. Parallel rna interference screens identify egfr activation as an escape mechanism in fgfr3-mutant cancer. Cancer Discover. 2013, 3, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, H.; Yun, M.R.; Kang, H.N.; Pyo, K.H.; Park, H.J.; Lee, J.M.; Choi, H.M.; Ellinghaus, P.; Ocker, M.; et al. Activation of the met kinase confers acquired drug resistance in fgfr-targeted lung cancer therapy. Oncogenesis 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.B.; Narasu, L.; Gundla, R.; Dayam, R.; J, A.R.P.S. Fibroblast growth factor receptor inhibitors. Curr. Pharm. Des. 2013, 19, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Latigo, J.; Jeffery, R.E.; Nye, E.; Poulsom, R.; Spencer-Dene, B.; Lemoine, N.R.; Stamp, G.W.; Aboagye, E.O.; Seckl, M.J. The fibroblast growth factor receptor inhibitor pd173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 2009, 69, 8645–8651. [Google Scholar] [CrossRef]
- Vibet, S.; Maheo, K.; Gore, J.; Dubois, P.; Bougnoux, P.; Chourpa, I. Differential subcellular distribution of mitoxantrone in relation to chemosensitization in two human breast cancer cell lines. Drug Metabol. Dispos. 2007, 35, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Gryczynski, I.; Gryczynski, Z.; Lakowicz, J.R.; Yang, D.; Burke, T.G. Fluorescence spectral properties of the anticancer drug topotecan by steady-state and frequency domain fluorometry with one-photon and multi-photon excitation. Photochem. Photobiol. 1999, 69, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Trummer, B.J.; Iyer, V.; Balu-Iyer, S.V.; O’Connor, R.; Straubinger, R.M. Physicochemical properties of epidermal growth factor receptor inhibitors and development of a nanoliposomal formulation of gefitinib. J. Pharm. Sci. 2012, 101, 2763–2776. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef] [PubMed]
- Englinger, B.; Kallus, S.; Senkiv, J.; Heilos, D.; Gabler, L.; van Schoonhoven, S.; Terenzi, A.; Moser, P.; Pirker, C.; Timelthaler, G.; et al. Intrinsic fluorescence of the clinically approved multikinase inhibitor nintedanib reveals lysosomal sequestration as resistance mechanism in fgfr-driven lung cancer. J. Exp. Clin. Cancer Res. 2017, 36. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updat. 2016, 24, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132. [Google Scholar] [CrossRef] [PubMed]
- Moinuddin, F.M.; Shinsato, Y.; Komatsu, M.; Mitsuo, R.; Minami, K.; Yamamoto, M.; Kawahara, K.; Hirano, H.; Arita, K.; Furukawa, T. Atp7b expression confers multidrug resistance through drug sequestration. Oncotarget 2016, 7, 22779–22790. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; Setinek, U.; Mohr, T.; Kindas-Mugge, I.; Vetterlein, M.; Dekan, G.; Eckersberger, F.; Caldas, C.; Micksche, M. Evidence for a role of fgf-2 and fgf receptors in the proliferation of non-small cell lung cancer cells. Int. J. Cancer 1999, 83, 415–423. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef] [Green Version]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Wynes, M.W.; Hinz, T.K.; Gao, D.; Martini, M.; Marek, L.A.; Ware, K.E.; Edwards, M.G.; Bohm, D.; Perner, S.; Helfrich, B.A.; et al. Fgfr1 mrna and protein expression, not gene copy number, predict fgfr tki sensitivity across all lung cancer histologies. Clin. Cancer Res. 2014, 20, 3299–3309. [Google Scholar] [CrossRef]
- Kallus, S.; Englinger, B.; Senkiv, J.; Laemmerer, A.; Heffeter, P.; Berger, W.; Kowol, C.R.; Keppler, B.K. Nanoformulations of anticancer fgfr inhibitors with improved therapeutic index. Nanomedicine 2018, 14, 2632–2643. [Google Scholar] [CrossRef]
- Fu, D.; Zhou, J.; Zhu, W.S.; Manley, P.W.; Wang, Y.K.; Hood, T.; Wylie, A.; Xie, X.S. Imaging the intracellular distribution of tyrosine kinase inhibitors in living cells with quantitative hyperspectral stimulated raman scattering. Nat. Chem. 2014, 6, 614–622. [Google Scholar] [CrossRef]
- Trapp, S.; Rosania, G.R.; Horobin, R.W.; Kornhuber, J. Quantitative modeling of selective lysosomal targeting for drug design. EBJ 2008, 37, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Dutta, D.; Donaldson, J.G. Search for inhibitors of endocytosis: Intended specificity and unintended consequences. Cell Logist. 2012, 2, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Es, H.H.; Renkema, H.; Aerts, H.; Schurr, E. Enhanced lysosomal acidification leads to increased chloroquine accumulation in cho cells expressing the pfmdr1 gene. Mol. Biochem. Parasitol. 1994, 68, 209–219. [Google Scholar] [CrossRef]
- Yoshimori, T.; Yamamoto, A.; Moriyama, Y.; Futai, M.; Tashiro, Y. Bafilomycin a1, a specific inhibitor of vacuolar-type h(+)-atpase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991, 266, 17707–17712. [Google Scholar] [PubMed]
