Abstract
In the decade since Yamanaka and colleagues described methods to reprogram somatic cells into a pluripotent state, human induced pluripotent stem cells (hiPSCs) have demonstrated tremendous promise in numerous disease modeling, drug discovery, and regenerative medicine applications. More recently, the development and refinement of advanced gene transduction and editing technologies have further accelerated the potential of hiPSCs. In this review, we discuss the various gene editing technologies that are being implemented with hiPSCs. Specifically, we describe the emergence of technologies including zinc-finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 that can be used to edit the genome at precise locations, and discuss the strengths and weaknesses of each of these technologies. In addition, we present the current applications of these technologies in elucidating the mechanisms of human development and disease, developing novel and effective therapeutic molecules, and engineering cell-based therapies. Finally, we discuss the emerging technological advances in targeted gene editing methods.
1. Introduction
The development of methods for the reprogramming of terminally differentiated somatic cells (e.g., skin fibroblasts, peripheral blood mononuclear cells) to a pluripotent state has provided new opportunities to study complex diseases in a simplified and accessible system as well as generate mature cell populations for regenerative medicine-based therapies. Since the initial reports of Yamanaka and colleagues generating induced pluripotent stem cells (iPSCs) from mouse [1] and subsequently human [2] cells by the ectopic expression of four transcription factors—KLF4, OCT4, SOX2, and cMYC—several improvements to iPSC generation methods have been reported including (i) generation of hiPSCs in the absence of the oncogene cMYC [3], (ii) development of ‘foot-print free’ episomal- [4], mRNA- [5], protein- [6], and chemical-based [7] reprograming methods that do not require genomic integration of the reprogramming factors, (iii) improvements in reprogramming efficiency through the use of small molecules [8,9] and microRNAs [10], and (iv) derivation and culture of hiPSC in completely defined, xeno-free conditions [11]. These technological advances have allowed for the development of hiPSC-based models of numerous neurodegenerative [12,13,14,15,16], psychiatric [17], cardiovascular [18,19], renal [20], pulmonary [21,22], hepatic [23], metabolic [24,25], and mitochondrial [26,27] diseases and disorders. In addition, progress in cellular reprogramming methods have allowed for further developments in therapies aimed to repair or replace diseased and dysfunctional cells in vivo [28]. In fact, in September 2014, Takahashi and colleagues began the first hiPSC-based clinical trial to treat wet age-related macular degeneration [29,30]. However, enhancing the research and clinical potential of hiPSCs will require the ability to modify (i.e., deletion, mutation, and insertion) their genome at precise locations. In this review, we discuss the current state of genome editing technologies in hiPSCs, with an emphasis on zinc-finger nuclease- (ZFN), transcription activator-like effector nuclease- (TALEN), and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-based methods.
2. Gene Targeting by Homologous Recombination (HR)
Homologous recombination (HR) has been extensively used to introduce foreign genetic material as a means to generate ‘knock-in’ or ‘knock-out’ animals (namely mice) to study gene function and interrogate the mechanisms of human disease [31,32,33]. Methods for HR take advantage of the endogenous DNA repair mechanisms of a cell to alter or replace a specific genomic locus with engineered transduced homologous genetic sequence. Broadly speaking, such targeting events can result in the deletion, addition, or mutation of one or a long sequence of nucleotides. Traditional methods for HR-based gene targeting in pluripotent stem cells utilize electroporation, nucleofection, or chemical-based transfection methods to introduce linearized plasmid DNA constructs with homology arms of ~5 kb flanking positive selection cassettes (e.g., Blasticidin, Geneticin, Hygromycin B, Puromycin, Zeocin), resulting in disruption or deletion of one or more exons of the targeted gene. While isogenic DNA constructs were demonstrated to be a key factor for efficient targeting in mouse embryonic stem cells [34], several studies have demonstrated that HR in human cells is efficient with non-isogenic DNA [35,36,37]. However, the comparatively low frequency of homologous recombination compared to random insertion necessitates the need for the addition of a negative selection marker. Typically, the Herpes simplex virus thymidine kinase (HSV-tk) is inserted distally to one of the homology arms to increase the selection of clones with targeted gene insertions. With such a positive–negative selection system, randomly targeted cells will retain HSV-tk (and therefore be sensitive to the presence of Gancyclovir), while cells that are Gancyclovir-resistant will not have integrated the HSV-tk gene, potentially representing a HR event when occurring in the presence of the positive selection drug. In comparison to mouse pluripotent stem cells (mPSCs), such conventional methods for gene targeting applied in hPSCs are extremely inefficient, with targeting efficiencies varying between <0.1% and 5% [36,38,39,40,41]. Several reasons for the limited efficiency of HR in hPSCs compared to mPSCs have been postulated, including a poor rate of single-cell survival [42,43] as well as differences in DNA repair processes [44,45] and developmental stage [46,47].
3. Endogenous Repair of Double-Stranded DNA Breaks (DSB)
Within a cell, DNA damage can occur due to endogenous replication errors or exogenous factors such as exposure to ionizing radiation, oxygen free radicals, or mechanical stress [48,49]. Exposure to such deleterious events is common and arises during normal cellular processes. Importantly, healthy cells are equipped with endogenous repair mechanisms to detect DNA damage and recruit machinery to the repair site [50]. In particular, following a double-stranded break (DSB), the cell can activate repair mechanisms through non-homologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ is a process by which the cell can repair nucleotide mismatches or stand breaks without the need for a homologous template. Often NHEJ results in insertion or deletion of nucleotides (termed indels), resulting in disruption (e.g., frameshift mutations, premature stop codons, deletion) of the affected gene (Figure 1). By contrast, in HDR, a sequence of DNA that is partially homologous (typically the undamaged homologous chromosome) serves as a template for repair (Figure 1). Alternatively, if a DSB occurs in the presence of an exogenously introduced homologous repair template, the addition of specific DNA sequences can occur [51] (Figure 1).
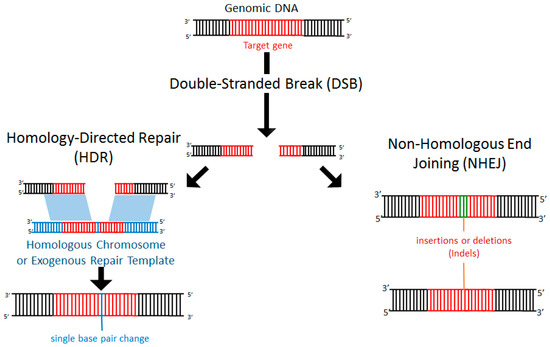
Figure 1.
Double-stranded breaks (DSBs) induce endogenous DNA repair mechanisms. DSBs can be repaired by non-homologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ often leads to deleterious insertions or deletions (indels), while HDR leads to high-fidelity DNA repair using the homologous chromosome or exogenously introduced DNA as a template.
Because it has been well established that DSB significantly enhance HR in human cells [52,53], significant effort has been focused on engineering nucleases that can induce DSB at targeted genomic locations. Two pioneering studies by Rouet and colleagues in the 1990s demonstrated that DSBs created by site-specific nucleases (SSNs) can be successfully repaired by NHEJ or through the endogenous homology-mediated mechanism using an exogenously provided repair template [54,55]. Later the same group showed that in mESCs a rare cutting endonuclease could induce site-specific DSBs, and such sites exhibited ‘two-sided’ homologous recombination, the first demonstration that targeted DSBs in pluripotent cells are recombinogenic [56]. Overall, these studies have served as a foundation for the development of designer nucleases, which will be discussed in the next section.
