Allogeneic CAR-T Cells: More than Ease of Access?


Abstract
1. Introduction
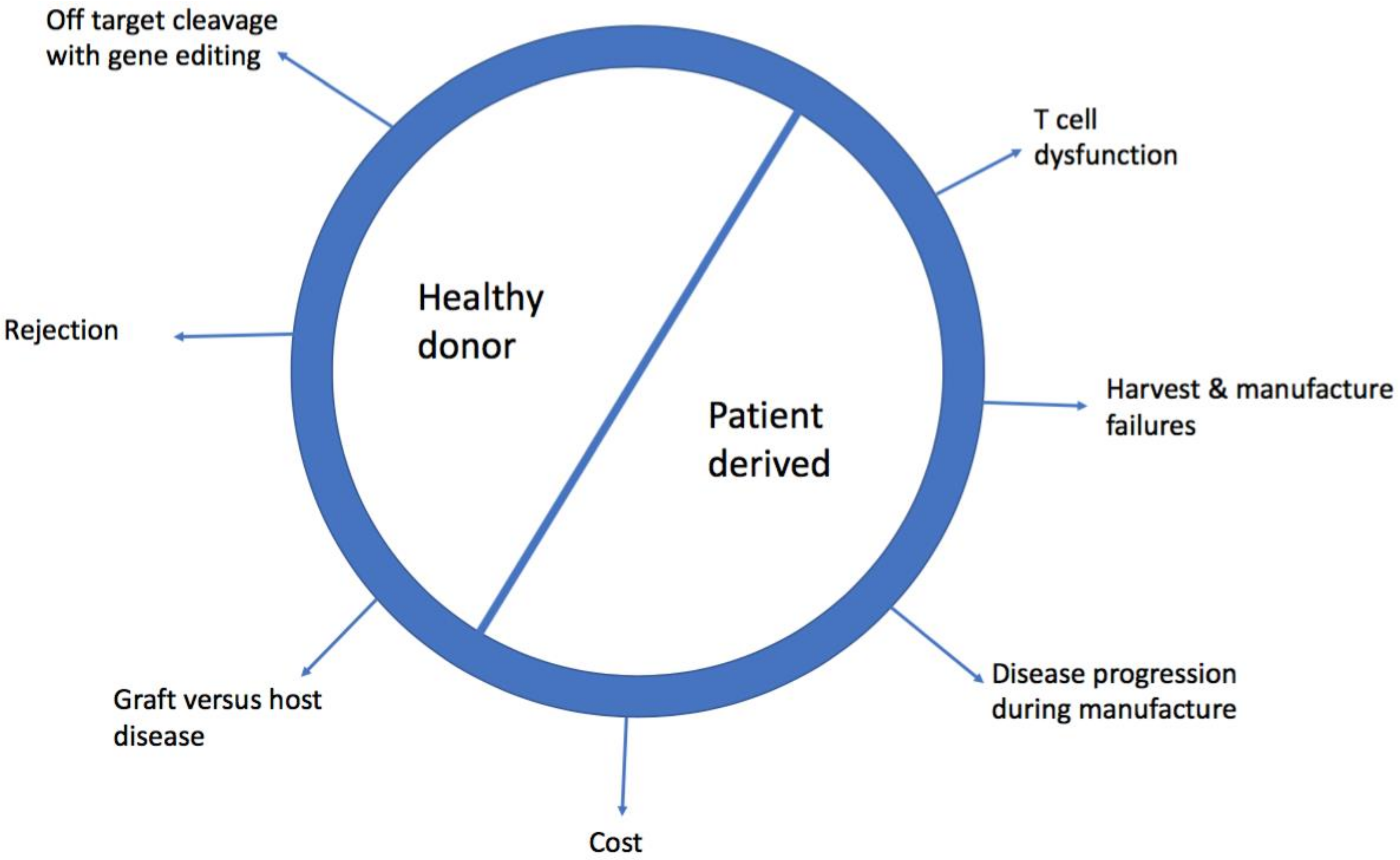
2. Limitations of Patient Derived, Autologous Chimeric Antigen Receptor-T (CAR-T Cells)
2.1. Cost
2.2. Harvest and Manufacturing Failures
2.3. Product Variability and Quality Control
2.4. Disease Progression During Manufacture
2.5. Contamination with Tumour Cells
2.6. T Cell Dysfunction
3. Barriers to Allogeneic, Healthy Donor CAR-T cells
3.1. Graft Versus Host Disease (GVHD)
3.2. Rejection of CAR-T Cells
4. Strategies to Deploy Allogeneic CAR-T Cells
4.1. Manufacture of CAR-T Cells from Previous Allogeneic Haematopoietic Stem Cell Transplant (HSCT) Donor
4.2. ‘Off the Shelf’ Virus Specific CAR-T Cells
4.3. Gene-Edited Healthy Donor CAR-T cells
4.4. Non Gene Edited Mismatch CAR-T Cells
4.5. To Prevent CAR-T Cell Rejection
4.6. Challenges of Gene Editing
4.7. Inducible Pluripotent Stem (iPS) Derived CAR-T Cells
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Harper, M. Novartis CEO’s Dilemma: Is $475,000 Too Much for a Leukaemia Breakthrough? Or Is It No Enough? 2017. Forbes Web site. Available online: https://www.forbes.com/sites/matthewherper/2017/08/30/novartis-ceos-dilemma-is-475000-too-much-for-a-leukemia-breakthrough-or-is-it-not-enough/-367b613e556e (accessed on 20 August 2017).