- Zhao, F.L.; Yang, G.H.; Xiang, S.; Gao, D.D.; Zeng, C. In silico analysis of the effect of mutation on epidermal growth factor receptor in non-small-cell lung carcinoma: From mutational analysis to drug designing. J. Biomol. Struct. Dyn. 2017, 35, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Ung, M.H.; Varn, F.S.; Cheng, C. In silico frameworks for systematic pre-clinical screening of potential anti-leukemia therapeutics. Expert Opin. Drug Discov. 2016, 11, 1213–1222. [Google Scholar] [CrossRef] [Green Version]
- Hossam, M.; Lasheen, D.S.; Abouzid, K.A. Covalent egfr inhibitors: Binding mechanisms, synthetic approaches, and clinical profiles. Arch. Pharm. (Weinheim) 2016, 349, 573–593. [Google Scholar] [CrossRef]
- Lazar-Molnar, E.; Scandiuzzi, L.; Basu, I.; Quinn, T.; Sylvestre, E.; Palmieri, E.; Ramagopal, U.A.; Nathenson, S.G.; Guha, C.; Almo, S.C. Structure-guided development of a high-affinity human programmed cell death-1: Implications for tumor immunotherapy. EBioMedicine 2017, 17, 30–44. [Google Scholar] [CrossRef]
- Lu, S.; Sung, T.; Lin, N.; Abraham, R.T.; Jessen, B.A. Lysosomal adaptation: How cells respond to lysosomotropic compounds. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Lee, R.F.S.; Theiner, S.; Meibom, A.; Koellensperger, G.; Keppler, B.K.; Dyson, P.J. Application of imaging mass spectrometry approaches to facilitate metal-based anticancer drug research. Metallomics 2017, 9, 365–381. [Google Scholar] [CrossRef]
- Rutherford, A.V.; Willingham, M.C. Ultrastructural localization of daunomycin in multidrug-resistant cultured cells with modulation of the multidrug transporter. J. Histochem. Cytochem. 1993, 41, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Ranftler, C.; Meisslitzer-Ruppitsch, C.; Stangl, H.; Rohrl, C.; Fruhwurth, S.; Neumuller, J.; Pavelka, M.; Ellinger, A. 2-deoxy-d-glucose treatment changes the golgi apparatus architecture without blocking synthesis of complex lipids. Histochem. Cell Biol. 2015, 143, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Thurber, G.M.; Yang, K.S.; Reiner, T.; Kohler, R.H.; Sorger, P.; Mitchison, T.; Weissleder, R. Single-cell and subcellular pharmacokinetic imaging allows insight into drug action in vivo. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Herlevsen, M.; Oxford, G.; Owens, C.R.; Conaway, M.; Theodorescu, D. Depletion of major vault protein increases doxorubicin sensitivity and nuclear accumulation and disrupts its sequestration in lysosomes. Mol. Cancer Ther. 2007, 6, 1804–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Adar, Y.; Stark, M.; Bram, E.E.; Nowak-Sliwinska, P.; van den Bergh, H.; Szewczyk, G.; Sarna, T.; Skladanowski, A.; Griffioen, A.W.; Assaraf, Y.G. Imidazoacridinone-dependent lysosomal photodestruction: A pharmacological trojan horse approach to eradicate multidrug-resistant cancers. Cell Death Dis. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Gyparaki, M.T.; Papavassiliou, A.G. Lysosome: The cell’s ’suicidal bag’ as a promising cancer target. Trends Mol. Med. 2014, 20, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Sandhoff, K. Cancer: Killing from the inside. Nature 2013, 502, 312–313. [Google Scholar] [CrossRef]
- Zhang, I.; Cui, Y.; Amiri, A.; Ding, Y.; Campbell, R.E.; Maysinger, D. Pharmacological inhibition of lipid droplet formation enhances the effectiveness of curcumin in glioblastoma. Eur. J. Pharm. Biopharm. 2016, 100, 66–76. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Laser (nM) | Bandpass Filter (nm) | Fluorescence Intensity (a.u. mean ± SD) a | Relative Fluorescence Increase b | |
|---|---|---|---|---|---|
| Control | PD173074 | ||||
| NCI-H1703 | 405 | 450/40 | 7.99 ± 0.08 | 4129.54 ± 43.02 | 516.8 *** |
| NCI-H520 | 405 | 450/40 | 11.34 ± 0.10 | 6210.24 ± 191.27 | 547.6 *** |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Englinger, B.; Kallus, S.; Senkiv, J.; Laemmerer, A.; Moser, P.; Gabler, L.; Groza, D.; Kowol, C.R.; Heffeter, P.; Grusch, M.; et al. Lysosomal Sequestration Impairs the Activity of the Preclinical FGFR Inhibitor PD173074. Cells 2018, 7, 259. https://doi.org/10.3390/cells7120259
Englinger B, Kallus S, Senkiv J, Laemmerer A, Moser P, Gabler L, Groza D, Kowol CR, Heffeter P, Grusch M, et al. Lysosomal Sequestration Impairs the Activity of the Preclinical FGFR Inhibitor PD173074. Cells. 2018; 7(12):259. https://doi.org/10.3390/cells7120259
Chicago/Turabian StyleEnglinger, Bernhard, Sebastian Kallus, Julia Senkiv, Anna Laemmerer, Patrick Moser, Lisa Gabler, Diana Groza, Christian R. Kowol, Petra Heffeter, Michael Grusch, and et al. 2018. "Lysosomal Sequestration Impairs the Activity of the Preclinical FGFR Inhibitor PD173074" Cells 7, no. 12: 259. https://doi.org/10.3390/cells7120259
APA StyleEnglinger, B., Kallus, S., Senkiv, J., Laemmerer, A., Moser, P., Gabler, L., Groza, D., Kowol, C. R., Heffeter, P., Grusch, M., & Berger, W. (2018). Lysosomal Sequestration Impairs the Activity of the Preclinical FGFR Inhibitor PD173074. Cells, 7(12), 259. https://doi.org/10.3390/cells7120259