Chronic Infections: A Possible Scenario for Autophagy and Senescence Cross-Talk

Abstract

1. Introduction
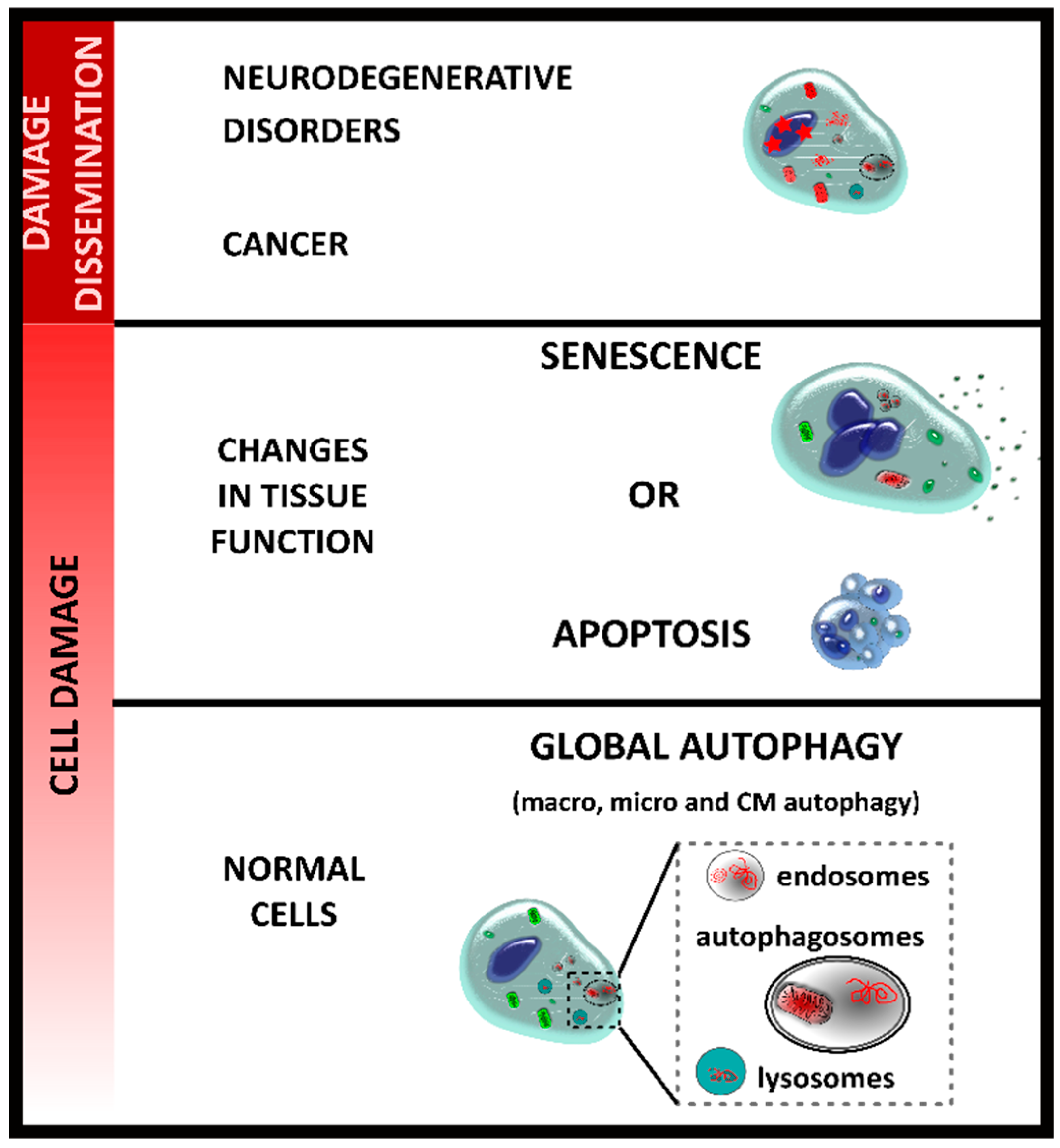
2. Autophagy and Senescence
3. The Aging of the Immune System
4. Senescence and Chronic Infections
4.1. Bacterial Infections and Senescence
4.2. The Chronic Trypanosoma Cruzi Infection
4.3. Persistent Human Cytomegalovirus Antigenic Stimulation and Immunosenescense: Possible Role for the Autophagy Pathway
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Vicencio, J.M.; Galluzzi, L.; Tajeddine, N.; Ortiz, C.; Criollo, A.; Tasdemir, E.; Morselli, E.; Ben Younes, A.; Maiuri, M.C.; Lavandero, S.; et al. Senescence, apoptosis or autophagy? Gerontology 2008, 54, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A. Selective autophagy in cellular quality control. In Protein Quality Control in Neurodegenerative Diseases; Springer Verlag: Berlin, Germany, 2013; pp. 63–75. ISBN 978-3-642-27927-0. [Google Scholar]
- Matsui, A.; Kamada, Y.; Matsuura, A. The Role of Autophagy in Genome Stability through Suppression of Abnormal Mitosis under Starvation. PLoS Genet. 2013, 9, e1003245. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Oh, S.; Li, D.; Ni, D.; Pirooz, S.D.; Lee, J.H.; Yang, S.; Lee, J.Y.; Ghozalli, I.; Costanzo, V.; et al. A Dual Role for UVRAG in Maintaining Chromosomal Stability Independent of Autophagy. Dev. Cell 2012, 22, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nie, T.; Xu, H.; Yang, J.; Yang, Q.; Mao, Z. Chaperone-mediated autophagy: Advances from bench to bedside. Neurobiol. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Salvador, N.; Aguado, C.; Horst, M.; Knecht, E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J. Biol. Chem. 2000, 275, 27447–27456. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of Cytosolic Proteins by Late Endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Martinez-Vicente, M.; MacIan, F.; Verkhusha, V.V.; Cuervo, A.M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Aguilera, M.O.; Colombo, M.I. Autophagy response: Manipulating the mTOR-controlled machinery by amino acids and pathogens. Amino Acids 2015, 47, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. The Pathophysiology of Mitochondrial Cell Death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the Autophagy-Inflammation-Cell Death Axis in Organismal Aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondria: Dynamic Organelles in Disease, Aging, and Development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-M.; Jung, Y.-K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.J.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Kwon, D.H.; Song, H.K. Structure biology of selective autophagy receptors. BMB Rep. 2016, 49, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Svenning, S.; Johansen, T. Selective autophagy. Essays Biochem. 2013, 55, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.H.; Song, H.K. A Structural View of Xenophagy, a Battle between Host and Microbes. Mol. Cells 2018, 41, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Cosío, G.; Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 2009, 7, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Thurston, T.L.M.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Perrin, A.J.; Jiang, X.; Birmingham, C.L.; So, N.S.; Brumell, J.H. Recognition of bacteria in the cytosol of mammalian cells by the ubiquitin system. Curr. Biol. 2004, 14, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Mansilla Pareja, M.E.; Colombo, M.I. Autophagic clearance of bacterial pathogens: Molecular recognition of intracellular microorganisms. Front. Cell. Infect. Microbiol. 