
Invariant Chain Complexes and Clusters as Platforms for MIF Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structure of Invariant Chain
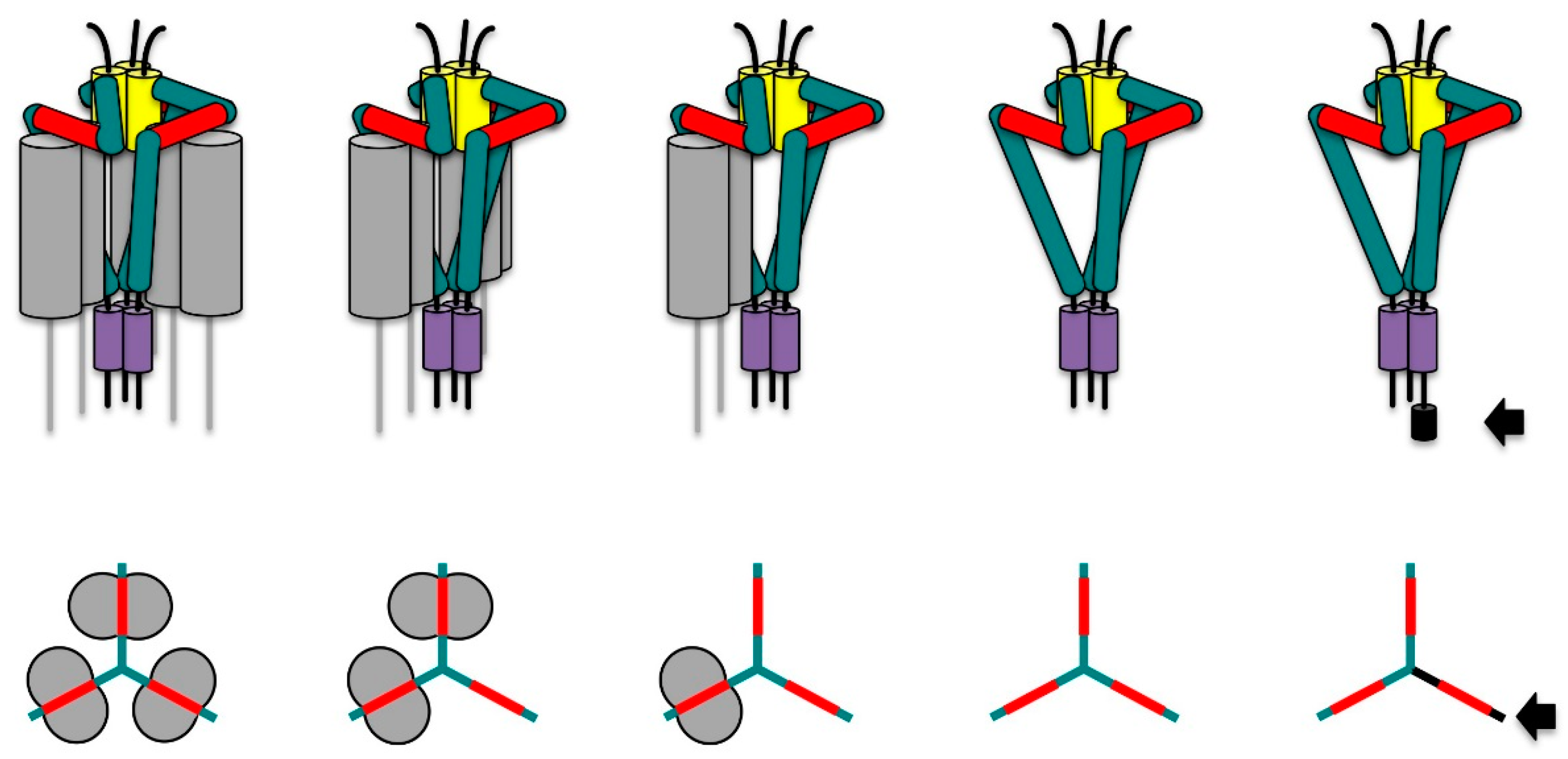
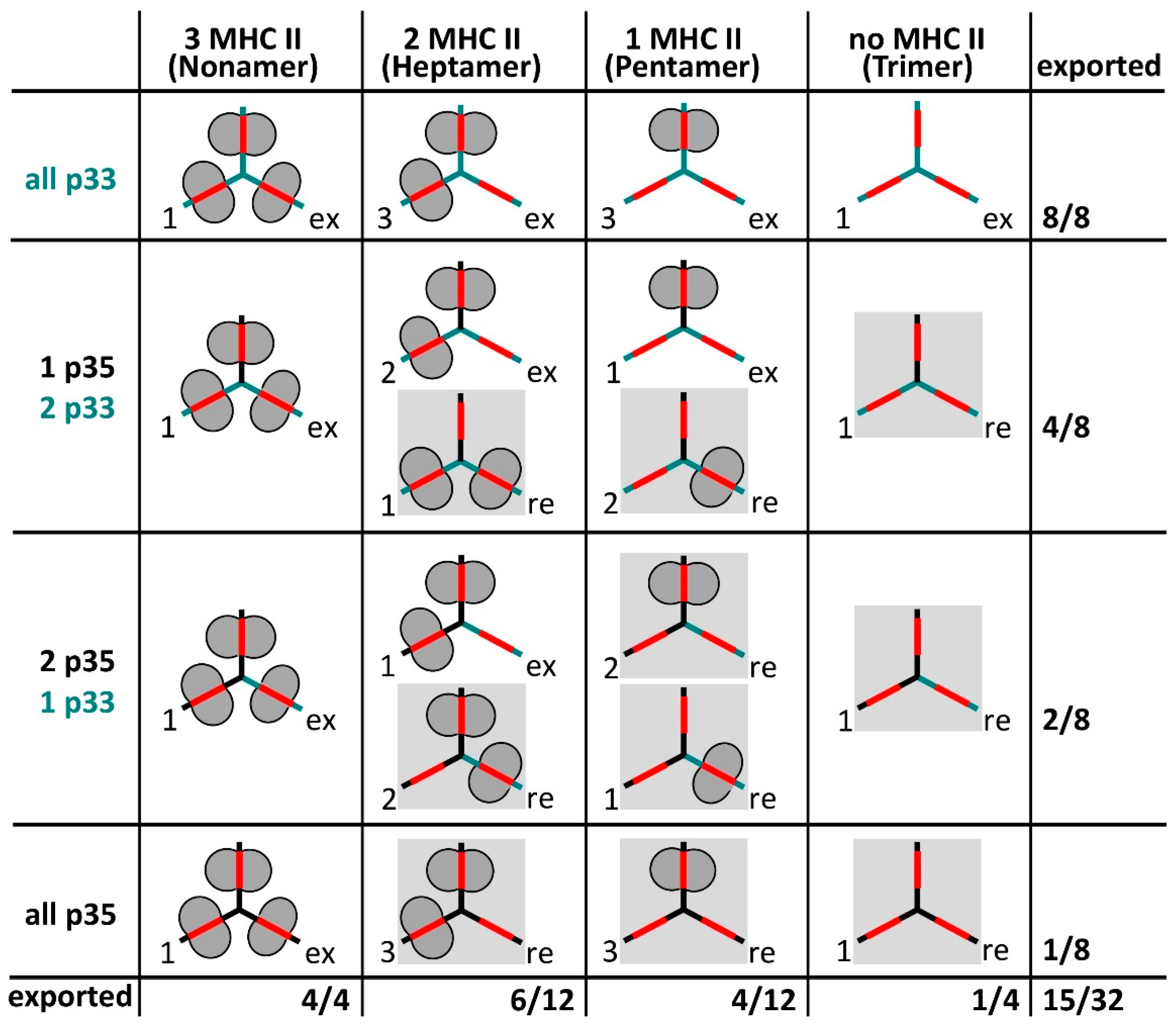
3. Invariant Chain as Chaperone and Transport Co-Factor for MHC II
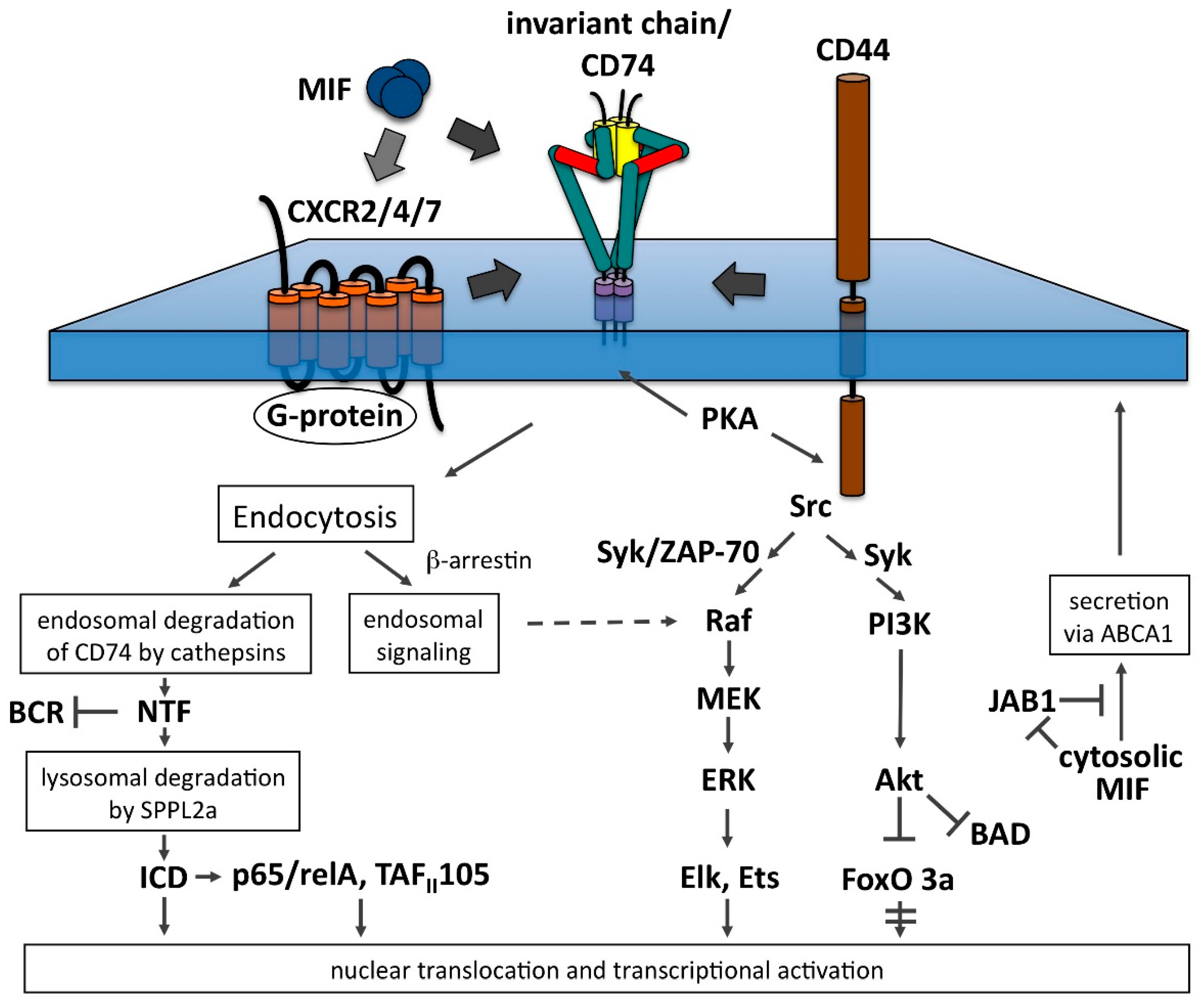
4. Invariant Chain as Receptor for Migration Inhibitory Factor (MIF)
5. MIF-Induced Signaling from Invariant Chain Complexes
5.1. The RIP Pathway
5.2. The PI3K-Akt Pathway
5.3. The MAPK/ERK Pathway
6. Ligand-Induced Signal Cluster Formation Enabled by Membrane Rafts
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Jones, P.P.; Murphy, D.B.; Hewgill, D.; McDevitt, H.O. Detection of a common polypeptide chain in I-A and I-E sub-region immunoprecipitates. Mol. Immunol. 1979, 16, 51–60. [Google Scholar] [CrossRef]
- Anderson, M.S.; Miller, J. Invariant chain can function as a chaperone protein for class II major histocompatibility complex molecules. Proc. Natl. Acad. Sci. USA 1992, 89, 2282–2286. [Google Scholar] [CrossRef] [PubMed]
- Roche, P.A.; Cresswell, P. Invariant chain association with HLA-DR molecules inhibits immunogenic peptide binding. Nature 1990, 345, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Busch, R.; Cloutier, I.; Sekaly, R.P.; Hämmerling, G.J. Invariant chain protects class II histocompatibility antigens from binding intact polypeptides in the endoplasmic reticulum. EMBO J. 1996, 15, 418–428. [Google Scholar] [PubMed]
- Lamb, C.A.; Yewdell, J.W.; Bennink, J.R.; Cresswell, P. Invariant chain targets HLA class II molecules to acidic endosomes containing internalized influenza virus. Proc. Natl. Acad. Sci. USA 1991, 88, 5998–6002. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.J.; Stollorz, V.; Peters, P.J.; Geuze, H.J.; Ploegh, H.L. The biosynthetic pathway of MHC class II but not class I molecules intersects the endocytic route. Cell 1990, 61, 171–183. [Google Scholar] [CrossRef]
- Lindner, R. Transient surface delivery of invariant chain-MHC II complexes via endosomes: A quantitative study. Traffic 2002, 3, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Dugast, M.; Toussaint, H.; Dousset, C.; Benaroch, P. AP2 clathrin adaptor complex, but not AP1, controls the access of the major histocompatibility complex (MHC) class II to endosomes. J. Biol. Chem. 2005, 280, 19656–19664. [Google Scholar] [CrossRef] [PubMed]
- McCormick, P.J.; Martina, J.A.; Bonifacino, J.S. Involvement of clathrin and AP-2 in the trafficking of MHC class II molecules to antigen-processing compartments. Proc. Natl. Acad. Sci. USA 2005, 102, 7910–7915. [Google Scholar] [CrossRef] [PubMed]
- Basha, G.; Omilusik, K.; Chavez-Steenbock, A.; Reinicke, A.T.; Lack, N.; Choi, K.B.; Jefferies, W.A. A CD74-dependent MHC class I endolysosomal cross-presentation pathway. Nat. Immunol. 2012, 13, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena-Wolf, J.; Benlagha, K.; Chiu, Y.H.; Mehr, R.; Bendelac, A. CD1d endosomal trafficking is independently regulated by an intrinsic CD1d-encoded tyrosine motif and by the invariant chain. Immunity 2001, 15, 897–908. [Google Scholar] [CrossRef]
- Kang, S.J.; Cresswell, P. Regulation of intracellular trafficking of human CD1d by association with MHC class II molecules. EMBO J. 2002, 21, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Zwart, W.; Peperzak, V.; de Vries, E.; Keller, A.M.; van der Horst, G.; Veraar, E.A.M.; Geumann, U.; Janssen, H.; Janssen, L.; Naik, S.H.; et al. The invariant chain transports TNF family member CD70 to MHC class II compartments in dendritic cells. J. Cell Sci. 2010, 123, 3817–3827. [Google Scholar] [CrossRef] [PubMed]