4. The Age of Designer Nucleases: ZFNs, TALENs, and CRISPR/Cas9
Over the past 15 years the three prominent genome editing tools that have emerged are zinc finger nucleases (ZFNs), transcription activator-like effector nuclease (TALEN), and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9. These technologies are similar in that they consist of two domains—a catalytic domain to initiate DSBs and a programmable domain which recognizes specific DNA sequences. In this section, we will discuss the basic principles of each of these technologies as well as their advantages and limitations (Figure 2).
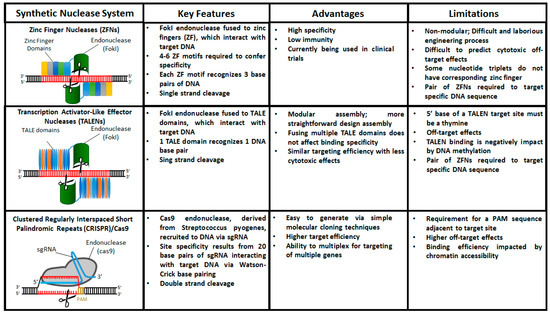
Figure 2.
Comparison of engineered nucleases used for targeted gene editing in hiPSCs.
4.1. Zinc Finger Nucleases (ZFNs)
Zinc finger nucleases (ZFNs) consist of a customized DNA binding domain and a DNA cleavage domain [57]. The DNA-binding domain contains a sequential fusion of multiple zinc finger domains, which bind to specific DNA target sequences [57]. Each zinc finger domain binds a three base pair sequence of DNA. As such, the DNA binding domains of ZFNs typically consist of an array of six linked zinc finger domains that confer specificity to a unique target sequence [58]. The DNA cleavage domain employs the type IIs restriction endonuclease FokI. Because the double-stranded cleavage activity of FokI requires dimerization, a pair of ZFNs that bind on opposite sides of the target site is used [59].
To date, there are many reports of ZFN-based genome editing of a variety of organisms, including human cells [57]. The first demonstration of a ZFN-targeted gene correction in human somatic cells was performed in 2003 by Proteus and Baltimore, where the authors used known ZFN recognition sites in a mutated GFP cassette to demonstrate successful gene correction and GFP expression [60]. More recently, the clinical application of ZFNs has rapidly advanced, including a Phase I clinical trial using ZFN-modified autologous CD4 T cells with a mutated CCR5 gene to treat HIV infection [61,62,63].
Despite the advances of ZFN-based methods in basic and translational research, ZFN technology has several limitations. First, because zinc finger domains have a higher affinity for G-rich sequences, there are several three-base nucleotide sequences that do not have associated zinc finger binding domains, which limits the robustness and specificity of ZFN-mediated cleavage [64]. Second, off-target cleavage by ZFNs can alter the function of unknown genes, introduce oncogenic translocations, and result in elevated cytotoxicity [65]. Finally, the generation of functional ZFNs requires a significant amount of expertise in protein engineering [66,67].
4.2. Transcription Activator-Like Effector Nucleases (TALENs)
Similar to ZFNs, transcription activator-like effector nucleases (TALENs) are a fusion protein consisting of nonspecific FokI endonuclease and an adaptable TALE DNA-binding domain. The TALE domains, derived from plant pathogenic bacteria [68], consist of a highly conserved sequence of 33–35 amino acid repeats with variable amino acids found at the 12th and 13th position [69,70]. The amino acids located at these positions, referred to as the repeat variable diresidues (RVD), confer sequence specificity to the TALENs. Specifically, there are four RVDs that contain the amino acid sequences Asn/Ile (NI), Asn/Asn (NN), Asn/Gly (NG), and His/Asp (HD), which recognize the nucleotides adenine, guanine, thymine, and cytosine, respectively [69,70]. Because of this one RVD to one base pair ratio, modular assembly methods can be used to engineer a series of TALE repeats that bind with high affinity to a single genomic locus [71,72]. Finally, similar to ZFNs, a pair of TALENs is required to target a specific stretch of DNA.
Overall, compared to ZFNs, TALENs have several advantages. Unlike ZFNs, in which the linking of multiple zinc finger domains can reduce DNA-binding specificity [73], fusing multiple TALE domains does not affect binding specificity [71,72,74]. Moreover, TALENs do not require the complex assembly processes that are required of ZFN construction [75]. In fact, high-throughput assembly methods [76] have allowed for the generation of a library of TALENs that span over 18,000 protein-coding genes in the human genome [77]. Additionally, compared to ZFNs, TALENs demonstrate similar targeting efficiency with less cytotoxic effects [78].
Even though TALEN-based methods have been used extensively to target numerous endogenous loci in human cells [72], there are still several shortcomings. Similar to ZFNs, deleterious undesired off-targets remain a concern with the use of TALENs [79]. Although TALENs do not have the same target-site restrictions that ZFNs have, the 5’ base of a TALEN target site must be a thymine [80], which may restrict applications where the targeting of specific single nucleotide is required. In addition, there are several reports that TALEN binding is negatively affected by DNA methylation [81], which may further impact successful targeting near gene regulatory regions, such as promoters, that are known to be highly methylated [82,83].
4.3. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9
Clustered regulatory interspaced short palindromic repeats (CRISPR) are adapted from the RNA-based immune system of prokaryotes, which confer resistance to bacteriophages through the CRISPR-associated protein (Cas) endonuclease-based destruction of foreign DNA [84,85]. As it relates to the in vitro targeting of genomic sequences in eukaryotic cells, the most common CRISPR/Cas system utilized is the Type II system from Streptococcus pyogenes [86,87]. The Type II CRISPR system consists of CRISPR associated protein 9 (Cas9) endonuclease and two short non-coding RNAs—a CRISPR (crRNA), which contains a seed sequence complimentary to the target DNA sequence (termed protospacer), and a transactivating crRNA (tracrRNA), which hybridizes with the crRNA to facilitate the endonuclease activity of the CRISPR/Cas9 complex [88]. When all three components are delivered to a target cell, a three base pair NGG protospacer-associated motif (PAM) on the target DNA strand guides Cas9 endonuclease to cleave three base pairs upstream of the PAM sequence [88,89,90]. The CRISPR/Cas9 system is highly adaptable as targeting Cas9 to a specific genomic locus can be achieved via cloning a 20 nucleotide sequence that is complementary to the protospacer into the crRNA construct. Moreover, to further facilitate the implementation of CRISPR/Cas9-based genome editing, more recent versions utilize a chimeric single-guide RNA (sgRNA), which consists of fused cRNA and tracrRNA [91].
Besides its simplicity and ease of use, the CRISPR/Cas9 system has several advantages over ZFN- and TALEN-based methods. Compared to TALENs, CRISPR/Cas9 exhibits lower cytotoxicity and higher targeting efficiencies [92]. Additionally, because of the small size and versatility of the sgRNA, several studies have demonstrated the simultaneous delivery of multiple sgRNAs to enable targeting of multiple genes [86,93]. Such multiplexed genome engineering strategies will greatly accelerate the study of the complex gene interactions that are the basis of multiple developmental programs and diseases.
Even though the CRISPR/Cas9 system is highly adaptable and easy to engineer, there are several concerns that may hinder its future applications. The requirement for a PAM sequence, which occurs roughly once every eight base pairs, may prevent the targeting of specific base pairs [94]. Another major impediment to the use of CRISPR/Cas9-based technologies for functional studies, as well as translational application, is the high frequency of off-target cleavage events [95,96,97]. An especially alarming study in human cells reported that off-target sites contained up to five mismatches and many off-target sites were mutated at frequencies comparable to or higher than target sites [96].