2013, 3, 54. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.A.; Kirby, R. Apoptosis: A review of pro-apoptotic and anti-apoptotic pathways and dysregulation in disease. J. Vet. Emerg. Crit. Care 2008, 18, 572–585. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Hongmei, Z. Extrinsic and Intrinsic Apoptosis Signal Pathway Review. In Apoptosis and Medicine; InTech: London, UK, 2012; pp. 3–22. ISBN 978-953-51-0701-9. [Google Scholar]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4337–4341. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; D’Adda Di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Rai, T.S.; Adams, P.D. Lessons from senescence: Chromatin maintenance in non-proliferating cells. BBA-Gene Regul. Mech. 2012, 1819, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular Senescence in Cancer and Aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory Networks during Cellular Senescence: Causes and Consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.O.; Lee, Y.K.; Kim, J.M.; Yoon, G. From cell senescence to age-related diseases: Differential mechanisms of action of senescence-associated secretory phenotypes. BMB Rep. 2015, 48, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006, 8, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.-Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [PubMed]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF Induces Senescence and Apoptosis through Pathways Mediated by the Secreted Protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, F.; Perico, N.; Cortinovis, M.; Remuzzi, G. Mesenchymal stromal cells in renal transplantation: Opportunities and challenges. Nat. Rev. Nephrol. 2016, 12, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Weisheit, C.K.; Engel, D.R.; Kurts, C. Dendritic cells and macrophages: Sentinels in the kidney. Clin. J. Am. Soc. Nephrol. 2015, 10, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, B.; Müller, S.; Walther, M.; Fischer, M.; Engelmann, R.; Müller-Hilke, B.; Pützer, B.M.; Kreutzer, M.; Nizze, H.; Jaster, R. Senescence determines the fate of activated rat pancreatic stellate cells. J. Cell. Mol. Med. 2012, 16, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Pitiyage, G.N.; Slijepcevic, P.; Gabrani, A.; Chianea, Y.G.; Lim, K.P.; Prime, S.S.; Tilakaratne, W.M.; Fortune, F.; Parkinson, E.K. Senescent mesenchymal cells accumulate in human fibrosis by a telomere-independent mechanism and ameliorate fibrosis through matrix metalloproteinases. J. Pathol. 2011, 223, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.F.M.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology 2013, 14, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD + elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Nopparat, C.; Sinjanakhom, P.; Govitrapong, P. Melatonin reverses H2O2-induced senescence in SH-SY5Y cells by enhancing autophagy via sirtuin 1 deacetylation of the RelA/p65 subunit of NF-κB. J. Pineal Res. 2017, 63, e12407. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Kaushik, S.; Zeng, M.; Bandyopadhyay, U.; Zhang, C.; Massey, A.C.; Martinez-Vicente, M.; Cuervo, A.M. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell Sci. 2007, 120, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Elledge, S.J. How autophagy both activates and inhibits cellular senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.J.