- Szaszák, M.; Chen, H.-D.; Chen, H.-C.; Baukal, A.; Hunyady, L.; Catt, K.J. Identification of the invariant chain (CD74) as an angiotensin AGTR1-interacting protein. J. Endocrinol. 2008, 199, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Tohmé, M.; Manoury, B. Invariant chain is a new chaperone for TLR7 in B cells. Mol. Immunol. 2015, 68, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Shachar, I.; Flavell, R.A. Requirement for invariant chain in B cell maturation and function. Science 1996, 274, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Matza, D.; Wolstein, O.; Dikstein, R.; Shachar, I. Invariant chain induces B cell maturation by activating a TAF(II)105-NF-κB-dependent transcription program. J. Biol. Chem. 2001, 276, 27203–27206. [Google Scholar] [CrossRef] [PubMed]
- Matza, D.; Kerem, A.; Medvedovsky, H.; Lantner, F.; Shachar, I. Invariant chain-induced B cell differentiation requires intramembrane proteolytic release of the cytosolic domain. Immunity 2002, 17, 549–560. [Google Scholar] [CrossRef]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF Signal Transduction Initiated by Binding to CD74. J. Exp. Med. 2003, 197, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 2006, 25, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Lue, H.; Kapurniotu, A.; Fingerle-Rowson, G.; Roger, T.; Leng, L.; Thiele, M.; Calandra, T.; Bucala, R.; Bernhagen, J. Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell. Signal. 2006, 18, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Lue, H.; Thiele, M.; Franz, J.; Dahl, E.; Speckgens, S.; Leng, L.; Fingerle-Rowson, G.; Bucala, R.; Lüscher, B.; Bernhagen, J. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene 2007, 26, 5046–5059. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, V.; Lue, H.; Kraemer, S.; Korbiel, J.; Krohn, R.; Ohl, K.; Bucala, R.; Weber, C.; Bernhagen, J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009, 583, 2749–2757. [Google Scholar] [CrossRef] [PubMed]
- Starlets, D.; Gore, Y.; Binsky, I.; Haran, M.; Harpaz, N.; Shvidel, L.; Becker-Herman, S.; Berrebi, A.; Shachar, I. Cell-surface CD74 initiates a signaling cascade leading to cell proliferation and survival. Blood 2006, 107, 4807–4816. [Google Scholar] [CrossRef] [PubMed]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Klasen, C.; Ohl, K.; Sternkopf, M.; Shachar, I.; Schmitz, C.; Heussen, N.; Hobeika, E.; Levit-Zerdoun, E.; Tenbrock, K.; Reth, M.; et al. MIF Promotes B Cell Chemotaxis through the Receptors CXCR4 and CD74 and ZAP-70 Signaling. J. Immunol. 2014, 192, 5273–5284. [Google Scholar] [CrossRef] [PubMed]
- Alampour-Rajabi, S.; El Bounkari, O.; Rot, A.; Müller-Newen, G.; Bachelerie, F.; Gawaz, M.; Weber, C.; Schober, A.; Bernhagen, J. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. 2015, 29, 4497–4511. [Google Scholar] [CrossRef] [PubMed]
- Subbannayya, T.; Variar, P.; Advani, J.; Nair, B.; Shankar, S.; Gowda, H.; Saussez, S.; Chatterjee, A.; Prasad, T.S.K. An integrated signal transduction network of macrophage migration inhibitory factor. J. Cell Commun. Signal. 2016, 10, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Gore, Y.; Starlets, D.; Maharshak, N.; Becker-Herman, S.; Kaneyuki, U.; Leng, L.; Bucala, R.; Shachar, I. Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J. Biol. Chem. 2008, 283, 2784–2792. [Google Scholar] [CrossRef] [PubMed]
- Schröder, B. The multifaceted roles of the invariant chain CD74—More than just a chaperone. Biochim. Biophys. Acta 2016, 1863, 1269–1281. [Google Scholar] [CrossRef] [PubMed]
- Strubin, M.; Berte, C.; Mach, B. Alternative splicing and alternative initiation of translation explain the four forms of the Ia antigen-associated invariant chain. EMBO J. 1986, 5, 3483–3488. [Google Scholar] [PubMed]
- Bevec, T.; Stoka, V.; Pungercic, G.; Dolenc, I.; Turk, V. Major histocompatibility complex class II-associated p41 invariant chain fragment is a strong inhibitor of lysosomal cathepsin L. J. Exp. Med. 1996, 183, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Mihelič, M.; Dobersek, A.; Guncar, G.; Turk, D. Inhibitory fragment from the p41 form of invariant chain can regulate activity of cysteine cathepsins in antigen presentation. J. Biol. Chem. 2008, 283, 14453–14460. [Google Scholar] [CrossRef] [PubMed]
