UBE2E1 Is Preferentially Expressed in the Cytoplasm of Slow-Twitch Fibers and Protects Skeletal Muscles from Exacerbated Atrophy upon Dexamethasone Treatment

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Constructs and Materials
2.2. Cell Culture and Knockdown Experiments
2.3. Animals and Knockdown Experiments
2.4. Immunohistochemistry
2.5. Protein Extraction
2.6. qRT-PCR
2.7. Statistical Analysis
3. Results and Discussion
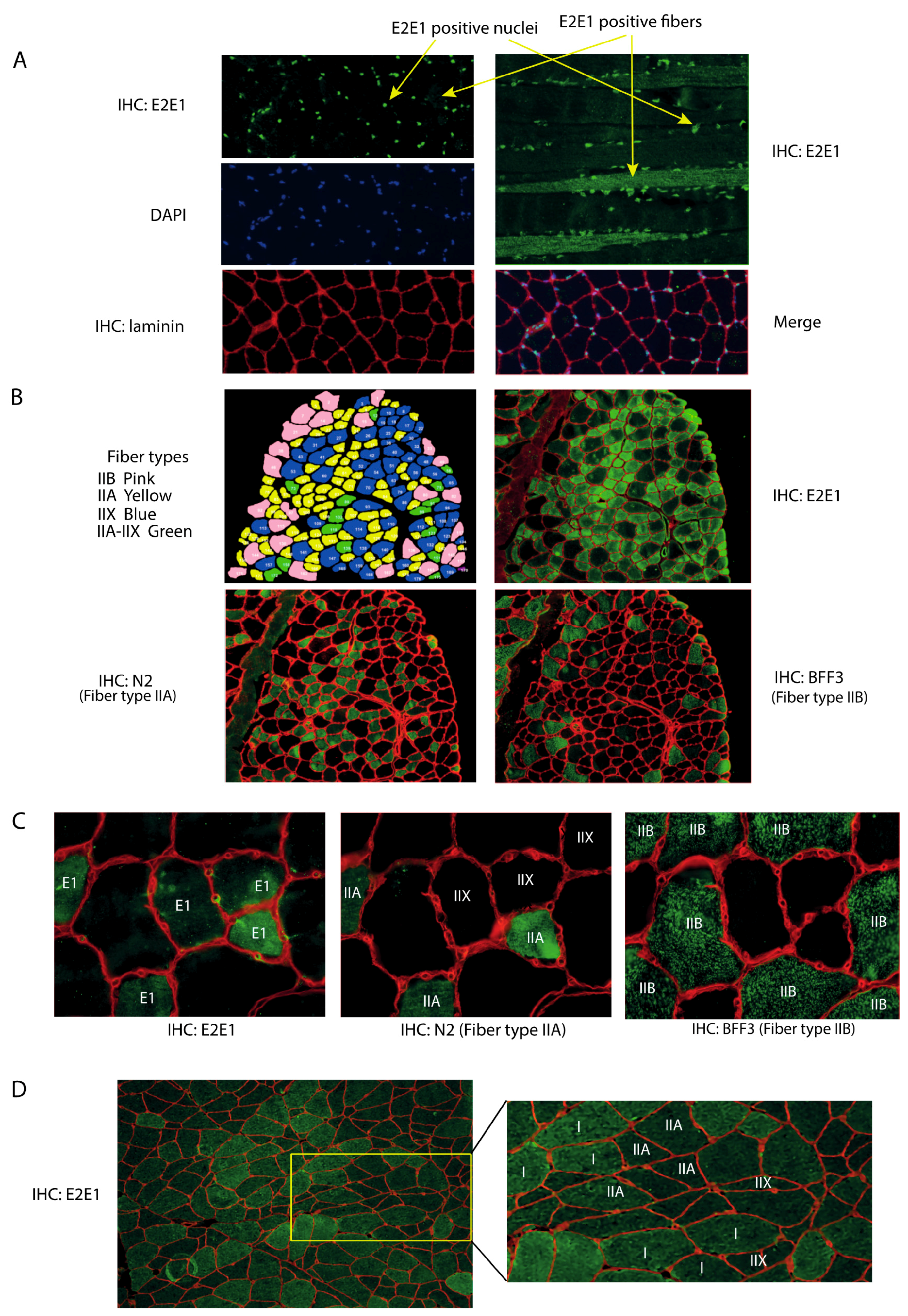
3.1. E2E1 Is Present in Both the Nuclei and the Cytoplasm of Mouse and Human Muscle Cells
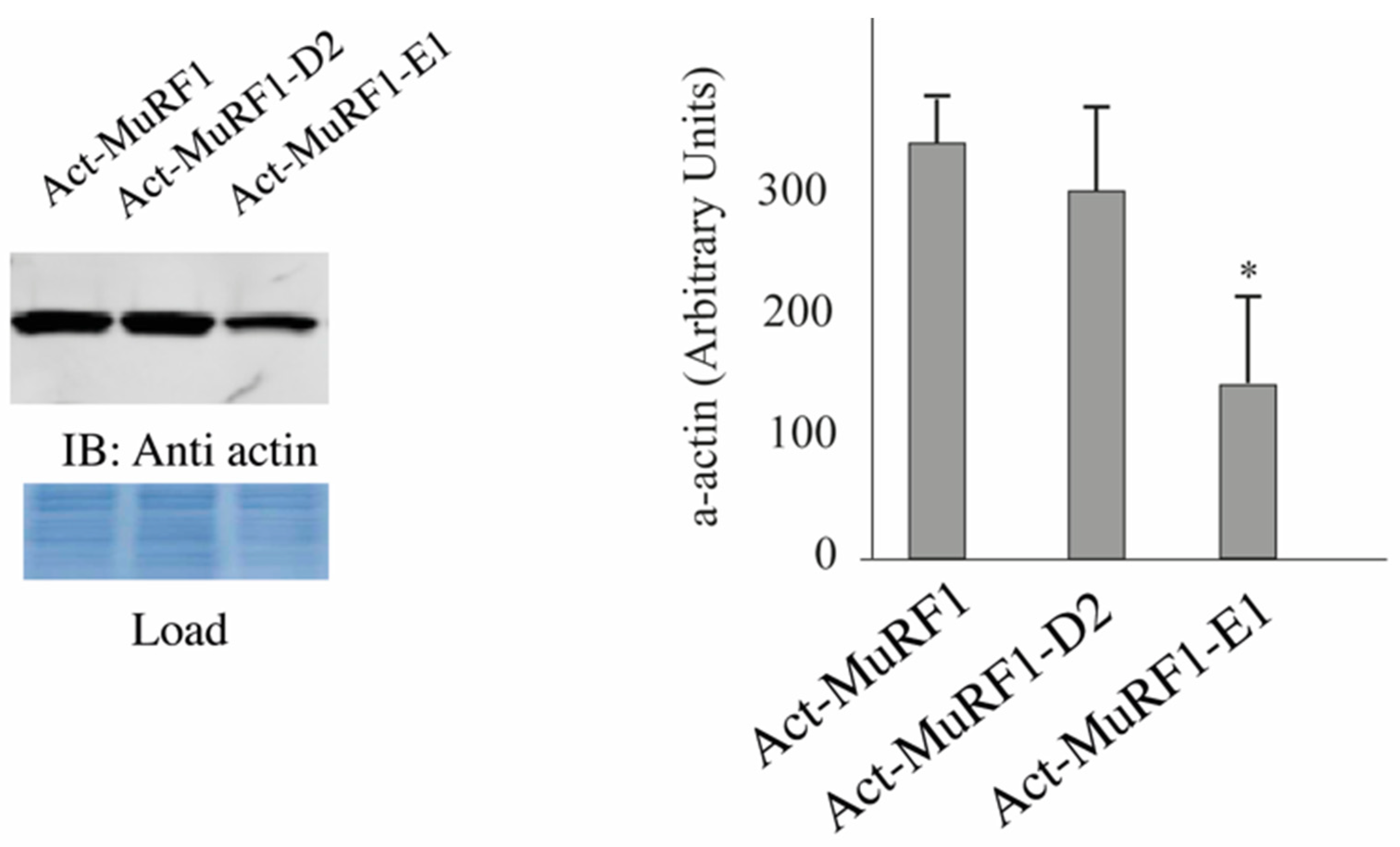
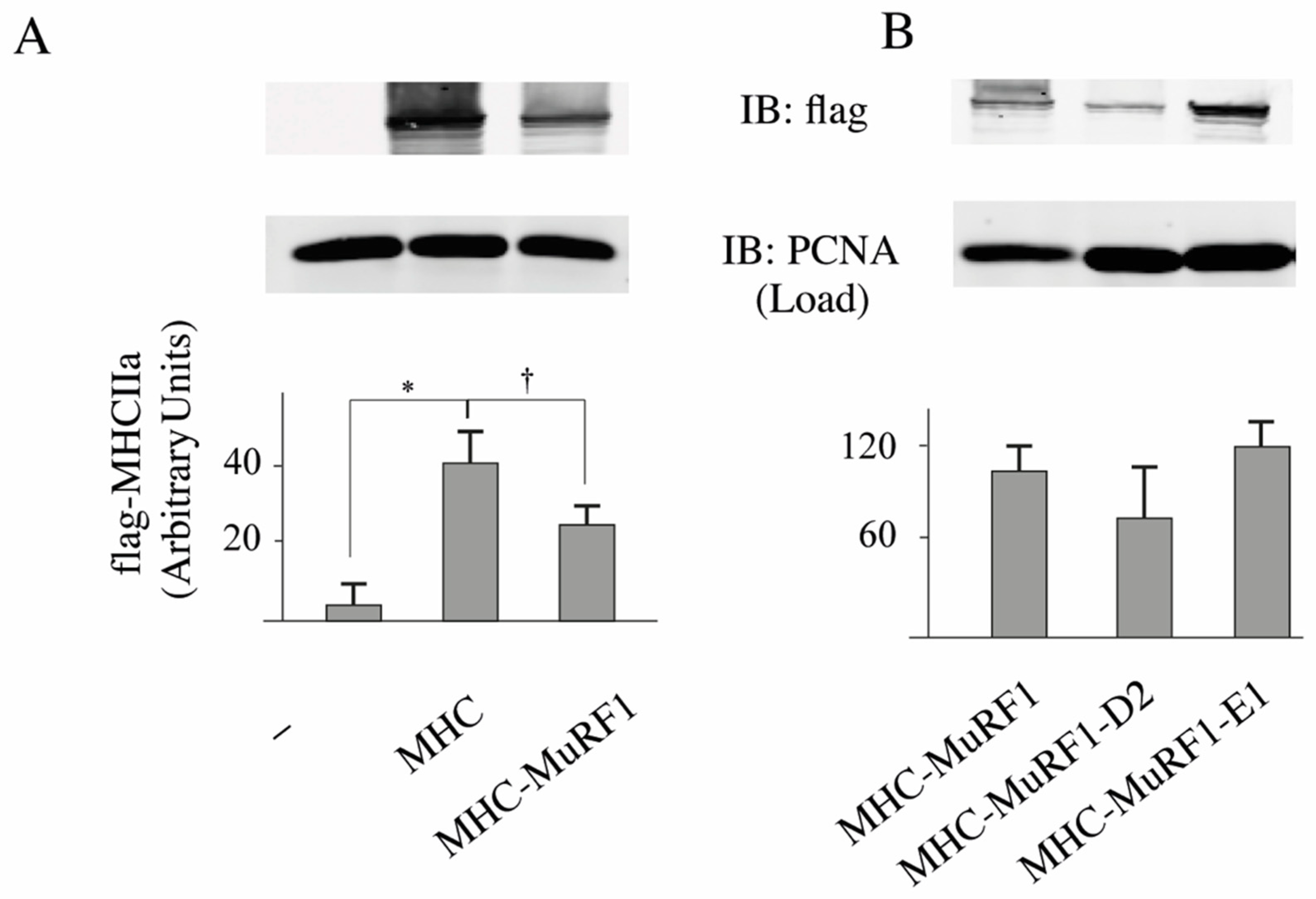
3.2. The MuRF1-E2E1 Couple Is Able to Target α-Actin But Not MHCIIa for Degradation in HEK293T Cells
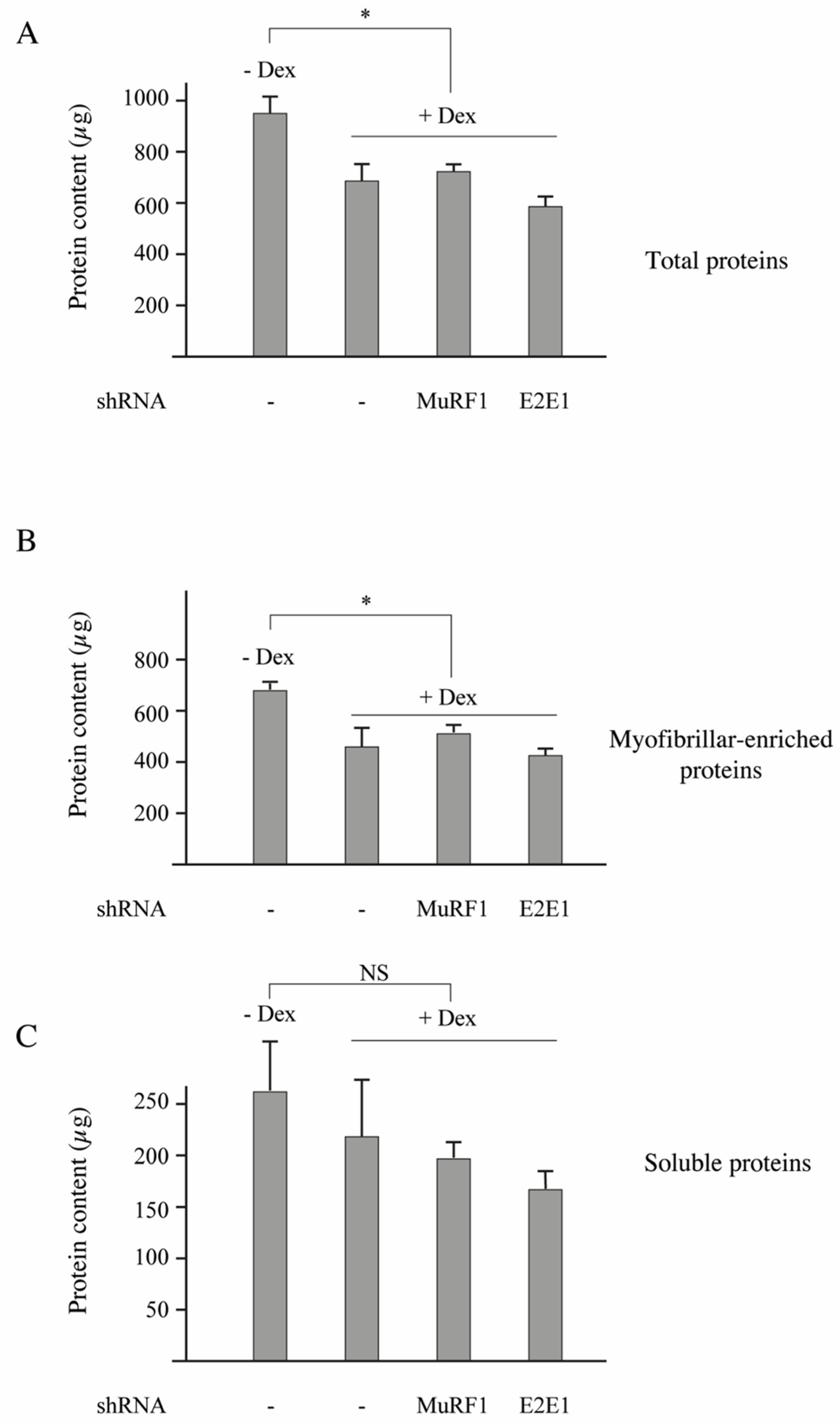
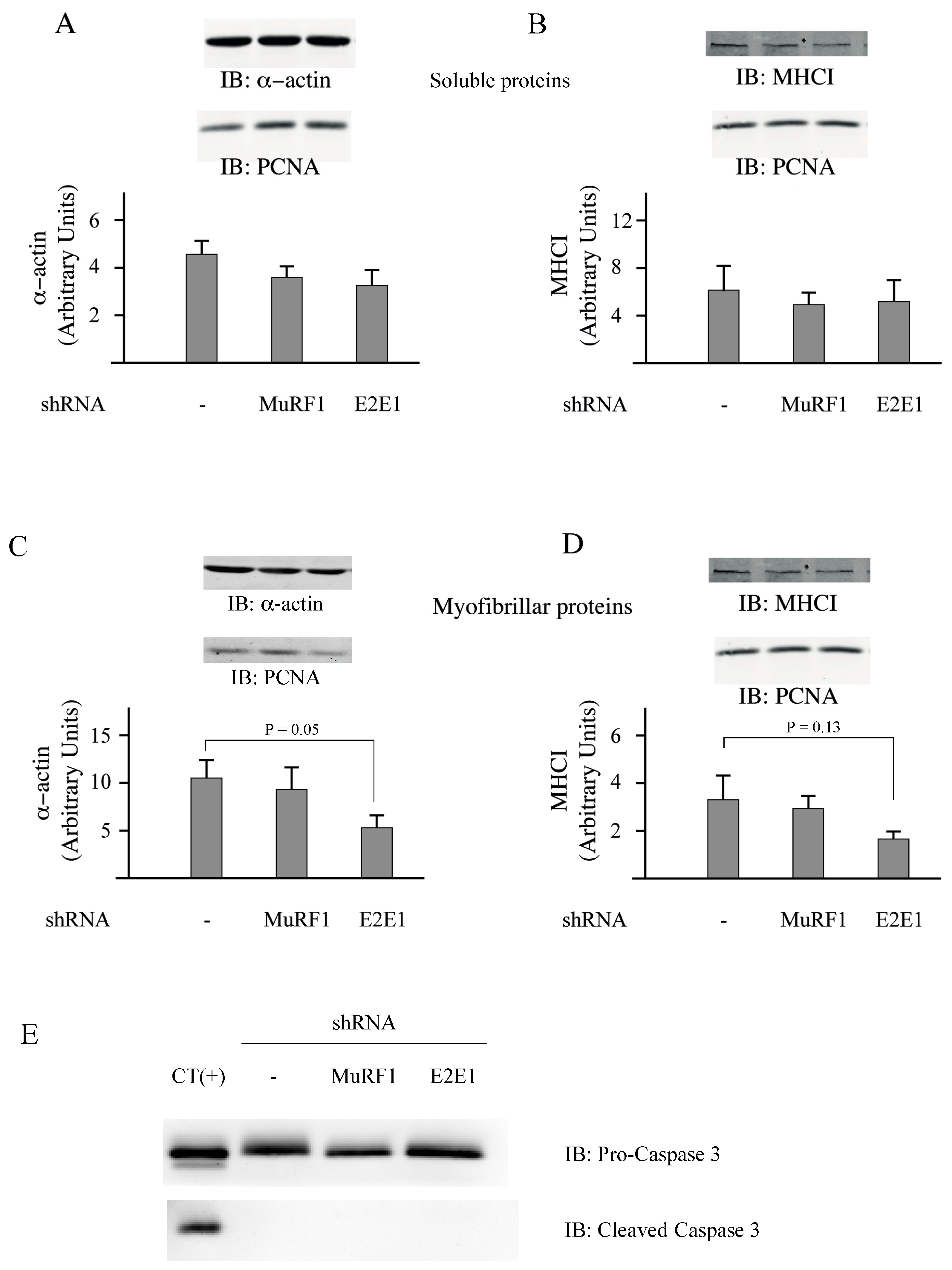
3.3. E2E1 Repression Decreased α-Actin Levels and Tended to Depress MHCI Levels in Catabolic C2C12 Myotubes
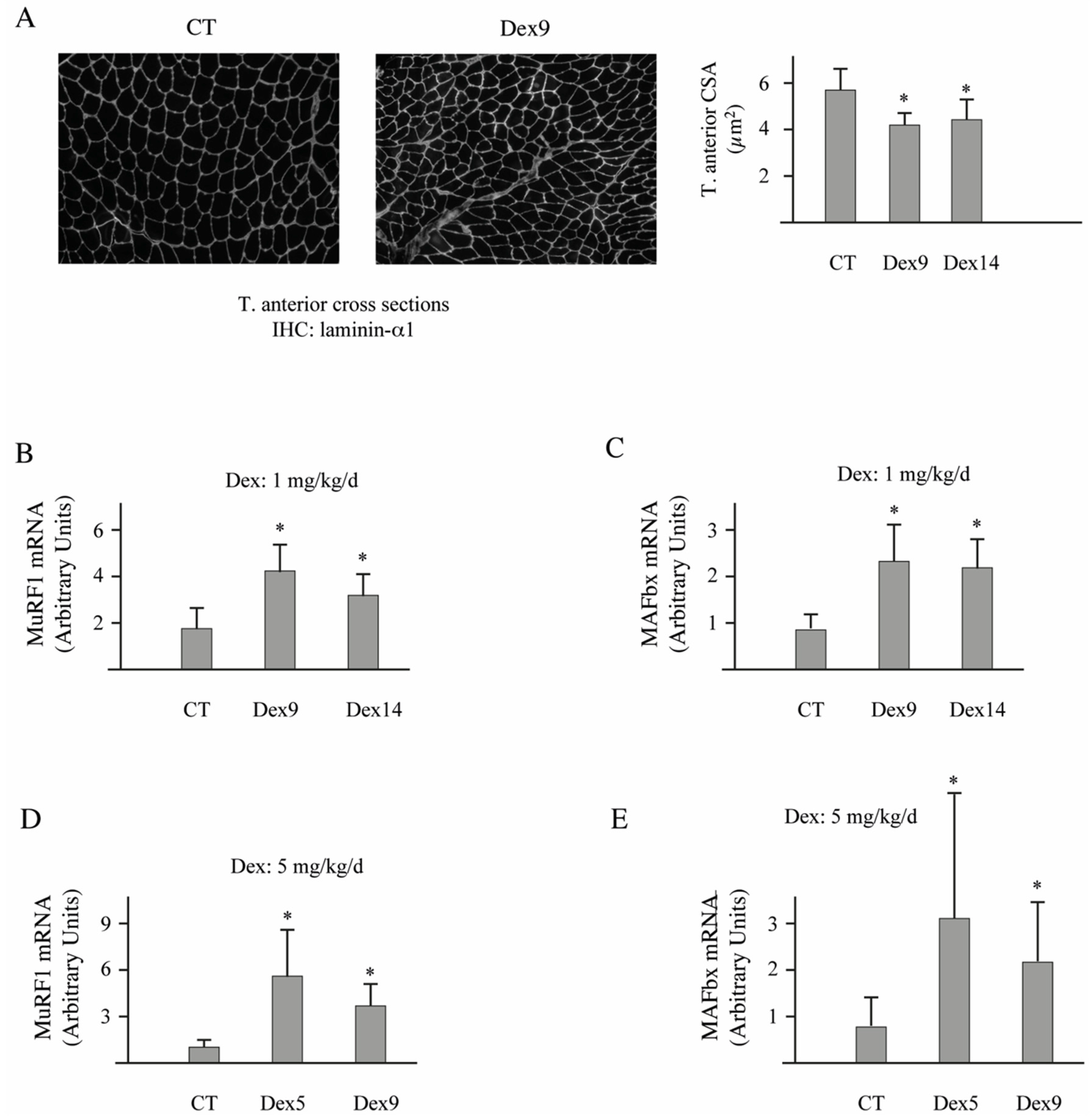
3.4. Dex-Treatment Induces Muscle Atrophy and Activates the UPS in Mice
3.5. E2E1 Knockdown Aggravates Muscle Atrophy in Dex-Treated Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fearon, K.; Arends, J.; Baracos, V. Understanding the mechanisms and treatment options in cancer cachexia. Nature reviews. Clin. Oncol. 2013, 10, 90–99. [Google Scholar]
- Von Haehling, S.; Anker, M.S.; Anker, S.D. Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: Facts and numbers update 2016. J. Cachexia Sarcopenia Muscle 2016, 7, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pessin, J.E. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.; Maves, L. Skeletal muscle fiber type: Using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley interdisciplinary reviews. Dev. Biol. 2016, 5, 518–534. [Google Scholar]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [PubMed]