- Kite’s Yescarta (Axicabtagene Ciloleucel) Becomes First CAR T Therapy Approved by the FDA for the Treatment of Adult Patients with Relapsed or Refractory Large B-Cell Lymphoma after Two or More Lines of Systemic Therapy. Available online: https://www.gilead.com/news/press-releases/2017/10/kites-yescarta-axicabtagene-ciloleucel-becomes-first-car-t-therapy-approved-by-the-fda-for-the-treatment-of-adult-patients-with-relapsed-or-refractory-large-bcell-lymphoma-after-two-or-more-lines-of-systemic-therapy (accessed on 20 July 2018).
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Meth. Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Lacey, S.F.; Xu, J.; Ruella, M.; Barrett, D.M.; Kulikovskaya, I.; Ambrose, D.E.; Patel, P.R.; Reich, T.; Scholler, J.; Nazimuddin, F.; et al. Cars in Leukemia: Relapse with Antigen-Negative Leukemia Originating from a Single B Cell Expressing the Leukemia-Targeting CAR. Blood 2016, 128, 281. [Google Scholar]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bragg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Totterman, T.H.; Carlsson, M.; Simonsson, B.; Bengtsson, M.; Nilsson, K. T-cell activation and subset patterns are altered in B-CLL and correlate with the stage of the disease. Blood 1989, 74, 786–792. [Google Scholar] [PubMed]
- Hoffmann, J.M.; Schubert, M.L.; Wang, L.; Huckelhoven, A.; Sellner, L.; Stock, S.; Schmitt, A.; Kleist, C.; Gern, U.; Loskog, A.; et al. Differences in Expansion Potential of Naive Chimeric Antigen Receptor T Cells from Healthy Donors and Untreated Chronic Lymphocytic Leukemia Patients. Front. Immunol. 2017, 8, 1956. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, I.; Kalland, I.; Kochenderfer, J.N.; Osterborg, A.; Uhlin, M.; Mattsson, J. CD19 Chimeric Antigen Receptor T Cells from Patients with Chronic Lymphocytic Leukaemia Display an Elevated IFN-g Production profile. J. Immunother. 2018, 41, 73–83. [Google Scholar] [PubMed]
- Trofe, J.; Buell, J.F.; First, M.R.; Hanaway, M.J.; Beebe, T.M.; Woodle, E.S. The role of immunosuppression in lymphoma. Recent Results Cancer Res. 2002, 159, 55–66. [Google Scholar] [PubMed]
- Rubinstein, P.G.; Aboulafia, D.M.; Zloza, A. Malignancies in HIV/AIDS: From epidemiology to therapeutic challenges. AIDS. 2014, 28, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Azzaoui, I.; Uhel, F.; Rossille, D.; Pangault, C.; Dulong, J.; Le Priol, J.; Lamy, T.; Houot, R.; Le Gouill, S.; Cartron, G.; et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 2016, 128, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Joshua, D.; Suen, H.; Brown, R.; Bryant, C.; Ho, P.J.; Hart, D.; Gibson, J. The T Cell in Myeloma. Clin. Lymphoma Myeloma Leuk. 2016, 16, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V.; Porter, D.L. Graft-versus-host disease after donor leukocyte infusions: Presentation and management. Best Pract. Res. Clin. Haematol. 2008, 21, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Smith, M.; James, S.E.; Davila, M.L.; Velardi, E.; Argyropoulos, K.V.; Gunset, G.; Perna, F.; Kreines, F.M.; Levy, E.R.; et al. Donor CD19 CART cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat. Med. 2017, 23, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Zakrzewski, J.; James, S.; Sadelain, M. Posttransplant chimeric antigen receptor therapy. Blood 2018, 131, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Leen, A.M.; Bollard, C.M.; Mendizabal, A.M.; Shpall, E.J.; Szabolcs, P.; Antin, J.H.; Kapoor, N.; Pai, S.Y.; Rowley, S.D.; Kebriaei, P.; et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood 2013, 121, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, R.J.; Prockop, S.; Hasan, A.N.; Koehne, G.; Doubrovina, E. Virus-specific T-cell banks for ‘off the shelf’ adoptive therapy of refractory infections. Bone Marrow Transplant. 