- Marks, M.S.; Blum, J.S.; Cresswell, P. Invariant chain trimers are sequestered in the rough endoplasmic reticulum in the absence of association with HLA class II antigens. J. Cell Biol. 1990, 111, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Jasanoff, A.; Park, S.J.; Wiley, D.C. Direct observation of disordered regions in the major histocompatibility complex class II-associated invariant chain. Proc. Natl. Acad. Sci. USA 1995, 92, 9900–9904. [Google Scholar] [CrossRef] [PubMed]
- Kukol, A.; Torres, J.; Arkin, I.T. A structure for the trimeric MHC class II-associated invariant chain transmembrane domain. J. Mol. Biol. 2002, 320, 1109–1117. [Google Scholar] [CrossRef]
- Dixon, A.M.; Stanley, B.J.; Matthews, E.E.; Dawson, J.P.; Engelman, D.M. Invariant chain transmembrane domain trimerization: A step in MHC class II assembly. Biochemistry 2006, 45, 5228–5234. [Google Scholar] [CrossRef] [PubMed]
- Motta, A.; Amodeo, P.; Fucile, P.; Castiglione Morelli, M.A.; Bremnes, B.; Bakke, O. A new triple-stranded alpha-helical bundle in solution: The assembling of the cytosolic tail of MHC-associated invariant chain. Structure 1997, 5, 1453–1464. [Google Scholar] [CrossRef]
- Bakke, O.; Dobberstein, B. MHC class II-associated invariant chain contains a sorting signal for endosomal compartments. Cell 1990, 63, 707–716. [Google Scholar] [CrossRef]
- Pieters, J.; Bakke, O.; Dobberstein, B. The MHC class II-associated invariant chain contains two endosomal targeting signals within its cytoplasmic tail. J. Cell Sci. 1993, 106, 831–846. [Google Scholar] [PubMed]
- Ghosh, P.; Amaya, M.; Mellins, E.; Wiley, D.C. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature 1995, 378, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Rudensky, A.Y.; Corper, A.L.; Teyton, L.; Wilson, I.A. Crystal Structure Of MHC Class II I-Ab in Complex with a Human CLIP Peptide: Prediction of an I-Ab Peptide-binding Motif. J. Mol. Biol. 2003, 326, 1157–1174. [Google Scholar] [CrossRef]
- Jasanoff, A.; Song, S.; Dinner, A.R.; Wagner, G.; Wiley, D.C. One of two unstructured domains of Ii becomes ordered in complexes with MHC class II molecules. Immunity 1999, 10, 761–768. [Google Scholar] [CrossRef]
- Jasanoff, A.; Wagner, G.; Wiley, D.C. Structure of a trimeric domain of the MHC class II-associated chaperonin and targeting protein Ii. EMBO J. 1998, 17, 6812–6818. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, J.R.; Carboy-Newcomb, C.; Cresswell, P. Trimeric interactions of the invariant chain and its association with major histocompatibility complex class IIabdimers. J. Biol. Chem. 1996, 271, 24249–24256. [Google Scholar] [CrossRef] [PubMed]
- Castellino, F.; Han, R.; Germain, R.N. The transmembrane segment of invariant chain mediates binding to MHC class II molecules in a CLIP-independent manner. Eur. J. Immunol. 2001, 31, 841–850. [Google Scholar] [CrossRef]
- Koch, N.; Hämmerling, G.J. The HLA-D-associated invariant chain binds palmitic acid at the cysteine adjacent to the membrane segment. J. Biol. Chem. 1986, 261, 3434–3440. [Google Scholar] [PubMed]
- Anderson, H.A.; Roche, P.A. Phosphorylation regulates the delivery of MHC class II invariant chain complexes to antigen processing compartments. J. Immunol. 1998, 160, 4850–4858. [Google Scholar] [PubMed]
- Anderson, H.A.; Bergstralh, D.T.; Kawamura, T.; Blauvelt, A.; Roche, P.A. Phosphorylation of the invariant chain by protein kinase C regulates MHC class II trafficking to antigen-processing compartments. J. Immunol. 1999, 163, 5435–5443. [Google Scholar] [PubMed]
- Neumann, J.; Schach, N.; Koch, N. Glycosylation signals that separate the trimerization from the mhc class II-binding domain control intracellular degradation of invariant chain. J. Biol. Chem. 2001, 276, 13469–13475. [Google Scholar] [CrossRef] [PubMed]
- Sant, A.J.; Cullen, S.E.; Schwartz, B.D. Biosynthetic relationships of the chondroitin sulfate proteoglycan with Ia and invariant chain glycoproteins. J. Immunol. 1985, 135, 416–422. [Google Scholar] [PubMed]
- Sant, A.J.; Cullen, S.E.; Giacoletto, K.S.; Schwartz, B.D. Invariant chain is the core protein of the Ia-associated chondroitin sulfate proteoglycan. J. Exp. Med. 1985, 162, 1916–1934. [Google Scholar] [CrossRef] [PubMed]