- Attaix, D.; Ventadour, S.; Codran, A.; Béchet, D.; Taillandier, D.; Combaret, L. The ubiquitin-proteasome system and skeletal muscle wasting. Essays Biochem. 2005, 41, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Fielitz, J.; Kim, M.-S.; Shelton, J.M.; Latif, S.; Spencer, J.A.; Glass, D.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Investig. 2007, 117, 2486–2495. [Google Scholar] [CrossRef] [PubMed]
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.H.; Rockman, H.A.; Patterson, C. Muscle-specific ring finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140. [Google Scholar] [CrossRef] [PubMed]
- Polge, C.; Cabantous, S.; Deval, C.; Claustre, A.; Hauvette, A.; Bouchenot, C.; Aniort, J.; Béchet, D.; Combaret, L.; Attaix, D.; et al. A muscle-specific MuRF1-E2 network requires stabilization of MuRF1-E2 complexes by telethonin, a newly identified substrate. J. Cachexia Sarcopenia Muscle 2018, 9, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Polge, C.; Heng, A.-E.; Jarzaguet, M.; Ventadour, S.; Claustre, A.; Combaret, L.; Bechet, D.; Matondo, M.; Uttenweiler-Joseph, S.; Monsarrat, B.; et al. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J. 2011, 25, 3790–3802. [Google Scholar] [CrossRef] [PubMed]
- Polge, C.; Attaix, D.; Taillandier, D. Role of E2-Ub-conjugating enzymes during skeletal muscle atrophy. Front. Physiol. 2015, 6, 59. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Wijk, S.J.; Timmers, H.T. The family of ubiquitin-conjugating enzymes (E2s): Deciding between life and death of proteins. FASEB J. 2010, 24, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Markson, G.; Kiel, C.; Hyde, R.; Brown, S.; Charalabous, P.; Bremm, A.; Semple, J.; Woodsmith, J.; Duley, S.; Salehi-Ashtiani, K.; et al. Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res. 2009, 19, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.R.; Wilson, G.; Day, C.L. The N-terminal extension of UBE2E ubiquitin-conjugating enzymes limits chain assembly. J. Mol. Biol. 2013, 425, 4099–4111. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Iwahara, S.; Saeki, Y.; Sasajima, H.; Yokosawa, H. Link between the ubiquitin conjugation system and the ISG15 conjugation system: ISG15 conjugation to the UbcH6 ubiquitin E2 enzyme. J. Biochem. 2005, 138, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Nowak, D.G.; Narula, N.; Robinson, B.; Watrud, K.; Ambrico, A.; Herzka, T.M.; Zeeman, M.E.; Minderer, M.; Zheng, W.; et al. The nuclear transport receptor Importin-11 is a tumor suppressor that maintains PTEN protein. J. Cell Biol. 2017, 216, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Plafker, K.S.; Starnes, A.; Cook, M.; Klevit, R.E.; Plafker, S.M. The ubiquitin-conjugating enzyme, UbcM2, is restricted to monoubiquitylation by a two-fold mechanism that involves backside residues of E2 and Lys48 of ubiquitin. Biochemistry 2014, 53, 4004–4014. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.E.; Brzovic, P.S.; Klevit, R.E. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat. Struct. Mol. Biol. 2007, 14, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Banka, P.A.; Behera, A.P.; Sarkar, S.; Datta, A.B. RING E3-Catalyzed E2 Self-Ubiquitination Attenuates the Activity of Ube2E Ubiquitin-Conjugating Enzymes. J. Mol. Biol. 2015, 427, 2290–2304. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.M.; Jaffray, E.G.; Hay, R.T.; Meroni, G. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem. J. 2011, 434, 309–319. [Google Scholar] [CrossRef] [PubMed]