In order to overcome some of these limitations, especially with respect to off-target mutations and indel formation, several Cas9 variants have been engineered (Table 1). For example, several reports describe the generation of a Cas9 nickase (Cas9n), which generates a single-strand DNA break (SSB) rather than the DSB typical of wild-type Cas9 (spCas9) [86,98]. Compared to DSBs, the nicked DNA is predominately repaired through the high-fidelity HDR mechanism, using the uncut complementary strand as the template [99]. The use of Cas9n has been shown to reduce off-target effects while retaining the targeting efficiency of spCas9 [86,98]. Moreover, double-stranded nicking facilitated by dual sgRNA Cas9n pairs (Cas9dn) showed increased target specificity with a 50- to 1500-fold reduction of off-target indel formation [100]. Along similar lines, the use of fused catalytically inactive Cas9 (dCas9) and FokI nuclease (fCas9), which can induce DSBs through FokI dimerization, can modify target DNA sites with a higher specificity and similar efficiency to that of Cas9dn [101]. More recently, Howden and colleagues reported the fusion of spCas9 to a peptide derivative of the human Geminin protein (spCas9-Gem) [102], facilitating the degradation of Cas9 in the phase of cell cycle during which error-prone NHEJ primary occurs [103]. This programmable editing system was shown to significantly decrease NHEJ-induced indels at the target locus [102]. Finally, Komor and colleagues implemented an engineered Cas9n fused to a cytidine deaminase and uracil DNA glycosylase inhibitor programmed with a sgRNA to facilitate the direct base pair conversion of a cytosine to thymine or, guanine to adenine, within a five base pair window in the protospacer [104]. The use of this base editing (BE) technology was reported to facilitate the direct base conversion with 15%–75% efficiency in human cells without creating a DSB. As such, direct base conversion eliminates the possibility of NHEJ events and has numerous implications in disease modeling applications that target point mutations.

Table 1.
Summary of engineered Cas9 variants for gene editing applications.
In addition to the common Cas9 endonuclease, Cpf1 is an RNA guided endonuclease derived from a class II (or type V) CRISPR system that has shown promise for gene editing applications in human cells [105]. Cpf1 differs from Cas9 in a few key ways including the requirement of only a single guide RNA without the need for a tracrRNA, single endonuclease domain, thymidine-rich PAM recognition sequences (e.g., 5’-TTTN-3’), and production of a 5-nt staggered DSB distal to the PAM sequence (in contrast with Cas9 which forms blunt end breaks) [106]. A recent analysis of Lachnospiraceae bacterium Cpf1 (LbCpf1) and Acidaminococcus sp. Cpf1 (AsCpf1) compared targeted editing efficiencies to spCas9 at ten chromosomal target sites that contain PAM sequences for both spCas9 and each Cpf1 variant. The study demonstrated that while spCas9 has the highest mutation rate (32% ± 4%), the LbCpf1 and AsCpf1 variants showed comparable mutation rates (19% ± 6% and 20% ± 5%, respectively) [105]. Although CRISPR-Cas Cpf1 technology is in its infancy, there are numerous advantages that may simplify multiplex gene editing. Multiplex gene editing using Cas9 nucleases require large constructs or multiple plasmid delivery. In contrast, Cpf1 alone is required for crRNA maturation where a single RNA pol III promoter can be used to drive expression of multiple crRNAs, allowing for targeting of up to four genes simultaneously in human cells [107].
4.4. Practical Considerations for Synthetic Nuclease Mediated HR
The use of synthetic nucleases in applications (see Section 5) that implement HDR requires the co-delivery of homology templates. Conventional approaches use a linearized or plasmid donor vector that contains ~10 kb (the upper limit of typical targeting vectors) of homologous DNA and an antibiotic resistance cassette. Still, such approaches are encumbered by low targeting efficiencies, even in the presence of a synthetic nuclease-induced DSB [108]. Because it has been reported that increasing the homology arms significantly increases targeting frequencies [109,110], bacteria artificial chromosomes (BACs) have been used as a method to create targeting vectors with homology arms in excess of 100 kb [111]. However, BAC-based methods require the use of recombineering technologies that may not be accessible to all researchers [112]. Alternatively, the delivery of homology templates via adeno-associated viruses (AAV) has been shown to facilitate improved gene targeting [113,114,115,116]. The mechanisms to explain this high targeting efficiency are not well understood, but it has been proposed that AAV is a single-stranded DNA virus and, upon infection and entry into the cell, this single-stranded piece mimics DNA damage and provides an ideal substrate for the endogenous DNA repair machinery, thereby significantly increasing gene-targeting efficiencies [117].
One primary concern with plasmid-, BAC-, and AAV-based methods for delivery of homology templates is that the identification of properly targeted cells requires the use of selection cassettes, which may affect neighboring gene expression and regulation [118,119]. To overcome this limitation, several groups have employed the Cre/loxP system to remove the selection transgenes after genome targeting [118,120]. With this system, selection cassettes flanked by two loxP sites are removed after Cre-mediated recombination [121]. Finally, the use of single-stranded oligonucleotides (ssODN), which are easily synthesized, offers a viable alternative for delivery of ‘scarless’ homology templates [122,123,124]. These ssODNs contain 60bp homology arms at a minimum and have been reported to more efficiently mediate SNP conversions via HDR [125,126,127,128] However, current ssODN-mediated methods still suffer from low efficiency (<1%), requiring high-throughput screening methods to identify targeted clones [122].
5. Applications of Gene Editing Technologies with hiPSCs
The various gene editing technologies discussed here have several applications as it relates to the research and clinical applications of hiPSCs. First, these methods can be used to engineer hiPSC lines with targeted reporter genes or selection markers. Such lines are useful to study the underlying mechanisms that regulate cell fate decisions, optimize protocols for differentiation into specific cells types, and isolate cell populations to be used in disease modeling and cell replacement therapies. Second, the generation of hiPSCs line with targeted mutations introduced into their genomes can be used to interrogate the onset and progression of various diseases. This is especially important with respect to the generation of isogenic hiPSCs as genetic and epigenetic variances between healthy control and disease lines may confound the interpretation of observed phenotypic differences. Finally, gene targeting strategies have important implications concerning the genetic modification of hiPSCs to be used in cell therapies. Although a discussion of all examples of these applications with hiPSCs is outside the scope of this review, we will discuss key examples in this section.
5.1. Generation of Targeted Reporter Lines
Several methods including chemical-based transfection reagents, electroporation, or viral infection have been successfully employed to randomly integrate reporter elements into the genome of hPSCs [128,129,130]. However, these approaches are hindered by poor characterization of regulatory elements known to control gene expression, positional effects that may lead to transgene silencing, and possible integration into sites that affect cell phenotype. On the other hand, the ability to insert reporter genes into specific genomic loci in hiPSCs offers the opportunity to faithfully reproduce patterns of gene expression with minimal effects on adjacent genes.
Several early studies reported the use of genome editing technologies to insert fluorescent proteins into loci associated with the undifferentiated state of hiPSCs [131,132,133]. For example, ZFNs [132], TALEN [131,134], and CRISPR/Cas9 [131,133] technologies have been used to insert green fluorescent protein (GFP) downstream of the last exon of OCT4. Importantly, not only did such lines faithfully recapitulate OCT4 expression to monitor hiPSC pluripotency, but also they did not impact the ability of targeted cells to differentiate into mature cell types, such as NKX6.1+SOX9+ pancreatic progenitor cells.