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.J.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Young, A.R.J.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Andò, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, I.; Fujita, K.; Jenkins, L.M.M.; Hiyoshi, Y.; Mondal, A.M.; Vojtesek, B.; Lane, D.P.; Appella, E.; Harris, C.C. Autophagic degradation of the inhibitory p53 isoform δ 133p53α as a regulatory mechanism for p53-mediated senescence. Nat. Commun. 2014, 5, 4706. [Google Scholar] [CrossRef] [PubMed]
- Gross, P.A. Current recommendations for the prevention and treatment of influenza in the older population. Drugs Aging 1991, 1, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Janssens, J.-P.; Krause, K.-H. Pneumonia in the very old. Lancet Infect. Dis. 2004, 4, 112–124. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottviani, E.; De Benedictis, G. Inflamm-aging: An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2006, 908, 244–254. [Google Scholar] [CrossRef]
- Aspinall, R.; Del Giudice, G.; Effros, R.B.; Grubeck-Loebenstein, B.; Sambhara, S. Challenges for vaccination in the elderly. Immun. Ageing 2007, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Fietta, A.; Merlini, C.; De Bernardi, P.M.; Gandola, L.; Piccioni, P.D.; Grassi, C. Non specific immunity in aged healthy subjects and in patients with chronic bronchitis. Aging Milano 1993, 5, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Martín, S.; Pérez, A.; Aldecoa, C. Sepsis and Immunosenescence in the Elderly Patient: A Review. Front. Med. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Vardi, M.; Ghanem-Zoubi, N.O.; Bitterman, H.; Abo-Helo, N.; Yurin, V.; Weber, G.; Laor, A. Sepsis in nonagenarians admitted to internal medicine departments: A comparative study of outcomes. QJM 2013, 106, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Weinberger, B.; Grubeck-Loebenstein, B. The aging of the immune system. Transpl. Int. 2009, 22, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Butcher, S.; Chahel, H.; Lord, J.M. Ageing and the neutrophil: No appetite for killing? Immunology 2000, 100, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Butcher, S.K.; Chahal, H.; Nayak, L.; Sinclair, A.; Henriquez, N.V.; Sapey, E.; O’Mahony, D.; Lord, J.M. Senescence in innate immune responses: Reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J. Leukoc. Biol. 2001, 70, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Sauce, D. Naive T cells: The crux of cellular immune aging? Exp. Gerontol. 2014, 54, 90–93. [Google Scholar] [CrossRef] [PubMed]
- McElhaney, J.E.; Effros, R.B. Immunosenescence: What does it mean to health outcomes in older adults? Curr. Opin. Immunol. 2009, 21, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. Age and immunity: What is “immunosenescence”? Exp. Gerontol. 2018, 105, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Stervbo, U.; Meier, S.; Mälzer, J.N.; Baron, U.; Bozzetti, C.; Jürchott, K.; Nienen, M.; Olek, S.; Rachwalik, D.; Schulz, A.R.; et al. Effects of aging on human leukocytes (part I): Immunophenotyping of innate immune cells. Age 2015, 37. [Google Scholar] [CrossRef] [PubMed]