- David, Y.; Ziv, T.; Admon, A.; Navon, A. The E2 ubiquitin-conjugating enzymes direct polyubiquitination to preferred lysines. J. Biol. Chem. 2010, 285, 8595–8604. [Google Scholar] [CrossRef] [PubMed]
- Sarkari, F.; Wheaton, K.; La Delfa, A.; Mohamed, M.; Shaikh, F.; Khatun, R.; Arrowsmith, C.H.; Frappier, L.; Saridakis, V.; Sheng, Y. Ubiquitin-specific protease 7 is a regulator of ubiquitin-conjugating enzyme UbE2E1. J. Biol. Chem. 2013, 288, 16975–16985. [Google Scholar] [CrossRef] [PubMed]
- David, Y.; Ternette, N.; Edelmann, M.J.; Ziv, T.; Gayer, B.; Sertchook, R.; Dadon, Y.; Kessler, B.M.; Navon, A. E3 ligases determine ubiquitination site and conjugate type by enforcing specificity on E2 enzymes. J. Biol. Chem. 2011, 286, 44104–44115. [Google Scholar] [CrossRef] [PubMed]
- Plafker, S.M.; Plafker, K.S.; Weissman, A.M.; Macara, I.G. Ubiquitin charging of human class III ubiquitin-conjugating enzymes triggers their nuclear import. J. Cell Biol. 2004, 167, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, K.; Sarkari, F.; Stanly Johns, B.; Davarinejad, H.; Egorova, O.; Kaustov, L.; Raught, B.; Saridakis, V.; Sheng, Y. UbE2E1/UBCH6 Is a Critical in Vivo E2 for the PRC1-catalyzed Ubiquitination of H2A at Lys-119. J. Biol. Chem. 2017, 292, 2893–2902. [Google Scholar] [CrossRef] [PubMed]
- Polge, C.; Koulmann, N.; Claustre, A.; Jarzaguet, M.; Serrurier, B.; Combaret, L.; Béchet, D.; Bigard, X.; Attaix, D.; Taillandier, D. UBE2D2 is not involved in MuRF1-dependent muscle wasting during hindlimb suspension. Int. J. Biochem. Cell Biol. 2016, 79, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Ventadour, S.; Jarzaguet, M.; Wing, S.S.; Chambon, C.; Combaret, L.; Bechet, D.; Attaix, D.; Taillandier, D. A new method of purification of proteasome substrates reveals polyubiquitination of 20 S proteasome subunits. J. Biol. Chem. 2007, 282, 5302–5309. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.J.; Cagnin, S.; Chemello, F.; Silvestrin, M.; Musaro, A.; De Pitta, C.; Lanfranchi, G.; Sandri, M. Involvement of microRNAs in the regulation of muscle wasting during catabolic conditions. J. Biol. Chem. 2014, 289, 21909–21925. [Google Scholar] [CrossRef] [PubMed]
- Gueugneau, M.; Coudy-Gandilhon, C.; Théron, L.; Meunier, B.; Barboiron, C.; Combaret, L.; Taillandier, D.; Polge, C.; Attaix, D.; Picard, B.; et al. Skeletal muscle lipid content and oxidative activity in relation to muscle fiber type in aging and metabolic syndrome. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Ochala, J.; Gustafson, A.-M.; Diez, M.L.; Renaud, G.; Li, M.; Aare, S.; Qaisar, R.; Banduseela, V.C.; Hedström, Y.; Tang, X.; et al. Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: Underlying mechanisms. J. Physiol. 2011, 589, 2007–2026. [Google Scholar] [CrossRef] [PubMed]
- McElhinny, A.S.; Kakinuma, K.; Sorimachi, H.; Labeit, S.; Gregorio, C.C. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J. Cell Biol. 2002, 157, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Knöll, R.; Linke, W.A.; Zou, P.; Miocic, S.; Kostin, S.; Buyandelger, B.; Ku, C.H.; Neef, S.; Bug, M.; Schäfer, K.; et al. Telethonin deficiency is associated with maladaptation to biomechanical stress in the mammalian heart. Circ. Res. 2011, 109, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S. Muscle fiber type diversity revealed by anti-myosin heavy chain antibodies. FEBS J. 2018, 385, 3688–3694. [Google Scholar] [CrossRef] [PubMed]
