The Effect of Ethanol on Telomere Dynamics and Regulation in Human Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Experimental System and Growth Conditions
2.2. Cell Viability Assay
2.3. Southern Blot and Mean TRF Measurement
2.4. Telomeres Repeat Amplification Protocol (TRAP)
2.5. DNA Methylation

2.6. Gene Expression Profiling
2.7. Western Blot
2.8. Chromatin Immunoprecipitation (ChIP) Dot Blot
2.9. Cell Exposure to Various Treatments
- 25 mM EtOH for two days.
- 25 mM EtOH for one week.
- 25 µM AcH for two days or one week.
- 25 mM EtOH + 2 mM 4-MP.
- Recovery: Comprised of a first week of standard EtOH treatment followed by a second week of EtOH-free medium.
3. Results
3.1. The Addition of 125 mM EtoH Does Not Affect Cell Viability
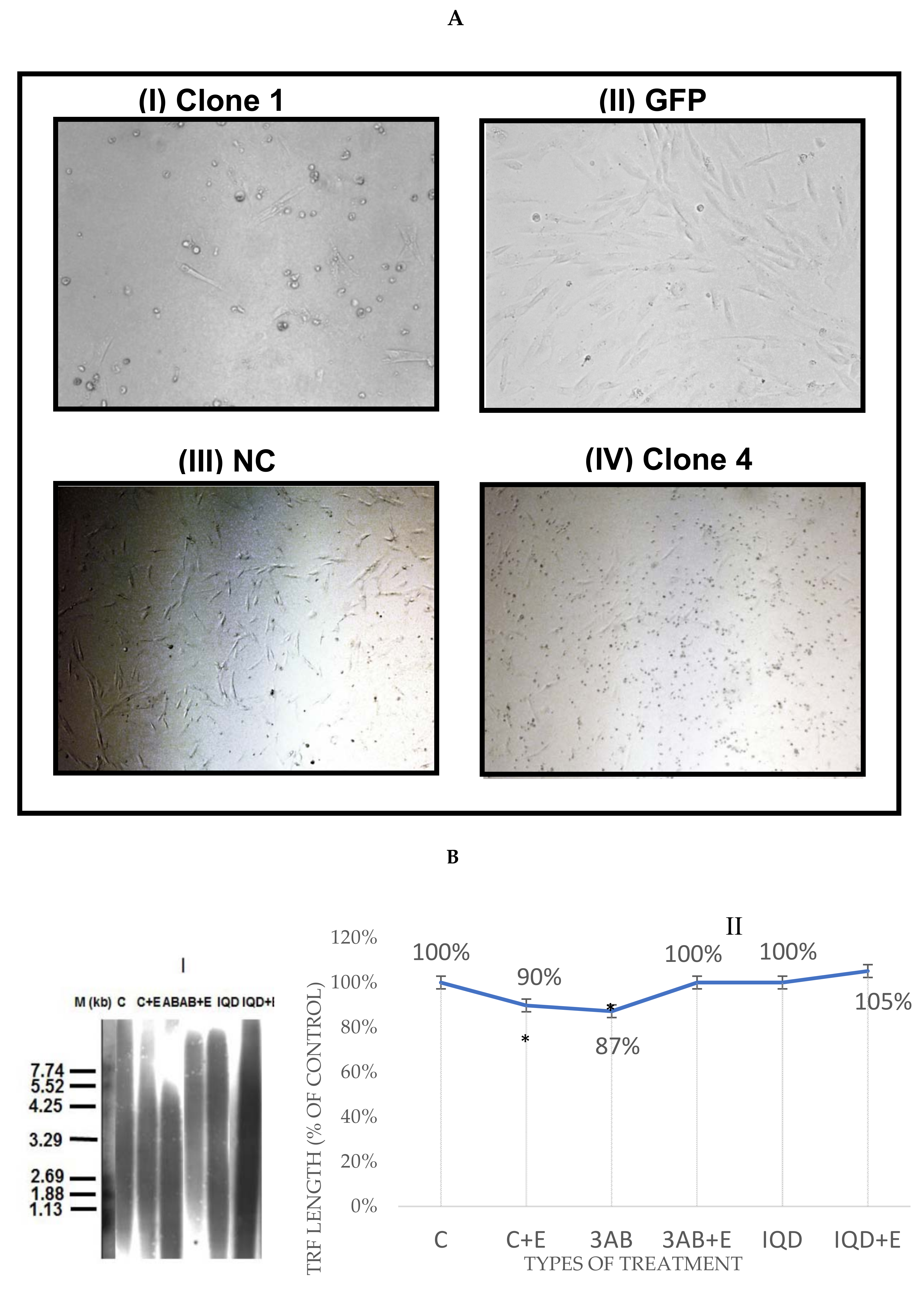
3.2. EtOH Shortens Telomeres
3.3. EtOH Effect on TL Length Is Not Mediated by Telomerase Activity
3.4. TRF2 and RAP1 Binding to Telomeric Regions Are Affected by EtOH
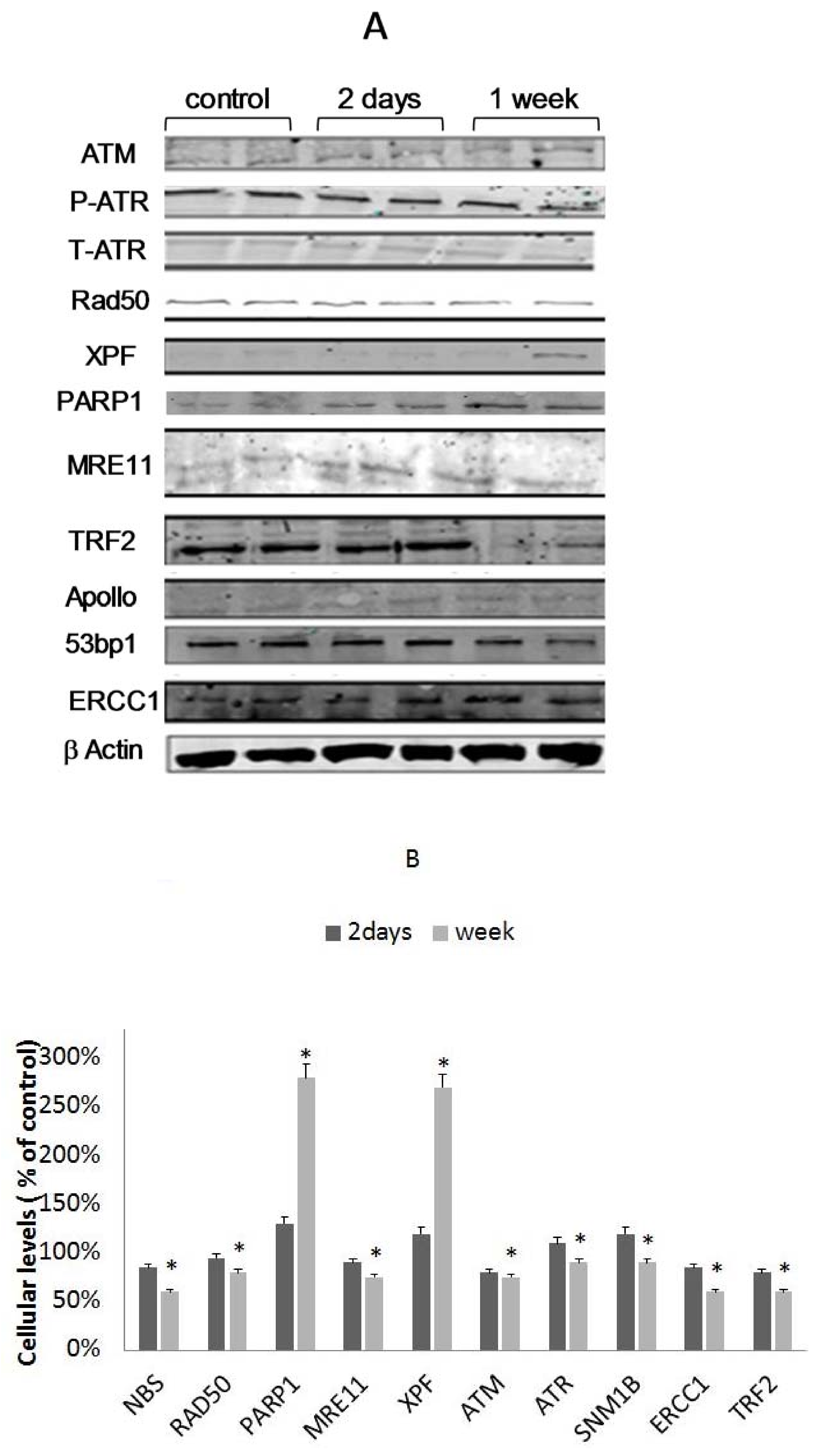
3.5. Differential Binding of TRF2-Associated Proteins to Telomeres
3.6. The Involvement of XPF in EtOH-Dependent Telomere Shortening
3.7. The Role of PARP1 in EtOH-Dependent Telomere Shortening
3.8. The Presence of DNA Double-Strand Breaks Was Not Apparent
3.9. Acetaldehyde May Have a Key Role in TL Shortening in Response to EtOH Exposure
3.10. Epigenetic Changes in Response to EtOH
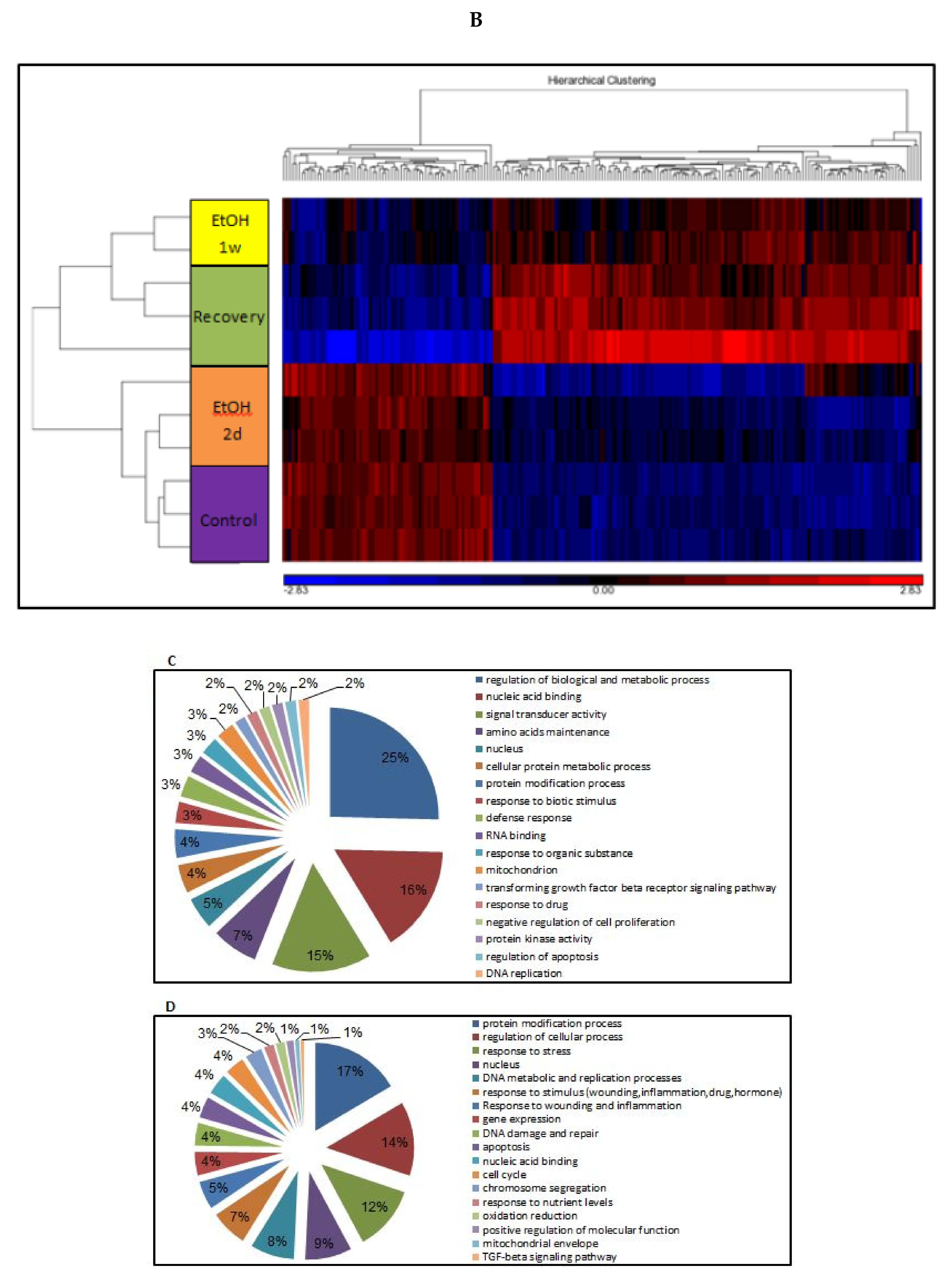
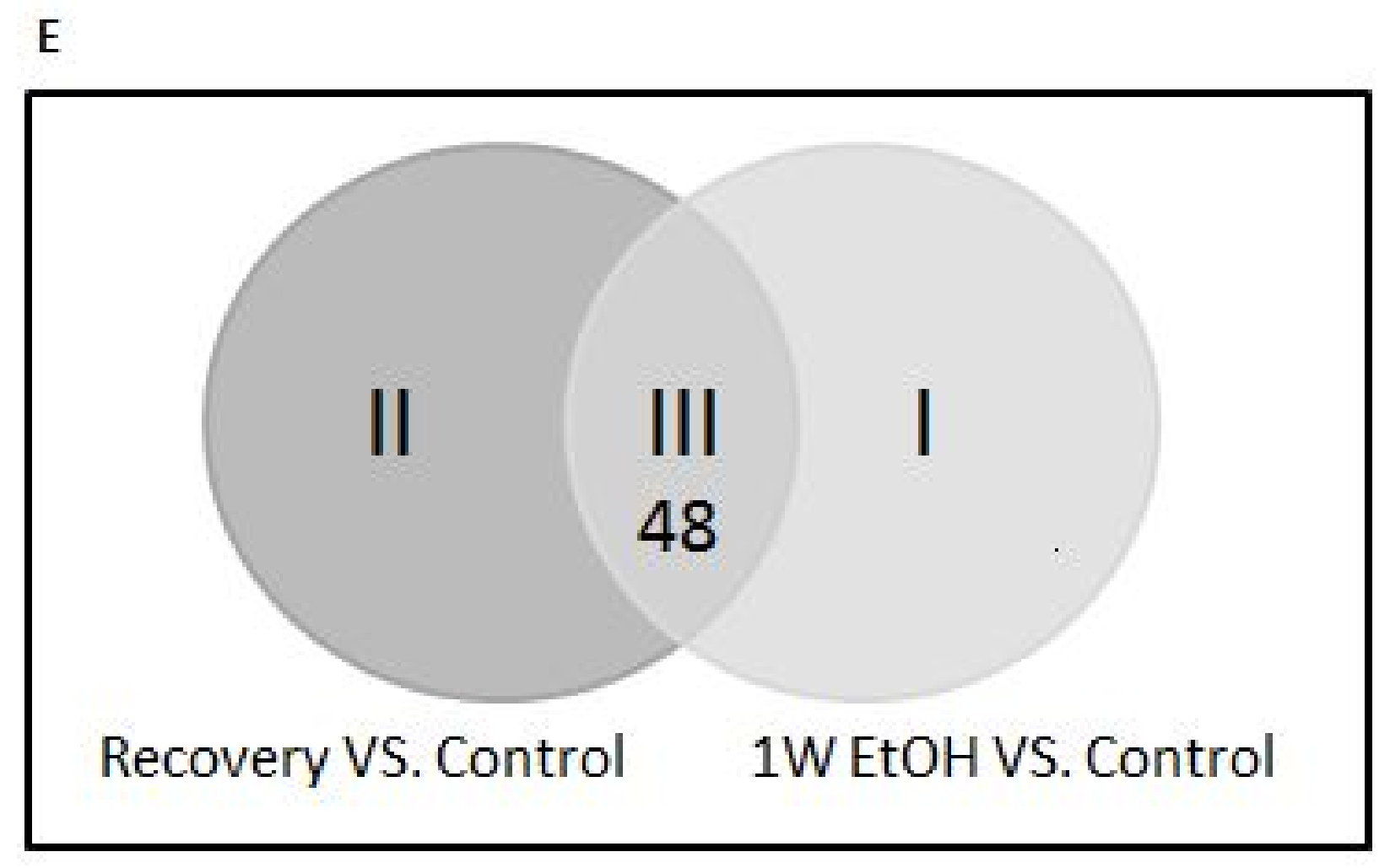
3.11. Changes of Genome-Wide Expression in Response to EtOH Treatment