Targeted lines have been developed to monitor the differentiation of hiPSCs and isolate various, often rare, therapeutically relevant cell types. Forster et al. used ZFNs to generate an endogenous LGR5-GFP reporter line [135]. Through the FACS-based isolation of rare LGR5-GFP+ cells from differentiating cells, the authors were able to generate intestinal organoids with a composition and organization that mimicked those of in vivo intestinal tissue. ZFN-mediated methods have also been employed to generate GFAP-GFP astrocyte reporter lines from one healthy control and two ALS patient hiPSC lines [136]. Subsequent analysis of GFAP-GFP+ cells demonstrate their functional maturity and ability to engraft into a rat spinal cord, thereby paving the way for future cell-based therapies to treat ALS.
In the same vein, CRISPR/Cas9dn was used to generate a BAC-based homology template as a means to insert a GFP reporter before the stop codon of the PAX7, an early myogenic marker [137]. A similar approach was used to generate a fluorescent reporter line to mark MYF5+ cells, one of the earliest myogenic determination genes in the developing somites [138]. Collectively, these studies hold important implications for the prospective isolation of myogenic cells to be used in the modeling and treatment of muscular dystrophy.
Several groups have also used ZFN [139,140,141] and TALEN [142,143] approaches to introduce fluorescent proteins into safe harbor loci. In one important example, Wang and colleagues used ZFNs to introduce red fluorescent protein (RFP), luciferase, and herpes simplex virus thymidine kinase reporter constructs into the AAVS1 locus [141]. Remarkably, cardiomyocytes and endothelial cells derived from these targeted lines were successfully tracked in vivo using non-invasive bioluminescence imaging over the course of four weeks after being transplanted into mouse hearts. The ability to track the survival, engraftment, and location of hiPSC-derived cell populations in real time, in vivo is critically important for their clinical translation.
5.2. Using Designer Nucleases to Generate hiPSC-Based Disease Models
Broadly speaking two approaches can be used to generate isogenic disease models in hiPSCs with synthetic nuclease—the introduction of disease mutation into healthy control hiPSCs or the correction of the disease-related mutation in patient hiPSCs. In both approaches, applications in which the targeted gene will simply be rendered non-functional via frameshift indel mutations or partial deletions do not require the co-delivery of homology templates whereas applications in which a specific disease-related mutation will be corrected or introduced will also require the introduction of donor template (see Section 4.4). For the sake of simplicity, the examples discussed in this subsection have been divided according to the specific engineered nuclease that was implemented.
5.2.1. ZFN
Two early examples of ZFN-generated hiPSC models relate to the study of sickle cell disease [144,145]. In both examples, the authors elegantly used ZFNs templates to precisely correct mutations in hiPSCs derived from patients with mutated β-globin alleles. ZFNs have also been extensively used in generating hiPSC-based models of neurodegenerative diseases [127,146,147,148]. In particular, Soldner and colleagues used ZFN approaches with both plasmid and ssODN donor templates to introduce Parkinson’s disease (PD) mutations into wild-type cells as well as to correct the same mutations in diseased hiPSCs [127]. In a related study, the phenotypic analysis of midbrain dopaminergic neurons (the neuronal subtype predominantly affected in PD) derived from these isogenic lines revealed altered transcriptional network activity related to mitochondrial dysfunction observed in PD hiPSC-derived neurons [146]. Moreover, the authors used a small-molecule high-throughput screening approach with these isogenic hiPSCs to identify isoxazole as a potential therapeutic compound. In another example, genome-wide expression analysis of motor neurons (the neuronal subtype predominantly affected in amyotrophic lateral sclerosis (ALS)) derived from ZFN-corrected SOD1 mutant hiPSCs revealed a unique, disease-dependent transcriptional signature indicative of increased oxidative stress, reduced mitochondrial function, altered subcellular transport, and activation of the ER stress [148]. Finally, ZFN-mediated gene editing was used to develop two sets of isogenic hiPSC lines—one with a TAU-A152T mutation corrected, and another with the same homozygous mutation introduced. Because tauopathies are characteristic of numerous neurodegenerative disorders including Alzheimer’s disease (AD), frontotemporal dementia (FTD), and progressive supranuclear palsy (PSP) [149,150,151], neurons derived from these hiPSCs will be useful for the study of disease mechanisms and evaluation of therapeutic compounds.
5.2.2. TALENs
Several groups have employed TALEN technologies to generate numerous hiPSC-models of disease including Duchenne muscular dystrophy [152]. Niemann–Pick Type C disease [153], Lesch–Nyhan syndrome [154], X-linked severe combined immunodeficiency [155], and various blood disorders [156,157,158,159]. One of the more well-designed applications of TALEN methods with hiPSC-based models was performed by Woodruff and colleagues to investigate the mechanisms by which mutations in presenilin-1 (PS1) contribute to familial Alzheimer’s disease (FAD) [160]. In this study the authors generated an allelic series of isogenic PS1 ΔE9 mutations, (WT/null, WT/ΔE9, ΔE9/ΔE9, ΔE9/null) in hiPSCs derived from J Craig Venter, a completely sequenced genomic background [161,162,163]. Phenotypic analysis of neurons generated from these isogenic lines revealed for the first time that FAD PS1 mutations do not act as through loss-of-function but rather exhibit gain of toxic activity. In a more recent study with these same cell lines, the authors determined that neurons with PS1 ΔE9 mutations displayed defective transcytosis of lipoproteins [163]. This novel finding may shed new light onto the mechanisms by which FAD mutations lead to neuronal degeneration in patients as neurons require extracellular cholesterol uptake from lipoprotein particles for axon elongation as well as synapse formation and maintenance [164,165,166].
5.2.3. CRISPR/Cas9
Despite being in its infancy CRISPR/Cas9 technology has been widely applied to generate hiPSC-based models of human disease. Similar to the other gene editing technologies discussed here, CRISPR/Cas9 methods have been extensively applied in hiPSC to develop models of immunodeficiency and blood diseases including β-Thalssemia [167,168], hemophilia [169], ICF syndrome [170], sickle cell disease [171], and severe combined immunodeficiency [172]. More recently, CRISPR/Cas9 techniques have been combined with advanced bioengineering culture techniques to create more accurate in vitro disease models [173,174]. For example, Firth et al. used an air–liquid interface culture system to generate complex, multicellular pulmonary epithelial cell cultures from hiPSCs derived from patients with cystic fibrosis (CF) [173]. Remarkably, the correction of CF-related mutations in these cultures via CRISPR/Cas9 resulted in the restoration of functional expression of CFTR protein and chloride channel function. In another example, CRISPR/Cas9 genome editing coupled with engineered 'heart-on-chip' methods, were employed to elucidate the pathological mechanisms related to the cardiomyopathy of Barth syndrome (BTHS) [174]. Specifically, the authors were able to use cardiomyocytes derived from isogenic sets of hiPSCs to directly link the metabolic, structural and functional phenotypes associated with Barth syndrome to mutation of the gene encoding tafazzin. Finally, in a pioneering study by Howden and colleagues, the authors implemented CRISPR/Cas9 systems to simultaneously reprogram and correct the disease-related mutation in fibroblasts from a patient with retinitis pigmentosa [175]. This ability to generate isogenic cell lines quickly, efficiently, and without the need for drug selection will significantly advance the personalized medicine application of hiPSCs.