- Menconi, M.; Gonnella, P.; Petkova, V.; Lecker, S.; Hasselgren, P.O. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J. Cell. Biochem. 2008, 105, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Naito, M.; Tsuruo, T. Caspase-mediated cleavage of cytoskeletal actin plays a positive role in the process of morphological apoptosis. Oncogene 1999, 18, 2423–2430. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, S.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xu, Y.; Zhu, B.; Liu, Q.; Yao, Q.; Zhao, G. Resveratrol induces apoptosis in SGC-7901 gastric cancer cells. Resveratrol induces apoptosis in SGC-7901 gastric cancer cells. Oncol. Lett. 2018, 16, 2949–2956. [Google Scholar] [PubMed]
- Umeki, D.; Ohnuki, Y.; Mototani, Y.; Shiozawa, K.; Suita, K.; Fujita, T.; Nakamura, Y.; Saeki, Y.; Okumura, S. Protective Effects of Clenbuterol against Dexamethasone-Induced Masseter Muscle Atrophy and Myosin Heavy Chain Transition. PLoS ONE 2015, 10, e0128263. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.K.; Rayavarapu, S.; Damsker, J.M.; Nagaraju, K. Glucocorticoid analogues: Potential therapeutic alternatives for treating inflammatory muscle diseases. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Nakao, R.; Yamamoto, S.; Yasumoto, Y.; Oishi, K. Dosing schedule-dependent attenuation of dexamethasone-induced muscle atrophy in mice. Chronobiol. Int. 2014, 31, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Protzek, A.O.P.; Costa-Júnior, J.M.; Rezende, L.F.; Santos, G.J.; Araújo, T.G.; Vettorazzi, J.F.; Ortis, F.; Carneiro, E.M.; Rafacho, A.; Boschero, A.C. Augmented beta-Cell Function and Mass in Glucocorticoid-Treated Rodents Are Associated with Increased Islet Ir-beta/AKT/mTOR and Decreased AMPK/ACC and AS160 Signaling. Int. J. Endocrinol. 2014, 2014, 983453. [Google Scholar] [CrossRef] [PubMed]