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Shay, J.W.; Zou, Y.; Hiyama, E.; Wright, W.E. Telomerase and cancer. Hum. Mol. Genet. 2001, 10, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, A.M. Telomeres, telomerase, and aging: Origin of the theory. Exp. Gerontol. 1996, 31, 443–448. [Google Scholar] [CrossRef]
- Law, M.; Wald, N. Why heart disease and mortality is low in France: The time lag explanation. BMJ 1999, 318, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Takakura, M.; Taira, T.; Kanaya, T.; Itoh, H.; Yutsudo, M.; Ariga, H.; Inoue, M. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res. 2000, 28, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Verdun, R.E.; Karlseder, J. Replication and protection of telomeres. Nature 2007, 447, 924–993. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Wallner, M.; Hanchar, H.J.; Olsen, R.W. Low-dose alcohol actions on alpha4beta3delta GABAA receptors are reversed by the behavioral alcohol antagonist Ro15-4513. Proc. Natl. Acad. Sci. USA 2006, 103, 8540–8545. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Dodd, P.R.; Buckley, S.T.; Eckert, A.L.; Foley, P.F.; Innes, D.J. Genes and gene expression in the brains of human alcoholics. Ann. N. Y. Acad. Sci. 2006, 1074, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Hendricson, A.W.; Maldve, R.E.; Salinas, A.G.; Theile, J.W.; Zhang, T.A.; Diaz, L.M.; Morrisett, R.A. Aberrant synaptic activation of N-methyl-d-aspartate receptors underlies ethanol withdrawal hyperexcitability. J. Pharmacol. Exp. Ther. 