- Le Page, A.; Dupuis, G.; Larbi, A.; Witkowski, J.M.; Fülöp, T. Signal transduction changes in CD4+ and CD8+ T cell subpopulations with aging. Exp. Gerontol. 2018, 105, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Larbi, A.; Dupuis, G.; Khalil, A.; Douziech, N.; Fortin, C.; Fülöp, T. Differential role of lipid rafts in the functions of CD4+ and CD8+ human T lymphocytes with aging. Cell. Signal. 2006, 18, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.N.; Henson, S.M.; Lanna, A. Senescence of T Lymphocytes: Implications for Enhancing Human Immunity. Trends Immunol. 2016, 37, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Di Mitri, D.; Azevedo, R.I.; Henson, S.M.; Libri, V.; Riddell, N.E.; Macaulay, R.; Kipling, D.; Soares, M.V.D.; Battistini, L.; Akbar, A.N. Reversible Senescence in Human CD4+CD45RA+CD27- Memory T Cells. J. Immunol. 2011, 187, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Long, H.M.; Leese, A.M.; Chagoury, O.L.; Connerty, S.R.; Quarcoopome, J.; Quinn, L.L.; Shannon-Lowe, C.; Rickinson, A.B. Cytotoxic CD4+ T Cell Responses to EBV Contrast with CD8 Responses in Breadth of Lytic Cycle Antigen Choice and in Lytic Cycle Recognition. J. Immunol. 2011, 187, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Libri, V.; Azevedo, R.I.; Jackson, S.E.; Di Mitri, D.; Lachmann, R.; Fuhrmann, S.; Vukmanovic-Stejic, M.; Yong, K.; Battistini, L.; Kern, F.; et al. Cytomegalovirus infection induces the accumulation of short-lived, multifunctional CD4+ CD45RA+ CD27− T cells: The potential involvement of interleukin-7 in this process. Immunology 2011, 132, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Colonna-Romano, G.; Bulati, M.; Aquino, A.; Pellicanò, M.; Vitello, S.; Lio, D.; Candore, G.; Caruso, C. A double-negative (IgD-CD27-) B cell population is increased in the peripheral blood of elderly people. Mech. Ageing Dev. 2009, 130, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Bhagirath, A.Y.; Li, Y.; Somayajula, D.; Dadashi, M.; Badr, S.; Duan, K. Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm. Med. 2016, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Lund-Palau, H.; Turnbull, A.R.; Bush, A.; Bardin, E.; Cameron, L.; Soren, O.; Wierre-Gore, N.; Alton, E.W.F.W.; Bundy, J.G.; Connett, G.; et al. Pseudomonas aeruginosa infection in cystic fibrosis: Pathophysiological mechanisms and therapeutic approaches. Expert Rev. Respir. Med. 2016, 10, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Cuppens, H.; Macek, M.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 2008, 7, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Li, Z.; Maitz, P.K.M. Pseudomonas pyocyanin inhibits wound repair by inducing premature cellular senescence: Role for p38 mitogen-activated protein kinase. Burns 2009, 35, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, R.; Cooksley, W.G.E.; Cooke, W.D.D. Improved growth and clinical, nutritional, and respiratory changes in response to nutritional therapy in cystic fibrosis. J. Pediatr. 1980, 97, 351–357. [Google Scholar] [CrossRef]
- Sadikot, R.T.; Blackwell, T.S.; Christman, J.W.; Prince, A.S. Pathogen-host interactions in pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 2005, 171, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.; Dockrell, D.H.; Pattery, T.; Lee, D.G.; Cornelis, P.; Hellewell, P.G.; Whyte, M.K.B. Pyocyanin Production by Pseudomonas aeruginosa Induces Neutrophil Apoptosis and Impairs Neutrophil-Mediated Host Defenses In Vivo. J. Immunol. 2005, 174, 3643–3649. [Google Scholar] [CrossRef] [PubMed]