- Naujokas, M.F.; Morin, M.; Anderson, M.S.; Peterson, M.; Miller, J. The chondroitin sulfate form of invariant chain can enhance stimulation of T cell responses through interaction with CD44. Cell 1993, 74, 257–268. [Google Scholar] [CrossRef]
- Machamer, C.E.; Cresswell, P. Biosynthesis and glycosylation of the invariant chain associated with HLA-DR antigens. J. Immunol. 1982, 129, 2564–2569. [Google Scholar] [PubMed]
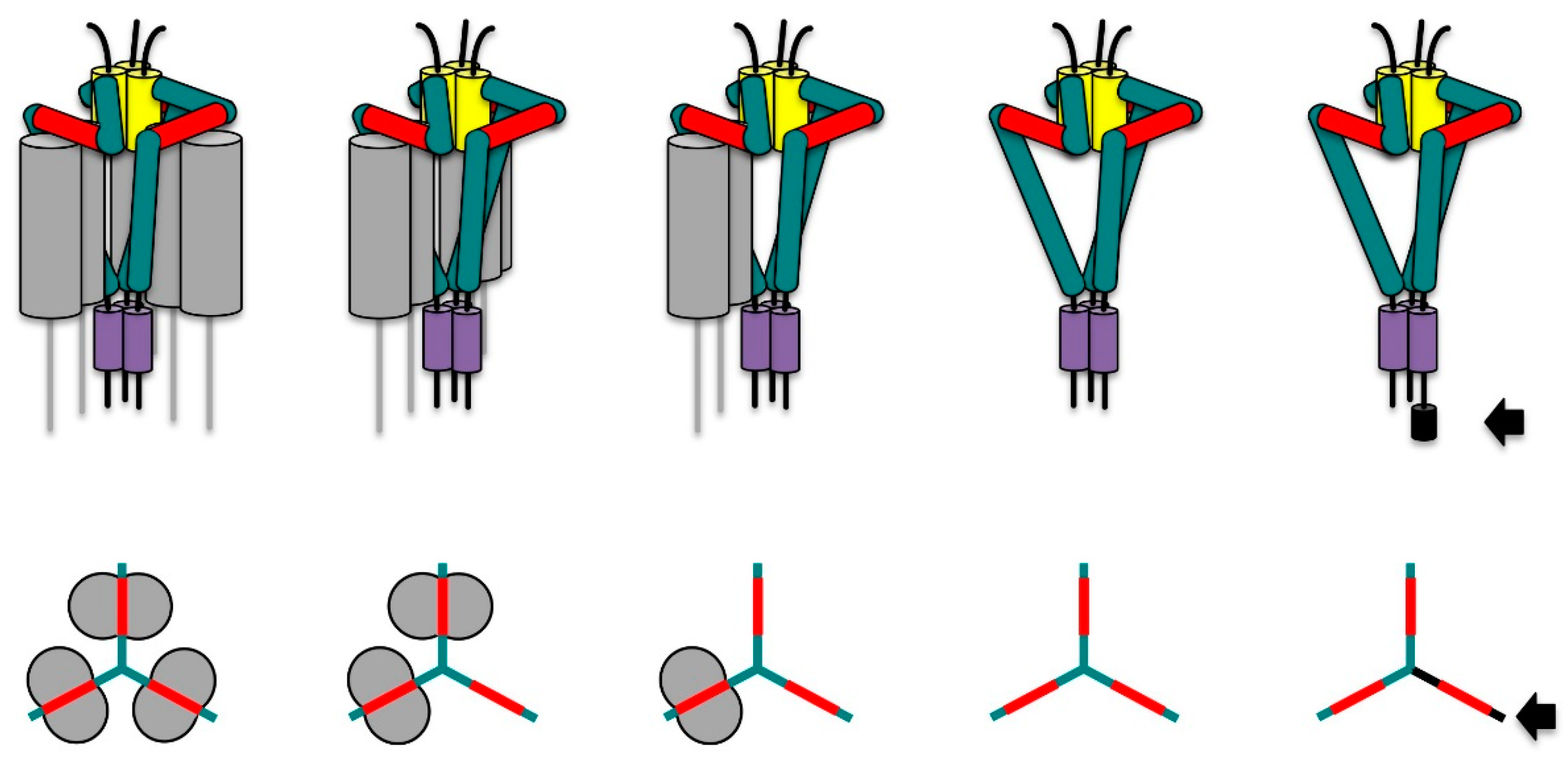
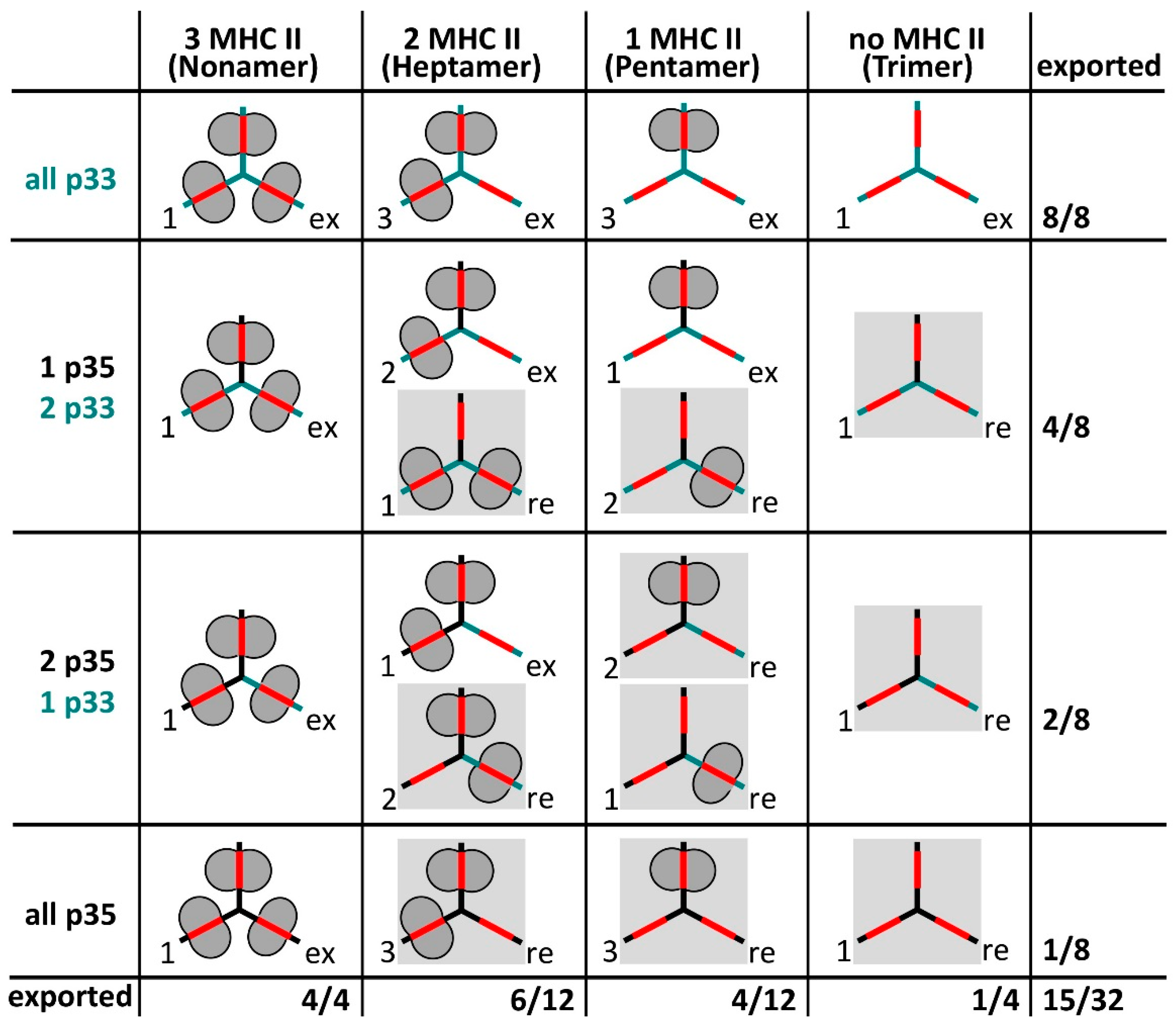
- Roche, P.A.; Marks, M.S.; Cresswell, P. Formation of a nine-subunit complex by HLA class II glycoproteins and the invariant chain. Nature 1991, 354, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Koch, N.; Zacharias, M.; Konig, A.; Temme, S.; Neumann, J.; Springer, S. Stoichiometry of HLA Class II-Invariant Chain Oligomers. PLoS ONE 2011, 6, e17257. [Google Scholar] [CrossRef] [PubMed]
- Cresswell, P.; Roche, P.A. Invariant chain-MHC class II complexes: Always odd and never invariant. Immunol. Cell Biol. 2014, 92, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, M.; Gauthier, C.; Fortin, J.-S.; Thibodeau, J. The invariant chain p35 isoform promotes formation of nonameric complexes with MHC II molecules. Immunol. Cell Biol. 2014, 92, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Brunet, A.; Saba, I.; Terra, R.; Sékaly, R.P.; Thibodeau, J. The MHC class II beta chain cytoplasmic tail overcomes the invariant chain p35-encoded endoplasmic reticulum retention signal. Int. Immunol. 2003, 15, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, M.; Gauthier, C.; Fortin, J.-S.; Genève, L.; Kim, K.; Gruenheid, S.; Kim, J.; Thibodeau, J. ER egress of invariant chain isoform p35 requires direct binding to MHCII molecules and is inhibited by the NleA virulence factor of enterohaemorrhagic Escherichia coli. Hum. Immunol. 2015, 76, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Majera, D.; Kristan, K.Č.; Neefjes, J.; Turk, D.; Mihelič, M. Expression, purification and assembly of soluble multimeric MHC class II-invariant chain complexes. FEBS Lett. 2012, 586, 1318–1324. [Google Scholar] [CrossRef] [PubMed]
- Peters, P.J.; Neefjes, J.J.; Oorschot, V.; Ploegh, H.L.; Geuze, H.J. Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for transport to lysosomal compartments. Nature 1991, 349, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, D.G.; Bakke, O. Medium chains of adaptor complexes AP-1 and AP-2 recognize leucine-based sorting signals from the invariant chain. J. Biol. Chem. 1998, 273, 6005–6008. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.N.; Kloster, M.M.; Rodionov, D.G.; Bakke, O. Re-routing of the invariant chain to the direct sorting pathway by introduction of an AP3-binding motif from LIMP II. Eur. J. Cell Biol. 2006, 85, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef] [PubMed]
- Odorizzi, C.G.; Trowbridge, I.S.; Xue, L.; Hopkins, C.R.; Davis, C.D.; Collawn, J.F. Sorting signals in the MHC class II invariant chain cytoplasmic tail and transmembrane region determine trafficking to an endocytic trafficking compartment. J. Cell Biol. 1994, 126, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Benaroch, P.; Yilla, M.; Raposo, G.; Ito, K.; Miwa, K.; Geuze, H.J.; Ploegh, H.L. How MHC class II molecules reach the endocytic pathway. EMBO J. 1995, 14, 37–49. [Google Scholar] [PubMed]
- Wraight, C.J.; van Endert, P.; Möller, P.; Lipp, J.; Ling, N.R.; MacLennan, I.C.M.; Koch, N.; Moldenhauer, G. Human major histocompatibility complex class II invariant chain is expressed on the cell surface. J. Biol. Chem. 1990, 265, 5787–5792. [Google Scholar] [PubMed]