- Arvaniti, K.; Ricquier, D.; Champigny, O.; Richard, D. Leptin and corticosterone have opposite effects on food intake and the expression of UCP1 mRNA in brown adipose tissue of lep(ob)/lep(ob) mice. Endocrinology 1998, 139, 4000–4003. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gounarides, J.S.; Korach-André, M.; Killary, K.; Argentieri, G.; Turner, O.; Laurent, D. Effect of dexamethasone on glucose tolerance and fat metabolism in a diet-induced obesity mouse model. Endocrinology 2008, 149, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Aniort, J.; Polge, C.; Claustre, A.; Combaret, L.; Béchet, D.; Attaix, D.; Heng, A.-E.; Taillandier, D. Upregulation of MuRF1 and MAFbx participates to muscle wasting upon gentamicin-induced acute kidney injury. Int. J. Biochem. Cell Biol. 2016, 79, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, G.; Thomas, M.; Langley, B.; Somers, W.; Patel, K.; Kemp, C.F.; Sharma, M.; Kambadur, R. Titin-cap associates with, and regulates secretion of, Myostatin. J. Cell. Physiol. 2002, 193, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Hormaechea-Agulla, D.; Kim, Y.; Song, M.S.; Song, S.J. New Insights into the Role of E2s in the Pathogenesis of Diseases: Lessons Learned from UBE2O. Mol. Cells 2018, 41, 168–178. [Google Scholar] [PubMed]
- Van Wijk, S.J.L.; de Vries, S.J.; Kemmeren, P.; Huang, A.; Boelens, R.; Bonvin, A.M.J.J.; Timmers, H.T.M. A comprehensive framework of E2-RING E3 interactions of the human ubiquitin-proteasome system. Mol. Syst. Biol. 2009, 5, 295. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment 1 | Experiment 2 | |||||
|---|---|---|---|---|---|---|
| Dex 1 mg/d/kg | Dex 5 mg/d/kg | |||||
| CT | Dex9 | Dex14 | CT | Dex5 | Dex9 | |
| Mice (g) | 26.01 ± 1.8 | 22.6 ± 1.4 * | 21.9 ± 2.2 * | 28.1 ± 1.8 | 24.6 ± 0.4 * | 24.2 ± 0.6 * |
| Gastrocnemius (mg) | 133.5 ± 8.0 | 112.0 ± 2.4 * | 94.9 ± 10.3 * | 163.7 ± 8.6 | 117.2 ± 5.4 * | 118.4 ± 7.5 * |