5.3. Development of Gene Edited hiPSCs for Cell-Based Therapies
The use of genome editing technologies with hiPSCs for regenerative medicine applications is rapidly accelerating. In one early example of such an approach, Yusa and colleagues used a ZFN approach for the bi-allelic targeted correction of a point mutation in the α1-antitrypsin (A1AT) gene that results in an α1-antitrypsin deficiency (A1ATD), the most common inherited liver metabolic disorder [176]. Remarkably, when these corrected hiPSCs were differentiated to hepatocyte-like cells and injected into a mouse model of liver disease, the cells not only incorporated into in vivo liver tissue but displayed functional activities, such as long-term secretion of human albumin. As such, this approach has great potential for the treatment of numerous genetic liver disease, for which the only current therapy is liver transplantation.
Based on the revolutionary studies that demonstrated a patient, the so-called ‘Berlin Patient’, had been successfully cured of HIV infection following allogeneic hematopoietic stem cell (HSCs) transplants from a homozygous CCR5Δ32 donor [177], there has been significant effort in generating similar cell types from hiPSCs [178]. To that end, Ye et al. used both TALEN- and CRISPR/Cas9-based methods to introduce bi-allelic CCR5Δ32 into wild-type hiPSCs [179]. Moreover, monocytes and macrophages differentiated from these mutated hiPSCs were resistant to in vitro HIV-1 infection. Overall, this approach has great promise to provide an unlimited source of HSCs as a means to functionally cure HIV patients.
6. Future Perspectives
As gene editing technologies continue to evolve, there are several emerging trends relating to their use in hiPSC-based efforts in disease modeling, drug discovery, and regenerative medicine. For example, in applications that require the use of HDR (see Section 4.4), the low targeting efficiencies remain problematic. To address this challenge, Yu et al. implemented a high-throughput screening approach to identify small molecules to enhance CRISPR/Cas9 genome editing in pluripotent stem cells [180]. The authors discovered that two small molecules, L755507 and Brefeldin A, could improve CRISPR/Cas9-facilitated HDR efficiency (3-fold for large DNA insertions and 9-fold for point mutations). In addition, the authors identified another compound, azidothymidine, that increased indel mutations mediated by NHEJ through the inhibition of HDR. Along similar lines, several studies have shown that epigenetic modifications, such as DNA and histone modifications, may negatively impact TALEN [81] and CRISPR/Cas9 [181]. The use of epigenetic modifiers such as histone deacetylases and DNA methyltransferases in conjunction with genome editing tools has shown promise to address such issues [81].
Another future application of the genome editing technologies discussed in this review relates to the immunogenicity of hiPSCs. Much of the excitement surrounding the development of hiPSC-based technologies was with regard to the potential of these cells to serve as an unlimited supply of autologous cells for cell-based therapies without the concerns about immune rejection that plagued the use of hESCs. However, there have been several conflicting reports regarding the immune tolerance of hiPSCs and their derivatives, with some reports suggesting that hiPSCs are completely immune privileged and others suggesting that hiPSCs are highly immunogenic [182,183,184,185,186]. One potential approach to address these immunological-related issues that has been previously applied to hESCs [187] is the use of gene editing technologies to introduce specific genes into safe harbor loci that confer immune protection to hiPSCs and their derivatives.
At the pace at which the genome editing field has been advancing over the past few years, it is likely that new technologies with characteristics superior to the methods discussed in this review are likely to emerge. For example, a novel method that employs a DNA-guided endonuclease derived from the Natronobacterium gregoryi Argonaute (NgAgo) has been shown to be advantageous in Cas9 methods as it does not require the presence of a PAM sequence and has fewer off-target effects [188,189]. However, this technology has yet to be applied to hiPSCs and there are concerns about its reproducibility for genome editing in human cells [190,191,192].
Finally, the use of gene-edited hiPSCs in basic science and translational applications will require overcoming additional technological bottlenecks unrelated to gene editing technologies per se. For example, issues related to clonal variation, the heterogeneity and impurity of differentiated cell populations, and scalable production methods [193,194,195,196,197] need to be overcome, which may limit the future application of hiPSCs in disease modeling, drug discovery, and cell-based therapies.
Acknowledgments
This work has been supported by funding from the Arizona Alzheimer’s Consortium and an ASU/Mayo Seed Grant.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Habib, O.; Habib, G.; Choi, H.W.; Hong, K.S.; Do, J.T.; Moon, S.H.; Chung, H.M. An improved method for the derivation of high quality iPSCs in the absence of c-myc. Exp. Cell Res. 2013, 319, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Su, R.J.; Baylink, D.J.; Neises, A.; Kiroyan, J.B.; Meng, X.; Payne, K.J.; Tschudy-Seney, B.; Duan, Y.; Appleby, N.; Kearns-Jonker, M.; et al. Efficient generation of integration-free ips cells from human adult peripheral blood using bcl-xl together with yamanaka factors. PLoS ONE 2013, 8, e64496. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ambasudhan, R.; Yuan, X.; Li, W.; Hilcove, S.; Abujarour, R.; Lin, X.; Hahm, H.S.; Hao, E.; Hayek, A.; et al. A chemical platform for improved induction of human iPSCs. Nat. Methods 2009, 6, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhu, X.; Liao, B.; Benda, C.; Zhuang, Q.; Pei, D.; Qin, B.; Esteban, M.A. Micrornas in somatic cell reprogramming. Curr. Opin. Cell Biol. 2013, 25, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Srivastava, A.; Srivastava, P.; Dhuriya, Y.K.; Pandey, A.; Kumar, D.; Rajpurohit, C.S. Advances in stem cell research- a ray of hope in better diagnosis and prognosis in neurodegenerative diseases. Front. Mol. Biosci. 2016, 3, 72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Jiao, B.; Zhou, M.; Zhou, T.; Shen, L. Modeling Alzheimer's disease with induced pluripotent stem cells: Current challenges and future concerns. Stem Cells Int. 2016, 2016, 7828049. [Google Scholar] [CrossRef] [PubMed]
- Preza, E.; Hardy, J.; Warner, T.; Wray, S. Review: Induced pluripotent stem cell models of frontotemporal dementia. Neuropathol. Appl. Neurobiol. 2016, 42, 497–520. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, S.; He, X.B.; Cheng, C.; Le, W. Induced pluripotent stem cells in Alzheimer's Disease: Applications for disease modeling and cell-replacement therapy. Mol. Neurodegener. 2016, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Mungenast, A.E.; Siegert, S.; Tsai, L.H. Modeling Alzheimer’s disease with human induced pluripotent stem (iPS) cells. Mol. Cell. Neurosci. 2016, 73, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Young-Pearse, T.L.; Morrow, E.M. Modeling developmental neuropsychiatric disorders with iPSC technology: Challenges and opportunities. Curr. Opin. Neurobiol. 2016, 36, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Matsa, E.; Ahrens, J.H.; Wu, J.C. Human induced pluripotent stem cells as a platform for personalized and precision cardiovascular medicine. Physiol. Rev. 2016, 96, 1093–1126. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Yuasa, S.; Node, K.; Fukuda, K. Cardiovascular disease modeling using patient-specific induced pluripotent stem cells. Int. J. Mol. Sci. 2015, 16, 18894–18922. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.S. Modeling kidney disease with ips cells. Biomark. Insights 2015, 10, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Mou, H.; Brazauskas, K.; Rajagopal, J. Personalized medicine for cystic fibrosis: Establishing human model systems. Pediatr. Pulmonol. 2015, 50 (Suppl. S40), S14–S23. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Wang, D. Clinical potentials of human pluripotent stem cells in lung diseases. Clin. Transl. Med. 2014, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Golas, M.M.; Sander, B. Use of human stem cells in huntington disease modeling and translational research. Exp. Neurol. 2016, 278, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Kawser Hossain, M.; Abdal Dayem, A.; Han, J.; Kumar Saha, S.; Yang, G.M.; Choi, H.Y.; Cho, S.G. Recent advances in disease modeling and drug discovery for diabetes mellitus using induced pluripotent stem cells. Int. J. Mol. Sci. 2016, 17, 256. [Google Scholar] [CrossRef] [PubMed]
- Balboa, D.; Otonkoski, T. Human pluripotent stem cell based islet models for diabetes research. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Goto, Y. Concise review: Heteroplasmic mitochondrial DNA mutations and mitochondrial diseases: Toward iPSC-based disease modeling, drug discovery, and regenerative therapeutics. Stem Cells 2016, 34, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Chen, C.T.; Wei, Y.H. Mitochondrial resetting and metabolic reprogramming in induced pluripotent stem cells and mitochondrial disease modeling. Biochim. Biophys. Acta. 2016, 1860, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Kamao, H.; Mandai, M.; Okamoto, S.; Sakai, N.; Suga, A.; Sugita, S.; Kiryu, J.; Takahashi, M. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Rep. 2014, 2, 205–218. [Google Scholar] [CrossRef] [PubMed]
- RIKEN; Japan. A Study of transplantation of autologous induced pluripotent stem cell (iPSC) derived retinal pigment epithelium (RPE) cell sheet in subjects with exudative age related macular degeneration. umin.ac.jp; Japan, 2013–16 December 2016. Available online: https://upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000013279. UMIN ID : UMIN000011929 (accessed on 2 March 2017).