- Munro, N.C.; Barker, A.; Rutman, A.; Taylor, G.; Watson, D.; McDonald-Gibson, W.J.; Towart, R.; Taylor, W.A.; Wilson, R.; Cole, P.J. Effect of pyocyanin and 1-hydroxyphenazine on in vivo tracheal mucus velocity. J. Appl. Physiol. 1989, 67, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Sykes, D.A.; Watson, D.; Rutman, A.; Taylor, G.W.; Cole, P.J. Measurement of Pseudomonas aeruginosa phenazine pigments in sputum and assessment of their contribution to sputum sol toxicity for respiratory epithelium. Infect. Immun. 1988, 56, 2515–2517. [Google Scholar] [PubMed]
- Denning, G.M.; Wollenweber, L.A.; Railsback, M.A.; Cox, C.D.; Stoll, L.L.; Bntigan, B.E.; Britigan, B.E. Pseudomonas Pyocyanin Increases Interleukin-8 Expression by Human Airway Pseudomonas Pyocyanin Increases Interleukin-8 Expression by Human Airway Epithelial Cells. Infect. Immun. 1998, 66, 5777–5784. [Google Scholar] [PubMed]
- Muller, M.; Sorrell, T.C. Production of leukotriene B4 and 5-hydroxyeicosatetraenoic acid by human neutrophils is inhibited by Pseudomonas aeruginosa phenazine derivatives. Infect. Immun. 1991, 59, 3316–3318. [Google Scholar] [PubMed]
- Cruickshank, C.N.; Lowbury, E.J. The effect of pyocyanin on human skin cells and leucocytes. Br. J. Exp. Pathol. 1953, 34, 583–587. [Google Scholar] [PubMed]
- Muller, M. Pyocyanin induces oxidative stress in human endothelial cells and modulates the glutathione redox cycle. Free Radic. Biol. Med. 2002, 33, 1527–1533. [Google Scholar] [CrossRef]
- O’Malley, Y.Q.; Abdalla, M.Y.; McCormick, M.L.; Reszka, K.J.; Denning, G.M.; Britigan, B.E.; O’Malley, Y.Q.; Abdalla, M.Y.; McCormick, M.L.; Reszka, K.J.; et al. Subcellular localization of Pseudomonas pyocyanin cytotoxicity in human lung epithelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2003, 284, L420–L430. [Google Scholar] [CrossRef] [PubMed]
- Muller, M. Premature cellular senescence induced by pyocyanin, a redox-active Pseudomonas aeruginosa toxin. Free Radic. Biol. Med. 2006, 41, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.S.; Ma, L.Q.; Zhu, K.; Yan, J.Y.; Bian, L.; Zhang, K.Q.; Zou, C.G. Pseudomonas toxin pyocyanin triggers autophagy: Implications for pathoadaptive mutations. Autophagy 2016, 12, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Cyktor, J.C.; Carruthers, B.; Stromberg, P.; Flaño, E.; Pircher, H.; Turnera, J. Killer cell lectin-like receptor G1 deficiency significantly enhances survival after Mycobacterium tuberculosis infection. Infect. Immun. 2013, 81, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, C.; Ngwenyama, N.; Schuetz, A.; Petrovas, C.; Reither, K.; Heeregrave, E.J.; Casazza, J.P.; Ambrozak, D.R.; Louder, M.; Ampofo, W.; et al. Preferential infection and depletion of Mycobacterium tuberculosis—Specific CD4 T cells after HIV-1 infection. J. Exp. Med. 2010, 207, 2869–2881. [Google Scholar] [CrossRef] [PubMed]
- Barathan, M.; Mohamed, R.; Vadivelu, J.; Chang, L.Y.; Vignesh, R.; Krishnan, J.; Sigamani, P.; Saeidi, A.; Ram, M.R.; Velu, V.; et al. CD8+ T cells of chronic HCV-infected patients express multiple negative immune checkpoints following stimulation with HCV peptides. Cell. Immunol. 2017, 313, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, A.; Buggert, M.; Che, K.F.; Kong, Y.Y.; Velu, V.; Larsson, M.; Shankar, E.M. Regulation of CD8+ T-cell cytotoxicity in HIV-1 infection. Cell. Immunol. 2015, 298, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Jagannath, C. Analysis of host-pathogen modulators of autophagy during Mycobacterium tuberculosis infection and therapeutic repercussions. Int. Rev. Immunol. 2017, 36, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brumell, J.H. Bacteria-autophagy interplay: A battle for survival. Nat. Rev. Microbiol. 2014, 12, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Murata-Kamiya, N.; Hirayama, T.; Ohba, Y.; Hatakeyama, M. Conversion of Helicobacter pylori CagA from senescence inducer to oncogenic driver through polarity-dependent regulation of p21. J. Exp. Med. 2010, 207, 2157–2174. [Google Scholar] [CrossRef] [PubMed]