2007, 321, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Matsubara, K.; Uezono, T.; Kimura, K.; Shiono, H. Reduced dorsal hippocampal glutamate release significantly correlates with the spatial memory deficits produced by benzodiazepines and ethanol. Neuroscience 1998, 83, 701–706. [Google Scholar] [CrossRef]
- Levitt, M.D.; Li, R.; DeMaster, E.G.; Elson, M.; Furne, J.; Levitt, D.G. Use of measurements of ethanol absorption from stomach and intestine to assess human ethanol metabolism. Am. J. Physiol. 1997, 273, G951–G957. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.J.; Cornell, N.W.; Veech, R.L. Alcohol dehydrogenase in cultured human skin fibroblasts. Human fibroblast alcohol dehydrogenase. Adv. Exp. Med. Biol. 1980, 132, 533–541. [Google Scholar] [PubMed]
- Tsukamoto, H.; Lu, S.C. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001, 15, 1335–1349. [Google Scholar] [CrossRef] [PubMed]
- Sprince, H.; Parker, C.M.; Smith, G.G.; Gonzales, L.J. Protection against acetaldehyde toxicity in the rat by L-cysteine, thiamin and L-2-methylthiazolidine-4-carboxylic acid. Agents Actions 1974, 4, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Stephens, R.; Ling, J.; Heffernan, T.M.; Heather, N.; Jones, K. A review of the literature on the cognitive effects of alcohol hangover. Alcohol Alcohol. 2008, 43, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Swift, R.; Davidson, D. Alcohol hangover: Mechanisms and mediators. Alcohol Health Res. World 1998, 22, 54–60. [Google Scholar] [PubMed]