| T. anterior (mg) | 42.9 ± 2.0 | 39.1 ± 0.7 * | 33.94 ± 3.8 * | 51.5 ± 1.5 | 40.2 ± 1.7 * | 43.7 ± 2.4 * |
| EDL (mg) | 9.9 ± 0.9 | 8.4 ± 0.4 * | 7.08 ± 1.0 * | 12 ± 0.7 | 8.8 ± 0.5 * | 9.1 ± 0.4 * |
| Soleus (mg) | 8.5 ± 1.5 | 7.4 ± 0.6 | 7.3 ± 0.6 | 9.3 ± 0.7 | 7.6 ± 0.3 * | 7.9 ± 0.6 * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polge, C.; Aniort, J.; Armani, A.; Claustre, A.; Coudy-Gandilhon, C.; Tournebize, C.; Deval, C.; Combaret, L.; Béchet, D.; Sandri, M.; et al. UBE2E1 Is Preferentially Expressed in the Cytoplasm of Slow-Twitch Fibers and Protects Skeletal Muscles from Exacerbated Atrophy upon Dexamethasone Treatment. Cells 2018, 7, 214. https://doi.org/10.3390/cells7110214
Polge C, Aniort J, Armani A, Claustre A, Coudy-Gandilhon C, Tournebize C, Deval C, Combaret L, Béchet D, Sandri M, et al. UBE2E1 Is Preferentially Expressed in the Cytoplasm of Slow-Twitch Fibers and Protects Skeletal Muscles from Exacerbated Atrophy upon Dexamethasone Treatment. Cells. 2018; 7(11):214. https://doi.org/10.3390/cells7110214
Chicago/Turabian StylePolge, Cécile, Julien Aniort, Andrea Armani, Agnès Claustre, Cécile Coudy-Gandilhon, Clara Tournebize, Christiane Deval, Lydie Combaret, Daniel Béchet, Marco Sandri, and et al. 2018. "UBE2E1 Is Preferentially Expressed in the Cytoplasm of Slow-Twitch Fibers and Protects Skeletal Muscles from Exacerbated Atrophy upon Dexamethasone Treatment" Cells 7, no. 11: 214. https://doi.org/10.3390/cells7110214
APA StylePolge, C., Aniort, J., Armani, A., Claustre, A., Coudy-Gandilhon, C., Tournebize, C., Deval, C., Combaret, L., Béchet, D., Sandri, M., Attaix, D., & Taillandier, D. (2018). UBE2E1 Is Preferentially Expressed in the Cytoplasm of Slow-Twitch Fibers and Protects Skeletal Muscles from Exacerbated Atrophy upon Dexamethasone Treatment. Cells, 7(11), 214. https://doi.org/10.3390/cells7110214