- Kalisperati, P.; Spanou, E.; Pateras, I.S.; Korkolopoulou, P.; Varvarigou, A.; Karavokyros, I.; Gorgoulis, V.G.; Vlachoyiannopoulos, P.G.; Sougioultzis, S. Inflammation, DNA damage, Helicobacter pylori and gastric tumorigenesis. Front. Genet. 2017, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Lewinska, A.; Wnuk, M. Helicobacter pylori-induced premature senescence of extragastric cells may contribute to chronic skin diseases. Biogerontology 2017, 18, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Terebiznik, M.R.; Raju, D.; Vázquez, C.L.; Torbricki, K.; Kulkarni, R.; Blanke, S.R.; Yoshimori, T.; Colombo, M.I.; Jones, N.L. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 2009, 5, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Galmiche, A.; Rassow, J.; Doye, A.; Cagnol, S.; Chambard, J.C.; Contamin, S.; de Thillot, V.; Just, I.; Ricci, V.; Solcia, E.; et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000. [Google Scholar] [CrossRef] [PubMed]
- Raju, D.; Hussey, S.; Ang, M.; Terebiznik, M.R.; Sibony, M.; Galindo-Mata, E.; Gupta, V.; Blanke, S.R.; Delgado, A.; Romero-Gallo, J.; et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 2012, 142, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Romano, P.S.; Cueto, J.A.; Casassa, A.F.; Vanrell, M.C.; Gottlieb, R.A.; Colombo, M.I. Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB Life 2012, 64, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Romano, P.S.; Arboit, M.A.; Vázquez, C.L.; Colombo, M.I. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy 2009, 5, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Onizuka, Y.; Takahashi, C.; Uematsu, A.; Shinjo, S.; Seto, E.; Nakajima-Shimada, J. Inhibition of autolysosome formation in host autophagy by Trypanosoma cruzi infection. Acta Trop. 2017, 170, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Andrade, D.V.; Gollob, K.J.; Dutra, W.O. Acute Chagas Disease: New Global Challenges for an Old Neglected Disease. PLoS Negl. Trop. Dis. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Sbaraglini, M.L.; Vanrell, M.C.; Bellera, C.L.; Benaim, G.; Carrillo, C.; Talevi, A.; Romano, P.S. Neglected Tropical Protozoan Diseases: Drug Repositioning as a Rational Option. Curr. Top. Med. Chem. 2016, 16, 2201–2222. [Google Scholar] [CrossRef] [PubMed]
- Albareda, M.C.; Olivera, G.C.; Laucella, S.A.; Alvarez, M.G.; Fernandez, E.R.; Lococo, B.; Viotti, R.; Tarleton, R.L.; Postan, M. Chronic human infection with Trypanosoma cruzi drives CD4+ T cells to immune senescence. J. Immunol. 2009, 183, 4103–4108. [Google Scholar] [CrossRef] [PubMed]
- Albareda, M.C.; Perez-Mazliah, D.; Natale, M.A.; Castro-Eiro, M.; Alvarez, M.G.; Viotti, R.; Bertocchi, G.; Lococo, B.; Tarleton, R.L.; Laucella, S.A. Perturbed T Cell IL-7 Receptor Signaling in Chronic Chagas Disease. J. Immunol. 2015, 194, 3883–3889. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, F.; Falcao, R.P.; Rossi, M.A.; Mengel, J. An age-related gamma-delta T cell suppressor activity correlates with the outcome of autoimmunity in experimental Trypanosoma cruzi infection. Eur. J. Immunol. 