2016, 51, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Kenderian, S.S. Next-Generation Chimeric Antigen Receptor T-Cell Therapy: Going off the Shelf. BioDrugs 2017, 31, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.R.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. 2017, 25, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, C.; Preece, R.; Nickolay, L.; Etuk, A.; Petrova, A.; Ladon, D.; Danyi, A.; Humphryes-Kirilov, N.; Ajetunmobi, A.; Kim, D.; et al. Long Terminal Repeat CRISPR-CAR-Coupled “Universal” T Cells Mediate Potent Anti-leukemic Effects. Mol. Ther. 2018, 26, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Gilham, D.E.; Michaux, A.; Breman, E.; Mauen, S.; Bolsée, J.; Huberty, F.; Marijsse, J.; Violle, B.; Jacques-Hespel, C.; Marchand, C.; et al. TCR inhibitory molecule as a promising allogeneic NKG2D CAR-t cell approach. In Proceedings of the 2018 ASCO Annual Meeting, Chicago, IL, USA, 5 June 2018. [Google Scholar]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Amara, S.C.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [PubMed]
- Valton, J.; Guyot, V.; Marechal, A.; Filhol, J.M.; Juillerat, A.; Duclert, A.; Duchateau, P.; Poirot, L. A Multidrug-resistant Engineered CAR T Cell for Allogeneic Combination Immunotherapy. Mol. Ther. 2015, 23, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef] [PubMed]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; Zhao, Y.B. CRISPR/Cas9 genome editing: Fueling the revolution in cancer immunotherapy. Curr. Res. Transl. Med. 2018, 66, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Torikai, H.; Reik, A.; Liu, P.Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovich, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef] [PubMed]
- Knipping, F.; Osborn, M.J.; Petri, K.; Tolar, J.; Glimm, H.; von Kalle, C.; Schmidt, M.; Gabriel, R.; et al. Genome-wide Specificity of Highly Efficient TALENs and CRISPR/Cas9 for T Cell Receptor Modification. Mol. Ther. Methods Clin. Dev. 2017, 4, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.J.; Webber, B.R.; Knipping, F.; Lonetree, C.L.; Tennis, N.; DeFeo, A.P.; McElroy, A.N.; Starker, C.G.; Lee, C.; Merkel, S.; et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol. Ther. 2016, 24, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Binding Region | Nuclease | Cutting Efficiency | Reference | |
|---|---|---|---|---|
| Zinc finger nucleases (ZFNs) | Protein | FOKI | 20–40% | [41] |
| Transcription activator-like effector nuclease (TALEN) | Protein | FOKI | 53.7% (double knockout) 78% TCR KO 78.8 & 81.2% | [34] [42] |
| CRISPR/Cas9 | gRNA | Cas9 | 60% | [43] |
| >80% | [38] | |||
| ~70% | [28] | |||
| 77% (CAR+ cells) | [30] | |||
| megaTAL Nucleases | Protein | Meganuclease | 75% | [43] |
| Engineered I-CreI Homing endonuclease | Protein | >60% | [29] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graham, C.; Jozwik, A.; Pepper, A.; Benjamin, R. Allogeneic CAR-T Cells: More than Ease of Access? Cells 2018, 7, 155. https://doi.org/10.3390/cells7100155
Graham C, Jozwik A, Pepper A, Benjamin R. Allogeneic CAR-T Cells: More than Ease of Access? Cells. 2018; 7(10):155. https://doi.org/10.3390/cells7100155
Chicago/Turabian StyleGraham, Charlotte, Agnieszka Jozwik, Andrea Pepper, and Reuben Benjamin. 2018. "Allogeneic CAR-T Cells: More than Ease of Access?" Cells 7, no. 10: 155. https://doi.org/10.3390/cells7100155
APA StyleGraham, C., Jozwik, A., Pepper, A., & Benjamin, R. (2018). Allogeneic CAR-T Cells: More than Ease of Access? Cells, 7(10), 155. https://doi.org/10.3390/cells7100155