- Roche, P.A.; Teletski, C.L.; Stang, E.; Bakke, O.; Long, E.O. Cell surface HLA-DR-invariant chain complexes are targeted to endosomes by rapid internalization. Proc. Natl. Acad. Sci. USA 1993, 90, 8581–8585. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, P.; Layet, C.; Yewdell, J.; Bakke, O.; Germain, R.N. Relationship between invariant chain expression and major histocompatibility complex class II transport into early and late endocytic compartments. J. Exp. Med. 1993, 177, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Landsverk, O.J.B.; Barois, N.; Gregers, T.F.; Bakke, O. Invariant chain increases the half-life of MHC II by delaying endosomal maturation. Immunol. Cell Biol. 2011, 89, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Lagaudriere-Gesbert, C.; Newmyer, S.L.; Gregers, T.F.; Bakke, O.; Ploegh, H.L. Uncoating ATPase Hsc70 is recruited by invariant chain and controls the size of endocytic compartments. Proc. Natl. Acad. Sci. USA 2002, 99, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Villadangos, J.A.; Bryant, R.A.; Deussing, J.; Driessen, C.; Lennon-Dumenil, A.M.; Riese, R.J.; Roth, W.; Saftig, P.; Shi, G.P.; Chapman, H.A.; et al. Proteases involved in MHC class II antigen presentation. Immunol. Rev. 1999, 172, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Mellins, E.D.; Stern, L.J. HLA-DM and HLA-DO, key regulators of MHC-II processing and presentation. Curr. Opin. Immunol. 2014, 26, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Boes, M.; Cerny, J.; Massol, R.; Op den Brouw, M.; Kirchhausen, T.; Chen, J.; Ploegh, H.L. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature 2002, 418, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.W.; Bernhagen, J.; Bucala, R.; Lolis, E. Crystal structure at 2.6-A resolution of human macrophage migration inhibitory factor. Proc. Natl. Acad. Sci. USA 1996, 93, 5191–5196. [Google Scholar] [CrossRef] [PubMed]
- Weiser, W.Y.; Temple, P.A.; Witek-Giannotti, J.S.; Remold, H.G.; Clark, S.C.; David, J.R. Molecular cloning of a cDNA encoding a human macrophage migration inhibitory factor. Proc. Natl. Acad. Sci. USA 1989, 86, 7522–7526. [Google Scholar] [CrossRef] [PubMed]
- Flieger, O.; Engling, A.; Bucala, R.; Lue, H.; Nickel, W.; Bernhagen, J. Regulated secretion of macrophage migration inhibitory factor is mediated by a non-classical pathway involving an ABC transporter. FEBS Lett. 2003, 551, 78–86. [Google Scholar] [CrossRef]
- Oddo, M.; Calandra, T.; Bucala, R.; Meylan, P.R.A. Macrophage migration inhibitory factor reduces the growth of virulent Mycobacterium tuberculosis in human macrophages. Infect. Immun. 2005, 73, 3783–3786. [Google Scholar] [CrossRef] [PubMed]
- Calandra, T.; Roger, T. Macrophage migration inhibitory factor: A regulator of innate immunity. Nat. Rev. Immunol. 2003, 3, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Shachar, I.; Haran, M. The secret second life of an innocent chaperone: The story of CD74 and B cell/chronic lymphocytic leukemia cell survival. Leuk. Lymphoma 2011, 52, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Merk, M.; Zierow, S.; Leng, L.; Das, R.; Du, X.; Schulte, W.; Fan, J.; Lue, H.; Chen, Y.; Xiong, H.; et al. The d-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc. Natl. Acad. Sci. USA 2011, 108, E577–E585. [Google Scholar] [CrossRef] [PubMed]
- Benedek, G.; Meza-Romero, R.; Andrew, S.; Leng, L.; Burrows, G.G.; Bourdette, D.; Offner, H.; Bucala, R.; Vandenbark, A.A. Partial MHC class II constructs inhibit MIF/CD74 binding and downstream effects. Eur. J. Immunol. 2013, 43, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Meza-Romero, R.; Benedek, G.; Yu, X.; Mooney, J.L.; Dahan, R.; Duvshani, N.; Bucala, R.; Offner, H.; Reiter, Y.; Burrows, G.G.; et al. HLA-DRα1 Constructs Block CD74 Expression and MIF Effects in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2014, 192, 4164–4173. [Google Scholar] [CrossRef] [PubMed]
- Meza-Romero, R.; Benedek, G.; Leng, L.; Bucala, R.; Vandenbark, A.A. Predicted structure of MIF/CD74 and RTL1000/CD74 complexes. Metab. Brain Dis. 2016, 31, 249–255. [Google Scholar] [CrossRef] [PubMed]
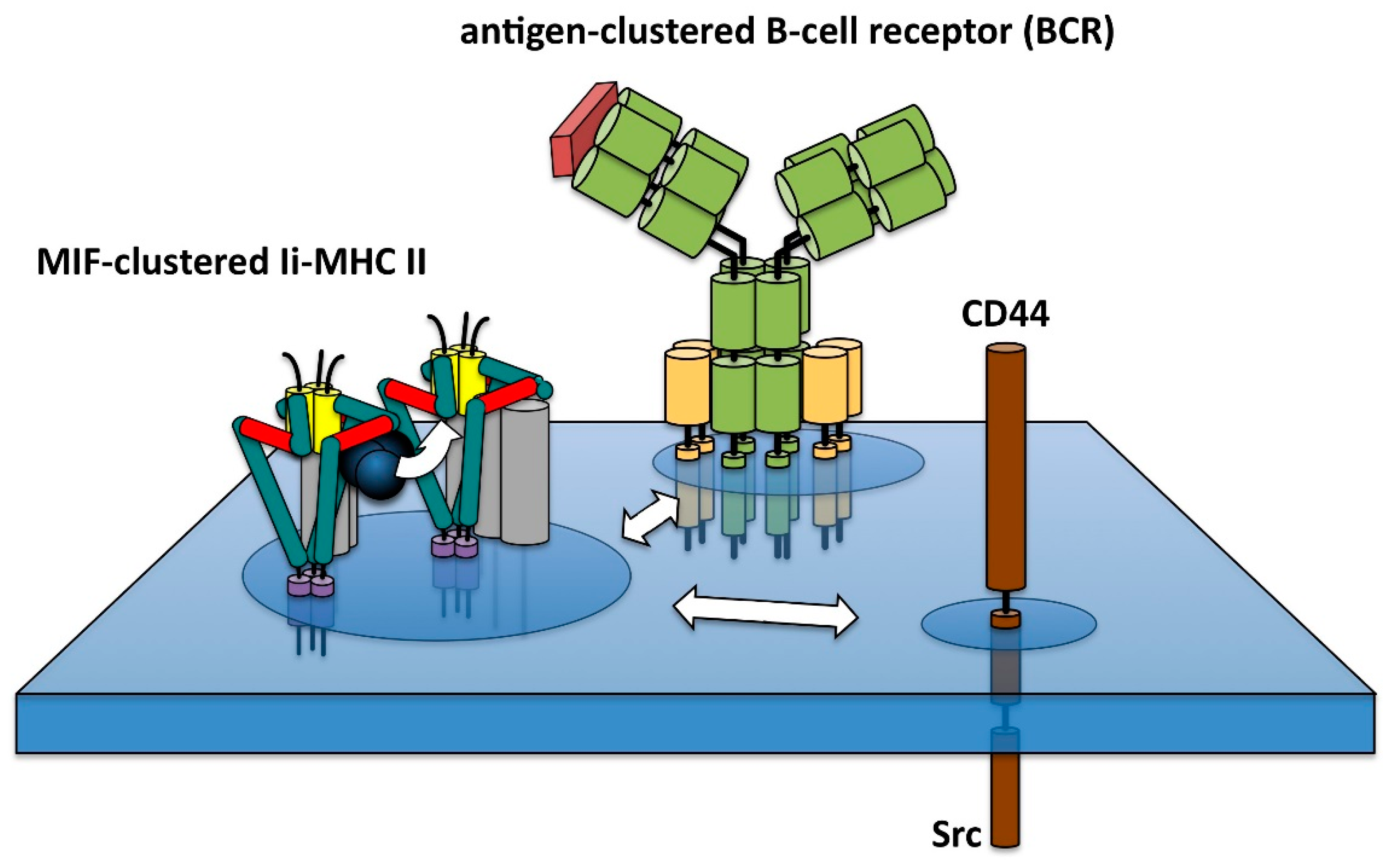
- Hauser, J.T.; Lindner, R. Coalescence of B cell receptor and invariant chain MHC II in a raft-like membrane domain. J. Leukoc. Biol. 2014, 96, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Dadaglio, G.; Nelson, C.A.; Deck, M.B.; Petzold, S.J.; Unanue, E.R. Characterization and quantitation of peptide-MHC complexes produced from hen egg lysozyme using a monoclonal antibody. Immunity 1997, 6, 727–738. [Google Scholar] [CrossRef]