1993, 23, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- González, F.B.; Calmon-Hamaty, F.; Nô Seara Cordeiro, S.; Fernández Bussy, R.; Spinelli, S.V.; D’Attilio, L.; Bottasso, O.; Savino, W.; Cotta-de-Almeida, V.; Villar, S.R.; et al. Trypanosoma cruzi Experimental Infection Impacts on the Thymic Regulatory T Cell Compartment. PLoS Negl. Trop. Dis. 2016, 10, e0004285. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, T.; Larbi, A.; Pawelec, G. Human T cell aging and the impact of persistent viral infections. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Chidrawar, S.; Khan, N.; Wei, W.; McLarnon, A.; Smith, N.; Nayak, L.; Moss, P. Cytomegalovirus-seropositivity has a profound influence on the magnitude of major lymphoid subsets within healthy individuals. Clin. Exp. Immunol. 2009, 155, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Karrer, U.; Mekker, A.; Wanke, K.; Tchang, V.; Haeberli, L. Cytomegalovirus and immune senescence: Culprit or innocent bystander? Exp. Gerontol. 2009, 44, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, B.; Lazuardi, L.; Weiskirchner, I.; Keller, M.; Neuner, C.; Fischer, K.H.; Neuman, B.; Würzner, R.; Grubeck-Loebenstein, B. Healthy Aging and Latent Infection with CMV Lead to Distinct Changes in CD8+ and CD4+ T-Cell Subsets in the Elderly. Hum. Immunol. 2007, 68, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Olsson, J.; Wikby, A.; Johansson, B.; Löfgren, S.; Nilsson, B.O.; Ferguson, F.G. Age-related change in peripheral blood T-lymphocyte subpopulations and cytomegalovirus infection in the very old: The Swedish longitudinal OCTO immune study. Mech. Ageing Dev. 2001, 121, 187–201. [Google Scholar] [CrossRef]
- Wikby, A.; Johansson, B.; Olsson, J.; Löfgren, S.; Nilsson, B.O.; Ferguson, F. Expansions of peripheral blood CD8 T-lymphocyte subpopulations and an association with cytomegalovirus seropositivity in the elderly: The Swedish NONA immune study. Exp. Gerontol. 2002, 37, 445–453. [Google Scholar] [CrossRef]
- Derhovanessian, E.; Theeten, H.; Hähnel, K.; Van Damme, P.; Cools, N.; Pawelec, G. Cytomegalovirus-associated accumulation of late-differentiated CD4 T-cells correlates with poor humoral response to influenza vaccination. Vaccine 2013, 31, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Diaz, A.; Romero, M.; Landin, A.M.; Blomberg, B.B. Cytomegalovirus (CMV) seropositivity decreases B cell responses to the influenza vaccine. Vaccine 2015, 33, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Trzonkowski, P.; Myśliwska, J.; Szmit, E.; Wiȩckiewicz, J.; Łukaszuk, K.; Brydak, L.B.; Machała, M.; Myśliwski, A. Association between cytomegalovirus infection, enhanced proinflammatory response and low level of anti-hemagglutinins during the anti-influenza vaccination- An impact of immunosenescence. Vaccine 2003, 21, 3826–3836. [Google Scholar] [CrossRef]
- Furman, D.; Jojic, V.; Sharma, S.; Shen-Orr, S.S.; Angel, C.J.; Onengut-Gumuscu, S.; Kidd, B.A.; Maecker, H.T.; Concannon, P.; Dekker, C.L.; et al. Cytomegalovirus infection enhances the immune response to influenza. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Selke, S.; Magaret, A.; Boeckh, M. Impact of human cytomegalovirus (CMV) infection on immune response to pandemic 2009 H1N1 influenza vaccine in healthy adults. J. Med. Virol. 2013, 85, 1557–1560. [Google Scholar] [CrossRef] [PubMed]
- Redeker, A.; Remmerswaal, E.B.M.; van der Gracht, E.T.I.; Welten, S.P.M.; Höllt, T.; Koning, F.; Cicin-Sain, L.; Nikolich-Žugich, J.; Ten Berge, I.J.M.; van Lier, R.A.W.; et al. The Contribution of Cytomegalovirus Infection to Immune Senescence Is Set by the Infectious Dose. Front. Immunol. 2017, 8, 1953. [Google Scholar] [CrossRef] [PubMed]