- Hall, B.; Limaye, A.; Kulkarni, A.B. Overview: Generation of gene knockout mice. Curr. Protoc. Cell Biol. 2009, 19, 11–17. [Google Scholar]
- Sakamoto, K.; Gurumurthy, C.B.; Wagner, K.U. Generation of conditional knockout mice. Methods Mol. Biol. 2014, 1194, 21–35. [Google Scholar] [PubMed]
- Tong, C.; Huang, G.; Ashton, C.; Li, P.; Ying, Q.L. Generating gene knockout rats by homologous recombination in embryonic stem cells. Nat. Protoc. 2011, 6, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, H.; Maandag, E.R.; Berns, A. Highly efficient gene targeting in embryonic stem cells through homologous recombination with isogenic DNA constructs. Proc. Natl. Acad. Sci. USA 1992, 89, 5128–5132. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, J.M. Gene targeting in human cells without isogenic DNA. Science 1999, 283, 9. [Google Scholar] [CrossRef]
- Urbach, A.; Schuldiner, M.; Benvenisty, N. Modeling for lesch-nyhan disease by gene targeting in human embryonic stem cells. Stem Cells 2004, 22, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, J.M.; Dutriaux, A. Gene targeting and somatic cell genetics--a rebirth or a coming of age? Trends Genet. 1999, 15, 88–90. [Google Scholar] [CrossRef]
- Ruby, K.M.; Zheng, B. Gene targeting in a hues line of human embryonic stem cells via electroporation. Stem Cells 2009, 27, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Zwaka, T.P.; Thomson, J.A. Homologous recombination in human embryonic stem cells. Nat. Biotechnol. 2003, 21, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, A.I.; Christodoulou, I.; Pells, S.C.; McWhir, J.; Thomson, A.J. Sequential genetic modification of the hprt locus in human escs combining gene targeting and recombinase-mediated cassette exchange. Cloning Stem Cells 2008, 10, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Irion, S.; Luche, H.; Gadue, P.; Fehling, H.J.; Kennedy, M.; Keller, G. Identification and targeting of the rosa26 locus in human embryonic stem cells. Nat. Biotechnol. 2007, 25, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Ueno, M.; Kamiya, D.; Nishiyama, A.; Matsumura, M.; Wataya, T.; Takahashi, J.B.; Nishikawa, S.; Nishikawa, S.; Muguruma, K.; et al. A rock inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007, 25, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Merkert, S.; Martin, U. Site-specific genome engineering in human pluripotent stem cells. Int. J. Mol. Sci. 2016, 17, 1000. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Suzuki, K.; Kim, N.Y.; Liu, G.H.; Izpisua Belmonte, J.C. A cut above the rest: Targeted genome editing technologies in human pluripotent stem cells. J. Biol. Chem. 2014, 289, 4594–4599. [Google Scholar] [CrossRef] [PubMed]
- Banuelos, C.A.; Banath, J.P.; MacPhail, S.H.; Zhao, J.; Eaves, C.A.; O’Connor, M.D.; Lansdorp, P.M.; Olive, P.L. Mouse but not human embryonic stem cells are deficient in rejoining of ionizing radiation-induced DNA double-strand breaks. DNA Repair (Amst.) 2008, 7, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- De Los Angeles, A.; Loh, Y.H.; Tesar, P.J.; Daley, G.Q. Accessing naive human pluripotency. Curr. Opin. Genet. Dev. 2012, 22, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Gafni, O.; Weinberger, L.; Mansour, A.A.; Manor, Y.S.; Chomsky, E.; Ben-Yosef, D.; Kalma, Y.; Viukov, S.; Maza, I.; Zviran, A.; et al. Derivation of novel human ground state naive pluripotent stem cells. Nature 2013, 504, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Argueso, J.L.; Westmoreland, J.; Mieczkowski, P.A.; Gawel, M.; Petes, T.D.; Resnick, M.A. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc. Natl. Acad. Sci. USA 2008, 105, 11845–11850. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Seah, Y.F.; El Farran, C.A.; Warrier, T.; Xu, J.; Loh, Y.H. Induced pluripotency and gene editing in disease modelling: Perspectives and challenges. Int. J. Mol. Sci. 2015, 16, 28614–28634. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Gomez-Gonzalez, B.; Aguilera, A. DNA repair in mammalian cells: DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef] [PubMed]
- Rouet, P.; Smih, F.; Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 1994, 14, 8096–8106. [Google Scholar] [CrossRef] [PubMed]
- Rouet, P.; Smih, F.; Jasin, M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. USA 1994, 91, 6064–6068. [Google Scholar] [CrossRef] [PubMed]
- Smih, F.; Rouet, P.; Romanienko, P.J.; Jasin, M. Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res. 1995, 23, 5012–5019. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.J.; Crotty, J.W.; Bhakta, M.S.; Barbas, C.F., 3rd; Horton, N.C. Structure of aart, a designed six-finger zinc finger peptide, bound to DNA. J. Mol. Biol. 2006, 363, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Mani, M.; Smith, J.; Kandavelou, K.; Berg, J.M.; Chandrasegaran, S. Binding of two zinc finger nuclease monomers to two specific sites is required for effective double-strand DNA cleavage. Biochem. Biophys. Res. Commun. 2005, 334, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 2003, 300, 763. [Google Scholar] [CrossRef] [PubMed]
- Hofer, U.; Henley, J.E.; Exline, C.M.; Mulhern, O.; Lopez, E.; Cannon, P.M. Pre-clinical modeling of CCR5 knockout in human hematopoietic stem cells by zinc finger nucleases using humanized mice. J. Infect. Dis 2013, 208 (Suppl. S2), S160–164. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.L.; et al. Establishment of hiv-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with hiv. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Isalan, M. Zinc-finger nucleases: How to play two good hands. Nat. Methods 2011, 9, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, R.; Lombardo, A.; Arens, A.; Miller, J.C.; Genovese, P.; Kaeppel, C.; Nowrouzi, A.; Bartholomae, C.C.; Wang, J.; Friedman, G.; et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011, 29, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.L.; Foley, J.E.; Wright, D.A.; Muller-Lerch, F.; Rahman, S.H.; Cornu, T.I.; Winfrey, R.J.; Sander, J.D.; Fu, F.; Townsend, J.A.; et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat. Methods 2008, 5, 374–375. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Thibodeau-Beganny, S.; Osiak, A.; Wright, D.A.; Anthony, R.M.; Eichtinger, M.; Jiang, T.; Foley, J.E.; Winfrey, R.J.; Townsend, J.A.; et al. Rapid "open-source" engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol. Cell. 2008, 31, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Bonas, U. Xanthomonas AvrBs3 family-type III effectors: Discovery and function. Annu. Rev. Phytopathol. 2010, 48, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of tal-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A tale nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cong, L.; Lodato, S.; Kosuri, S.; Church, G.M.; Arlotta, P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 2011, 29, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Pattanayak, V.; Reyon, D.; Tsai, S.Q.; Sander, J.D.; Joung, J.K.; Liu, D.R. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods 2014, 11, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Briggs, A.W.; Rios, X.; Chari, R.; Yang, L.; Zhang, F.; Mali, P.; Church, G.M. Iterative capped assembly: Rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res. 2012, 40, e117. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Burgk, J.L.; Schmidt, T.; Kaiser, V.; Honing, K.; Hornung, V. A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes. Nat. Biotechnol. 2013, 31, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kweon, J.; Kim, A.; Chon, J.K.; Yoo, J.Y.; Kim, H.J.; Kim, S.; Lee, C.; Jeong, E.; Chung, E.; et al. A library of tal effector nucleases spanning the human genome. Nat. Biotechnol. 2013, 31, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Morbitzer, R.; Lutge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel tale nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef] [PubMed]