- Binsky, I.; Haran, M.; Starlets, D.; Gore, Y.; Lantner, F.; Harpaz, N.; Leng, L.; Goldenberg, D.M.; Shvidel, L.; Berrebi, A.; et al. IL-8 secreted in a macrophage migration-inhibitory factor- and CD74-dependent manner regulates B cell chronic lymphocytic leukemia survival. Proc. Natl. Acad. Sci. USA 2007, 104, 13408–13413. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Siegler, K.L.; Leifheit, E.C.; Vera, P.L. Inhibition of macrophage migration inhibitory factor decreases proliferation and cytokine expression in bladder cancer cells. BMC Cancer 2004, 4, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taher, T.E.; Smit, L.; Griffioen, A.W.; Schilder-Tol, E.J.; Borst, J.; Pals, S.T. Signaling through CD44 is mediated by tyrosine kinases. Association with p56lck in T lymphocytes. J. Biol. Chem. 1996, 271, 2863–2867. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, V.; Krüttgen, A.; Weis, J.; Weber, C.; Ostendorf, T.; Lue, H.; Bernhagen, J. Role for CD74 and CXCR4 in clathrin-dependent endocytosis of the cytokine MIF. Eur. J. Cell Biol. 2012, 91, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, D.; Gröning, S.; Schmitz, C.; Zierow, S.; Drucker, N.; Bakou, M.; Kohl, K.; Mertens, A.; Lue, H.; Weber, C.; et al. Macrophage Migration Inhibitory Factor-CXCR4 Receptor Interactions: Evidence for partial allosteric agonism in comparison with Cxcl12 chemokine. J. Biol. Chem. 2016, 291, 15881–15895. [Google Scholar] [CrossRef] [PubMed]
- Matza, D.; Lantner, F.; Bogoch, Y.; Flaishon, L.; Hershkoviz, R.; Shachar, I. Invariant chain induces B cell maturation in a process that is independent of its chaperonic activity. Proc. Natl. Acad. Sci. USA 2002, 99, 3018–3023. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, N.; Madsen, L.; Fugger, L.; Benoist, C.; Mathis, D. Toxic MHC class II beta chains. Immunity 1999, 11, 515–516. [Google Scholar] [CrossRef]
- Maehr, R.; Kraus, M.; Ploegh, H.L. Mice deficient in invariant-chain and MHC class II exhibit a normal mature B2 cell compartment. Eur. J. Immunol. 2004, 34, 2230–2236. [Google Scholar] [CrossRef] [PubMed]
- Lipp, J.; Dobberstein, B. The membrane-spanning segment of invariant chain (I gamma) contains a potentially cleavable signal sequence. Cell 1986, 46, 1103–1112. [Google Scholar] [CrossRef]
- Becker-Herman, S.; Arie, G.; Medvedovsky, H.; Kerem, A.; Shachar, I. CD74 is a member of the regulated intramembrane proteolysis-processed protein family. Mol. Biol. Cell 2005, 16, 5061–5069. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, H.; Yabas, M.; Short, A.; Miosge, L.; Barthel, N.; Teh, C.E.; Roots, C.M.; Bull, K.R.; Jeelall, Y.; Horikawa, K.; et al. B cell survival, surface BCR and BAFFR expression, CD74 metabolism, and CD8- dendritic cells require the intramembrane endopeptidase SPPL2A. J. Exp. Med. 2013, 210, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Beisner, D.R.; Langerak, P.; Parker, A.E.; Dahlberg, C.; Otero, F.J.; Sutton, S.E.; Poirot, L.; Barnes, W.; Young, M.A.; Niessen, S.; et al. The intramembrane protease Sppl2a is required for B cell and DC development and survival via cleavage of the invariant chain. J. Exp. Med. 2013, 210, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Schneppenheim, J.; Dressel, R.; Hüttl, S.; Lüllmann-Rauch, R.; Engelke, M.; Dittmann, K.; Wienands, J.; Eskelinen, E.-L.; Hermans-Borgmeyer, I.; Fluhrer, R.; et al. The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J. Exp. Med. 2013, 210, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Schneppenheim, J.; Hüttl, S.; Kruchen, A.; Fluhrer, R.; Müller, I.; Saftig, P.; Schneppenheim, R.; Martin, C.L.; Schröder, B. Signal-peptide-peptidase-like 2a is required for CD74 intramembrane proteolysis in human B cells. Biochem. Biophys. Res. Commun. 2014, 451, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Hüttl, S.; Kläsener, K.; Schweizer, M.; Schneppenheim, J.; Oberg, H.-H.; Kabelitz, D.; Reth, M.; Saftig, P.; Schröder, B. Processing of CD74 by the Intramembrane Protease SPPL2a Is Critical for B Cell Receptor Signaling in Transitional B Cells. J. Immunol. 2015, 195, 1548–1563. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Lantner, F.; Starlets, D.; Gore, Y.; Flaishon, L.; Yamit-Hezi, A.; Dikstein, R.; Leng, L.; Bucala, R.; Machluf, Y.; Oren, M.; et al. CD74 induces TAp63 expression leading to B-cell survival. Blood 2007, 110, 4303–4311. [Google Scholar] [CrossRef] [PubMed]