- Wiese, J.G.; Shlipak, M.G.; Browner, W.S. The alcohol hangover. Ann. Intern. Med. 2000, 132, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; De Lorgeril, M. Wine, alcohol, and the French paradox for coronary heart disease. Lancet 1992, 339, 1523–1525. [Google Scholar] [CrossRef]
- Rimm, E.B.; Klatsky, A.; Grobbee, D.; Stampfer, M.J. Review of moderate alcohol consumption and reduced risk of coronary heart disease: Is the effect due to beer, wine, or spirits? BMJ 1996, 312, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.; Maiani, G.; Ferro-Luzzi, A. Alcohol-free red wine enhances plasma antioxidant capacity in humans. J. Nutr. 1998, 128, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.A. Alcohol and heart disease. Circulation 1996, 94, 3023–3025. [Google Scholar] [CrossRef] [PubMed]
- Siris, E.S. Identification and fracture outcomes of undiagnosed low bone density in postmenopausal women: Results from the National Osteoporosis Risk Assessment. JAMA 2001, 286, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
- Osie, K.S.; Kalkwarf, H.J. Bone density in relation to alcohol intake among men and women in the United States. Osteoporos. Int. 2007, 18, 391–400. [Google Scholar]
- Orgogozo, J.M.; Dartigues, J.F.; Lafont, S.; Letenneur, L.; Commenges, D.; Salamon, R.; Renaud, S.; Breteler, M.B. Wine consumption and dementia in the elderly: A prospective community study in the Bordeaux area. Rev. Neurol. 1997, 153, 185–192. [Google Scholar] [PubMed]