- Grau, J.; Boch, J.; Posch, S. TALENoffer: Genome-wide TALEN off-target prediction. Bioinformatics 2013, 29, 2931–2932. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Bultmann, S.; Morbitzer, R.; Schmidt, C.S.; Thanisch, K.; Spada, F.; Elsaesser, J.; Lahaye, T.; Leonhardt, H. Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer tales and inhibition of epigenetic modifiers. Nucleic Acids Res. 2012, 40, 5368–5377. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D'Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010, 11, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Jore, M.M.; Datsenko, K.A.; Semenova, A.; Westra, E.R.; Wanner, B.; van der Oost, J.; Brouns, S.J.; Severinov, K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. USA 2011, 108, 10098–10103. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. Elife. 2013, 2, e00471. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Regan, S.N.; Xia, Y.; Oostrom, L.A.; Cowan, C.A.; Musunuru, K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 2013, 12, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Musunuru, K. Genome engineering tools for building cellular models of disease. FEBS J. 2016, 283, 3222–3231. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.T.; Ryu, B.Y.; Annis, J.E.; Garibov, M.; Jarjour, J.; Rawlings, D.J.; Scharenberg, A.M. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods 2011, 8, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to foki nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Howden, S.E.; McColl, B.; Glaser, A.; Vadolas, J.; Petrou, S.; Little, M.H.; Elefanty, A.G.; Stanley, E.G. A Cas9 variant for efficient generation of indel-free knockin or gene-corrected human pluripotent stem cells. Stem Cell Rep. 2016, 7, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, J.; Hur, J.K.; Been, K.W.; Yoon, S.H.; Kim, J.S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR-Cpf1 using a single crrna array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Bernitz, J.M.; Lee, D.F.; Lemischka, I.R. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev. 2014, 23, 2673–2686. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Rivera-Perez, J.; Bradley, A. The length of homology required for gene targeting in embryonic stem cells. Mol. Cell. Biol. 1991, 11, 5586–5591. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Capecchi, M.R. Reexamination of gene targeting frequency as a function of the extent of homology between the targeting vector and the target locus. Mol. Cell. Biol. 1992, 12, 3365–3371. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Chung, S.K.; Xu, Y. Modeling disease in human escs using an efficient BAC-based homologous recombination system. Cell Stem Cell 2010, 6, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.G.; Jenkins, N.A.; Court, D.L. Recombineering: A powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001, 2, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Bartel, M.A.; Schaffer, D.V. Enhanced gene targeting of adult and pluripotent stem cells using evolved adeno-associated virus. Methods Mol. Biol. 2014, 1114, 169–179. [Google Scholar] [PubMed]
- Khan, I.F.; Hirata, R.K.; Russell, D.W. AAV-mediated gene targeting methods for human cells. Nat. Protoc. 2011, 6, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.F.; Hirata, R.K.; Wang, P.R.; Li, Y.; Kho, J.; Nelson, A.; Huo, Y.; Zavaljevski, M.; Ware, C.; Russell, D.W. Engineering of human pluripotent stem cells by AAV-mediated gene targeting. Mol. Ther. 2010, 18, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Brafman, D.A.; Moya, N.; Allen-Soltero, S.; Fellner, T.; Robinson, M.; McMillen, Z.L.; Gaasterland, T.; Willert, K. Analysis of SOX2-expressing cell populations derived from human pluripotent stem cells. Stem Cell Rep. 2013, 1, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Vasileva, A.; Jessberger, R. Precise hit: Adeno-associated virus in gene targeting. Nat. Rev. Microbiol. 2005, 3, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Nolden, L.; Edenhofer, F.; Peitz, M.; Brustle, O. Stem cell engineering using transducible cre recombinase. Methods Mol. Med. 2007, 140, 17–32. [Google Scholar] [PubMed]
- Miyaoka, Y.; Chan, A.H.; Judge, L.M.; Yoo, J.; Huang, M.; Nguyen, T.D.; Lizarraga, P.P.; So, P.L.; Conklin, B.R. Isolation of single-base genome-edited human ips cells without antibiotic selection. Nat. Methods 2014, 11, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Thibaud, E.; Salomon-Bernard, Y.; Rappaport, R. Prepubertal genital hemorrhage. Study of 50 cases. Ann. Pediatr. (Paris) 1984, 31, 195–198. [Google Scholar] [PubMed]
- Bouhassira, E.E.; Westerman, K.; Leboulch, P. Transcriptional behavior of lcr enhancer elements integrated at the same chromosomal locus by recombinase-mediated cassette exchange. Blood 1997, 90, 3332–3344. [Google Scholar] [PubMed]
- Yang, L.; Guell, M.; Byrne, S.; Yang, J.L.; de Los Angeles, A.; Mali, P.; Aach, J.; Kim-Kiselak, C.; Briggs, A.W.; Rios, X.; et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013, 41, 9049–9061. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, I.; Simons, J.P.; Owen, J.S. Oligonucleotide-directed gene-editing technology: Mechanisms and future prospects. Expert Opin. Biol. Ther. 2012, 12, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Radecke, F.; Peter, I.; Radecke, S.; Gellhaus, K.; Schwarz, K.; Cathomen, T. Targeted chromosomal gene modification in human cells by single-stranded oligodeoxynucleotides in the presence of a DNA double-strand break. Mol. Ther. 2006, 14, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Pruett-Miller, S.M.; Huang, Y.; Gjoka, M.; Duda, K.; Taunton, J.; Collingwood, T.N.; Frodin, M.; Davis, G.D. High-frequency genome editing using ssdna oligonucleotides with zinc-finger nucleases. Nat. Methods 2011, 8, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Laganiere, J.; Cheng, A.W.; Hockemeyer, D.; Gao, Q.; Alagappan, R.; Khurana, V.; Golbe, L.I.; Myers, R.H.; Lindquist, S.; et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell 2011, 146, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhang, S.C. Genetic modification of human embryonic stem cells. Biotechnol. Genet. Eng. Rev. 2007, 24, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, R. Lentivirus-mediated modification of pluripotent stem cells. Methods Mol. Biol. 2011, 767, 315–331. [Google Scholar] [PubMed]