- Mentrup, T.; Häsler, R.; Fluhrer, R.; Saftig, P.; Schröder, B. A Cell-based assay reveals nuclear translocation of intracellular domains released by SPPL proteases. Traffic 2015, 16, 871–892. [Google Scholar] [CrossRef] [PubMed]
- Gil-Yarom, N.; Radomir, L.; Sever, L.; Kramer, M.P.; Lewinsky, H.; Bornstein, C.; Blecher-Gonen, R.; Barnett-Itzhaki, Z.; Mirkin, V.; Friedlander, G.; et al. CD74 is a novel transcription regulator. Proc. Natl. Acad. Sci. USA 2016, 114, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Abe, M.; Asano, J.; Hara, T.; Kitazoe, K.; Sekimoto, E.; Tanaka, Y.; Shibata, H.; Hashimoto, T.; Ozaki, S.; et al. Myeloma cells suppress bone formation by secreting a soluble Wnt inhibitor, sFRP-2. Blood 2005, 106, 3160–3165. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16, S17–S27. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.A.; Haas, C.S.; Zhu, K.; Mansfield, P.J.; Kim, M.J.; Lackowski, N.P.; Koch, A.E. Migration inhibitory factor up-regulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 via Src, PI3 kinase, and NFκB. Blood 2006, 107, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.A.; Metz, C.N.; Peng, T.; Bucala, R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J. Biol. Chem. 1999, 274, 18100–18106. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, R.; Hausser, A.; Geiger, G.; Mischke, R.; Burger-Kentischer, A.; Flieger, O.; Johannes, F.J.; Roger, T.; Calandra, T.; Kapurniotu, A.; et al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature 2000, 408, 211–216. [Google Scholar] [PubMed]
- Swant, J.D.; Rendon, B.E.; Symons, M.; Mitchell, R.A. Rho GTPase-dependent signaling is required for macrophage migration inhibitory factor-mediated expression of cyclin D1. J. Biol. Chem. 2005, 280, 23066–23072. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Qiao, X.; Wu, Y.; Tang, J. β-Arrestin1 mediates the endocytosis and functions of macrophage migration inhibitory factor. PLoS ONE 2011, 6, e16428. [Google Scholar] [CrossRef] [PubMed]
- Eichel, K.; Jullié, D.; von Zastrow, M. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol. 2016, 18, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Karacsonyi, C.; Knorr, R.; Fülbier, A.; Lindner, R. Association of major histocompatibility complex II with cholesterol- and sphingolipid-rich membranes precedes peptide loading. J. Biol. Chem. 2004, 279, 34818–34826. [Google Scholar] [CrossRef] [PubMed]
- Poloso, N.J.; Muntasell, A.; Roche, P.A. MHC class II molecules traffic into lipid rafts during intracellular transport. J. Immunol. 2004, 173, 4539–4546. [Google Scholar] [CrossRef] [PubMed]
- Karacsonyi, C.; Bedke, T.; Hinrichsen, N.; Schwinzer, R.; Lindner, R. MHC II molecules and invariant chain reside in membranes distinct from conventional lipid rafts. J. Leukoc. Biol. 2005, 78, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts defined: A report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [PubMed]
- Lindner, R.; Naim, H.Y. Domains in biological membranes. Exp. Cell Res. 2009, 315, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Tolar, P.; Sohn, H.W.; Pierce, S.K. Viewing the antigen-induced initiation of B-cell activation in living cells. Immunol. Rev. 2008, 221, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Harwood, N.E.; Batista, F.D. Early events in B cell activation. Annu. Rev. Immunol. 2010, 28, 185–210. [Google Scholar] [CrossRef] [PubMed]