- Chaumorcel, M.; Lussignol, M.; Mouna, L.; Cavignac, Y.; Fahie, K.; Cotte-Laffitte, J.; Geballe, A.; Brune, W.; Beau, I.; Codogno, P.; et al. The Human Cytomegalovirus Protein TRS1 Inhibits Autophagy via Its Interaction with Beclin 1. J. Virol. 2012, 86, 2571–2584. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, S.; Aitken, J.; Sutherland, J.S.; Nicholl, M.J.; Preston, V.G.; Preston, C.M. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J. Virol. 2011, 85, 4212–4221. [Google Scholar] [CrossRef] [PubMed]
- Mouna, L.; Hernandez, E.; Bonte, D.; Brost, R.; Amazit, L.; Delgui, L.R.; Brune, W.; Geballe, A.P.; Beau, I.; Esclatine, A. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy 2016, 12, 327–342. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Liu, R.; Dong, X.; Zhong, Q. Beclin orthologs: Integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 2015, 25, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix Protein 2 of Influenza A Virus Blocks Autophagosome Fusion with Lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A.; Gobeil, P.A.M. Herpes simplex virus γ34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. MBio 2012, 3, e00267-12. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Finke-Isami, J.; Hopper-Chidlaw, A.C.; Schwerk, P.; Thompson, A.; Tedin, K. Salmonella co-opts host cell chaperone-mediated autophagy for intracellular growth. J. Biol. Chem. 2017, 292, 1847–1864. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Immunity | Cell Type | Senescent Phenotype | |
|---|---|---|---|
| Innate immunity | Neutrophils | Phagocytic activity Superoxide generation |  |
| Macrophages | Phagocytic capabilities Superoxide production MHC Levels | | |
| Dendritic cells | Migration capabilities | | |
| Alterations in phagocytic activity of apoptotic cells | |||
| Adaptive immunity | T-cells | CD4(+) and CD8(+) CD28(+) | |
| Proportion CD8(+) CD28(+) | | ||
| Blood naïve CD8(+) | | ||
| Proliferative activity | | ||
| B-cells | IgD(−) CD27(−) | | |
| Basal levels of IL-10 and TNF-α | | ||
| Pathogen | Autophagy | Senescence |
|---|---|---|
| Pseudomonas aeruginosa | Activated (pyocyanin) | Activated (pyocyanin) |
| Mycobacterium tuberculosis | Blocked autophagic flux (ESAT-6) | Activated (immunosenescence) |
| Helicobacter pylori | Blocked autophagic flux (VacA) | Activated (CagA) |
| Trypanosoma cruzi | Activated Blocked autophagic flux | Activated |
| HCMV | Activated Blocked autophagic flux (TRS1 and IRS1) | Activated (immunosenescence) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilera, M.O.; Delgui, L.R.; Romano, P.S.; Colombo, M.I. Chronic Infections: A Possible Scenario for Autophagy and Senescence Cross-Talk. Cells 2018, 7, 162. https://doi.org/10.3390/cells7100162
Aguilera MO, Delgui LR, Romano PS, Colombo MI. Chronic Infections: A Possible Scenario for Autophagy and Senescence Cross-Talk. Cells. 2018; 7(10):162. https://doi.org/10.3390/cells7100162
Chicago/Turabian StyleAguilera, Milton O., Laura R. Delgui, Patricia S. Romano, and María I. Colombo. 2018. "Chronic Infections: A Possible Scenario for Autophagy and Senescence Cross-Talk" Cells 7, no. 10: 162. https://doi.org/10.3390/cells7100162
APA StyleAguilera, M. O., Delgui, L. R., Romano, P. S., & Colombo, M. I. (2018). Chronic Infections: A Possible Scenario for Autophagy and Senescence Cross-Talk. Cells, 7(10), 162. https://doi.org/10.3390/cells7100162