- Ruitenberg, A.; van Swieten, J.C.; Witteman, J.C.; Mehta, K.M.; van Duijn, C.M.; Hofman, A.; Breteler, M.M. Alcohol consumption and risk of dementia: The Rotterdam Study. Lancet 2002, 359, 281–286. [Google Scholar] [CrossRef]
- Rashidkhani, B.; Åkesson, A.; Lindblad, P.; Wolk, A. Alcohol consumption and risk of renal cell carcinoma: A prospective study of Swedish women. Int. J. Cancer 2005, 117, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Lim, U.; Morton, L.M.; Subar, A.F.; Baris, D.; Stolzenberg-Solomon, R.; Leitzmann, M.; Kipnis, V.; Mouw, T.; Carroll, L.; Schatzkin, A.; et al. Alcohol, smoking, and body size in relation to incident Hodgkin’s and non-Hodgkin’s lymphoma risk. Am. J. Epidemiol. 2007, 166, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Nieters, A.; Deeg, E.; Becker, N. Tobacco and alcohol consumption and risk of lymphoma: Results of a population-based case-control study in Germany. Int. J. Cancer 2006, 118, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Llen, N.E.; Beral, V.; Casabonne, D.; Kan, S.W.; Reeves, G.K.; Brown, A.; Green, J. Million Women Study Collaborators Moderate alcohol intake and cancer incidence in women. J. Natl. Cancer Inst. 2009, 101, 296–305. [Google Scholar]
- Pikaar, N.A.; Wedel, M.; Hermus, R.J. Influence of several factors on blood alcohol concentrations after drinking alcohol. Alcohol Alcohol. 1988, 23, 289–297. [Google Scholar] [PubMed]