- Batista, F.D.; Iber, D.; Neuberger, M.S. B cells acquire antigen from target cells after synapse formation. Nature 2001, 411, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Tolar, P.; Hanna, J.; Krueger, P.D.; Pierce, S.K. The constant region of the membrane immunoglobulin mediates B cell-receptor clustering and signaling in response to membrane antigens. Immunity 2009, 30, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, Y.R.; Batista, F.D. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity 2007, 27, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Egen, J.G.; Huang, A.Y.; Germain, R.N. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science 2006, 312, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Qin, D.; Burton, G.F.; Szakal, A.K.; Tew, J.G. Follicular dendritic cell-derived antigen and accessory activity in initiation of memory IgG responses in vitro. J. Immunol. 1996, 157, 3404–3411. [Google Scholar] [PubMed]
- Tolar, P.; Sohn, H.W.; Pierce, S.K. The initiation of antigen-induced B cell antigen receptor signaling viewed in living cells by fluorescence resonance energy transfer. Nat. Immunol. 2005, 6, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Donatello, S.; Babina, I.S.; Hazelwood, L.D.; Hill, A.D.K.; Nabi, I.R.; Hopkins, A.M. Lipid raft association restricts CD44-ezrin interaction and promotion of breast cancer cell migration. Am. J. Pathol. 2012, 181, 2172–2187. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Xia, L.; Ling, P.; Waxman, S.; Jing, Y. CD44 ligation with A3D8 antibody induces apoptosis in acute myeloid leukemia cells through binding to CD44s and clustering lipid rafts. Cancer Biol. Ther. 2012, 13, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, H.; Yoshii, H.; Tanaka, Y.; Sato, H.; Yamamoto, N.; Kubo, Y. Raft localization of CXCR4 is primarily required for X4-tropic human immunodeficiency virus type 1 infection. Virology 2009, 386, 23–31. [Google Scholar] [CrossRef] [PubMed]
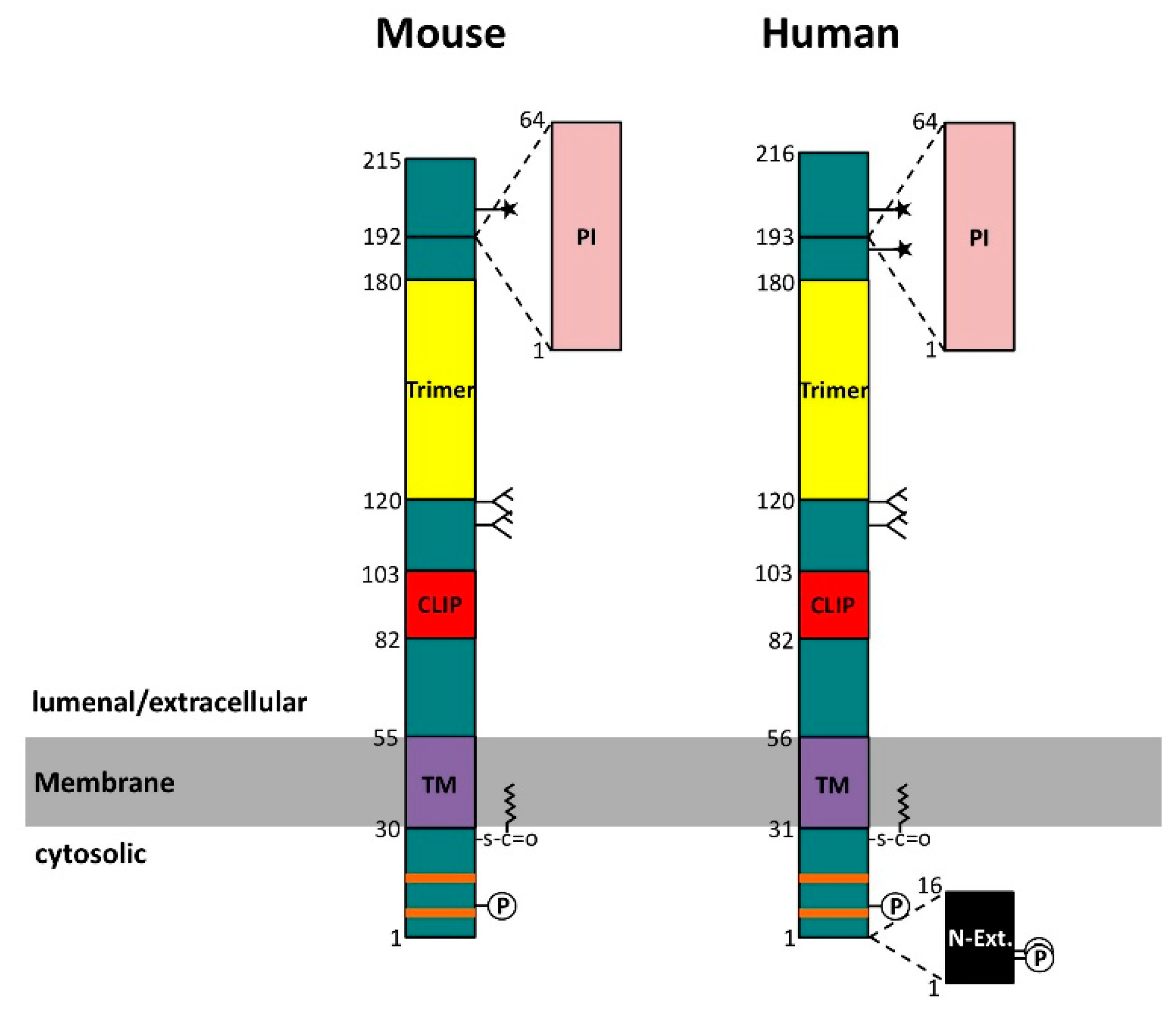
 : palmitoylation site; TM: transmembrane region; CLIP: class II-associated invariant chain peptide, i.e., the primary major histocompatibility complex class II (MHC II) binding segment; Trimer: trimerization domain; PI: thyroglobulin type 1 protease inhibitor domain; N-Ext.: N-terminal extension, due to alternative translation initiation;
: palmitoylation site; TM: transmembrane region; CLIP: class II-associated invariant chain peptide, i.e., the primary major histocompatibility complex class II (MHC II) binding segment; Trimer: trimerization domain; PI: thyroglobulin type 1 protease inhibitor domain; N-Ext.: N-terminal extension, due to alternative translation initiation;  : N-glycosylation site;
: N-glycosylation site;  : O-glycosylation site.
: palmitoylation site; TM: transmembrane region; CLIP: class II-associated invariant chain peptide, i.e., the primary major histocompatibility complex class II (MHC II) binding segment; Trimer: trimerization domain; PI: thyroglobulin type 1 protease inhibitor domain; N-Ext.: N-terminal extension, due to alternative translation initiation; : N-glycosylation site; : O-glycosylation site.
: O-glycosylation site.
: palmitoylation site; TM: transmembrane region; CLIP: class II-associated invariant chain peptide, i.e., the primary major histocompatibility complex class II (MHC II) binding segment; Trimer: trimerization domain; PI: thyroglobulin type 1 protease inhibitor domain; N-Ext.: N-terminal extension, due to alternative translation initiation; : N-glycosylation site; : O-glycosylation site.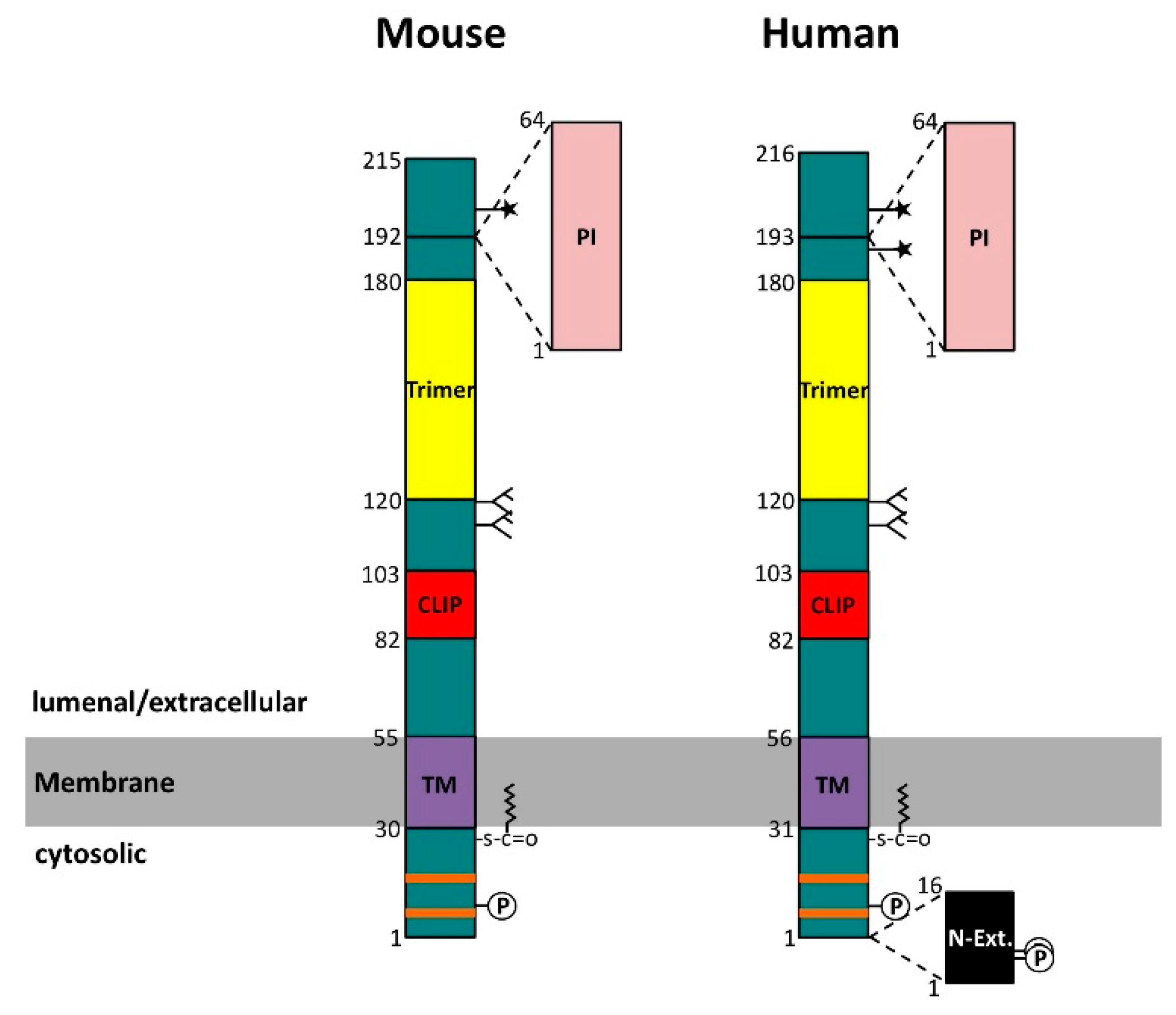

© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindner, R. Invariant Chain Complexes and Clusters as Platforms for MIF Signaling. Cells 2017, 6, 6. https://doi.org/10.3390/cells6010006
Lindner R. Invariant Chain Complexes and Clusters as Platforms for MIF Signaling. Cells. 2017; 6(1):6. https://doi.org/10.3390/cells6010006
Chicago/Turabian StyleLindner, Robert. 2017. "Invariant Chain Complexes and Clusters as Platforms for MIF Signaling" Cells 6, no. 1: 6. https://doi.org/10.3390/cells6010006
APA StyleLindner, R. (2017). Invariant Chain Complexes and Clusters as Platforms for MIF Signaling. Cells, 6(1), 6. https://doi.org/10.3390/cells6010006