- International Center for Alcohol Policies. ICAP Reports 14; International Center for Alcohol Policies: London, UK, 2003. [Google Scholar]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Romano, G.H.; Harari, Y.; Yehuda, T.; Podhorzer, A.; Rubinstein, L.; Shamir, R.; Gottlieb, A.; Silberberg, Y.; Pe’er, D.; Ruppin, E.; et al. Environmental stresses disrupt telomere length homeostasis. PLoS Genet. 2013, 9, e1003721. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 3011–3015. [Google Scholar] [CrossRef]
- Hou, M.; Xu, D.; Björkholm, M.; Gruber, A. Real-time quantitative telomeric repeat amplification protocol assay for the detection of telomerase activity. Clin. Chem. 2001, 47, 519–524. [Google Scholar] [PubMed]
- De Lange, T.; Shiue, L.; Myers, R.M.; Cox, D.R.; Naylor, S.L.; Killery, A.M.; Varmus, H.E. Structure and variability of human chromosome ends. Mol. Cell. Biol. 1990, 10, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, S.; Funk, W.E.; Hou, L. Republished: Environmental and occupational exposure to chemicals and telomere length in human studies. Postgrad. Med. J. 2013, 89, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Marcon, F.; Siniscalchi, E.; Crebelli, R.; Saieva, C.; Sera, F.; Fortini, P.; Simonelli, V.; Palli, D. Diet-related telomere shortening and chromosome stability. Mutagenesis 2012, 27, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Presented at the 40th annual scientific meeting of the Research Society on Alcoholism, Denver, CO, USA, 24–28 June 2017. Available online: https://www.emedevents.com/c/medical-conferences-2017/research-society-on-alcoholism-rsa-40th-annual-scientific-meeting (accessed on 5 May 2018).
- Novitskiy, G.; Traore, K.; Wang, L.; Trush, M.A.; Mezey, E. Effects of ethanol and acetaldehyde on reactive oxygen species production in rat hepatic stellate cells. Alcohol. Clin. Exp. Res. 2006, 30, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J.; Theruvathu, J.A. DNA adducts from acetaldehyde: Implications for alcohol-related carcinogenesis. Alcohol 2005, 35, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J. DNA damage, DNA repair, and alcohol toxicity—A review. Alcohol. Clin. Exp. Res. 1997, 21, 1073–1082. [Google Scholar] [PubMed]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Oikawa, S. Mechanism of telomere shortening by oxidative stress. Ann. N. Y. Acad. Sci. 2004, 1019, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Tchirkov, A.; Lansdorp, P.M. Role of oxidative stress in telomere shortening in cultured fibroblasts from normal individuals and patients with ataxia-telangiectasia. Hum. Mol. Genet. 2003, 12, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Richter, T.; von Zglinicki, T. A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Exp. Gerontol. 2007, 42, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Rageul, J.; Frëmin, C.; Ezan, F.; Baffet, G.; Langouët, S. The knock-down of ERCC1 but not of XPF causes multinucleation. DNA Repair 2011, 10, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Boesten, D.M.; de Vos-Houben, J.M.; Timmermans, L.; den Hartog, G.J.; Bast, A.; Hageman, G.J. Accelerated Aging during Chronic Oxidative Stress: A Role for PARP-1. Oxid. Med. Cell. Longev. 2013, 680414. [Google Scholar] [CrossRef] [PubMed]
- Gurung, R.L.; Balakrishnan, L.; Bhattacharjee, R.N.; Manikandan, J.; Swaminathan, S.; Hande, M.P. Inhibition of poly (ADP-Ribose) polymerase-1 in telomerase deficient mouse embryonic fibroblasts increases arsenite-induced genome instability. Genome Integr. 2010, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.Z.; Guan, W.P.; Maeda, T.; Makino, N. The subtelomere of Short telomeres is hypermethylated in Alzheimer’s disease. Aging Dis. 2012, 3, 164–170. [Google Scholar] [PubMed]
- Epel, E.S.; Blackburn, E.H.; Lin, J.; Dhabhar, F.S.; Adler, N.E.; Morrow, J.D.; Cawthon, R.M. Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. USA 2004, 101, 17312–17315. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, Z.; Chen, J. MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J. Cell Biol. 2011, 193, 267–273. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harpaz, T.; Abumock, H.; Beery, E.; Edel, Y.; Lahav, M.; Rozovski, U.; Uziel, O. The Effect of Ethanol on Telomere Dynamics and Regulation in Human Cells. Cells 2018, 7, 169. https://doi.org/10.3390/cells7100169
Harpaz T, Abumock H, Beery E, Edel Y, Lahav M, Rozovski U, Uziel O. The Effect of Ethanol on Telomere Dynamics and Regulation in Human Cells. Cells. 2018; 7(10):169. https://doi.org/10.3390/cells7100169
Chicago/Turabian StyleHarpaz, Tomer, Heba Abumock, Einat Beery, Yonatan Edel, Meir Lahav, Uri Rozovski, and Orit Uziel. 2018. "The Effect of Ethanol on Telomere Dynamics and Regulation in Human Cells" Cells 7, no. 10: 169. https://doi.org/10.3390/cells7100169
APA StyleHarpaz, T., Abumock, H., Beery, E., Edel, Y., Lahav, M., Rozovski, U., & Uziel, O. (2018). The Effect of Ethanol on Telomere Dynamics and Regulation in Human Cells. Cells, 7(10), 169. https://doi.org/10.3390/cells7100169