- Siemen, H.; Nolden, L.; Terstegge, S.; Koch, P.; Brustle, O. Nucleofection of human embryonic stem cells. Methods Mol. Biol. 2008, 423, 131–138. [Google Scholar] [PubMed]
- Krentz, N.A.; Nian, C.; Lynn, F.C. TALEN/CRISPR-mediated eGFP knock-in add-on at the OCT4 locus does not impact differentiation of human embryonic stem cells towards endoderm. PLoS ONE 2014, 9, e114275. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient targeting of expressed and silent genes in human escs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Verma, N.; Gonzalez, F.; Shi, Z.D.; Huangfu, D. A CRISPR/Cas-mediated selection-free knockin strategy in human embryonic stem cells. Stem Cell Rep. 2015, 4, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Wang, H.; Kiani, S.; Lai, C.S.; Gao, Q.; Cassady, J.P.; Cost, G.J.; Zhang, L.; Santiago, Y.; Miller, J.C.; et al. Genetic engineering of human pluripotent cells using tale nucleases. Nat. Biotechnol. 2011, 29, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Forster, R.; Chiba, K.; Schaeffer, L.; Regalado, S.G.; Lai, C.S.; Gao, Q.; Kiani, S.; Farin, H.F.; Clevers, H.; Cost, G.J.; et al. Human intestinal tissue with adult stem cell properties derived from pluripotent stem cells. Stem Cell Rep. 2014, 2, 838–852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.W.; Haidet-Phillips, A.M.; Pham, J.T.; Lee, Y.; Huo, Y.; Tienari, P.J.; Maragakis, N.J.; Sattler, R.; Rothstein, J.D. Generation of gfap::Gfp astrocyte reporter lines from human adult fibroblast-derived iPS cells using zinc-finger nuclease technology. Glia 2016, 64, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hunt, S.D.; Xue, H.; Liu, Y.; Darabi, R. Generation and validation of PAX7 reporter lines from human iPS cells using CRISPR/Cas9 technology. Stem Cell Res. 2016, 16, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hunt, S.D.; Xue, H.; Liu, Y.; Darabi, R. Generation and characterization of a MYF5 reporter human iPS cell line using CRISPR/Cas9 mediated homologous recombination. Sci. Rep. 2016, 6, 18759. [Google Scholar] [CrossRef] [PubMed]
- DeKelver, R.C.; Choi, V.M.; Moehle, E.A.; Paschon, D.E.; Hockemeyer, D.; Meijsing, S.H.; Sancak, Y.; Cui, X.; Steine, E.J.; Miller, J.C.; et al. Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010, 20, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Tay, F.C.; Tan, W.K.; Goh, S.L.; Ramachandra, C.J.; Lau, C.H.; Zhu, H.; Chen, C.; Du, S.; Phang, R.Z.; Shahbazi, M.; et al. Targeted transgene insertion into the AAVS1 locus driven by baculoviral vector-mediated zinc finger nuclease expression in human-induced pluripotent stem cells. J. Gene Med. 2013, 15, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.Y.; Hu, S.; Lan, F.; Lee, A.S.; Huber, B.; Lisowski, L.; Liang, P.; Huang, M.; de Almeida, P.E.; et al. Genome editing of human embryonic stem cells and induced pluripotent stem cells with zinc finger nucleases for cellular imaging. Circ. Res. 2012, 111, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lau, C.H.; Goh, S.L.; Liang, Q.; Chen, C.; Du, S.; Phang, R.Z.; Tay, F.C.; Tan, W.K.; Li, Z.; et al. Baculoviral transduction facilitates TALEN-mediated targeted transgene integration and Cre/LoxP cassette exchange in human-induced pluripotent stem cells. Nucleic Acids Res. 2013, 41, e180. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, C.; Cerbini, T.; San, H.; Lin, Y.; Chen, G.; Rao, M.S.; Zou, J. Stable enhanced green fluorescent protein expression after differentiation and transplantation of reporter human induced pluripotent stem cells generated by AAVS1 transcription activator-like effector nucleases. Stem Cells Transl. Med. 2014, 3, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Mali, P.; Huang, X.; Dowey, S.N.; Cheng, L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 2011, 118, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Sebastiano, V.; Maeder, M.L.; Angstman, J.F.; Haddad, B.; Khayter, C.; Yeo, D.T.; Goodwin, M.J.; Hawkins, J.S.; Ramirez, C.L.; Batista, L.F.; et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 2011, 29, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson's model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Fong, H.; Wang, C.; Knoferle, J.; Walker, D.; Balestra, M.E.; Tong, L.M.; Leung, L.; Ring, K.L.; Seeley, W.W.; Karydas, A.; et al. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Rep. 2013, 1, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Miller, B.L. New approaches to the treatment of frontotemporal lobar degeneration. Curr. Opin. Neurol. 2008, 21, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from niemann-pick type c patient-specific iPS cells. Stem Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Skryabin, B.V.; Greber, B. A modified TALEN-based system for robust generation of knock-out human pluripotent stem cell lines and disease models. BMC Genomics 2013, 14, 773. [Google Scholar] [CrossRef] [PubMed]
- Menon, T.; Firth, A.L.; Scripture-Adams, D.D.; Galic, Z.; Qualls, S.J.; Gilmore, W.B.; Ke, E.; Singer, O.; Anderson, L.S.; Bornzin, A.R.; et al. Lymphoid regeneration from gene-corrected SCID-X1 subject-derived iPSCs. Cell Stem Cell 2015, 16, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhao, H. Seamless correction of the sickle cell disease mutation of the hbb gene in human induced pluripotent stem cells using TALENs. Biotechnol. Bioeng. 2014, 111, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, H.; Kagoya, Y.; Kataoka, K.; Yoshimi, A.; Miyauchi, M.; Taoka, K.; Kumano, K.; Yamamoto, T.; Hotta, A.; Arai, S.; et al. Targeted gene correction of RUNX1 in induced pluripotent stem cells derived from familial platelet disorder with propensity to myeloid malignancy restores normal megakaryopoiesis. Exp. Hematol. 2015, 43, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Garate, Z.; Quintana-Bustamante, O.; Crane, A.M.; Olivier, E.; Poirot, L.; Galetto, R.; Kosinski, P.; Hill, C.; Kung, C.; Agirre, X.; et al. Generation of a high number of healthy erythroid cells from gene-edited pyruvate kinase deficiency patient-specific induced pluripotent stem cells. Stem Cell Rep. 2015, 5, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Kim, J.; Kweon, J.; Son, J.S.; Lee, J.S.; Yoo, J.E.; Cho, S.R.; Kim, J.H.; Kim, J.S.; Kim, D.W. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc. Natl. Acad. Sci. USA 2014, 111, 9253–9258. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, G.; Young, J.E.; Martinez, F.J.; Buen, F.; Gore, A.; Kinaga, J.; Li, Z.; Yuan, S.H.; Zhang, K.; Goldstein, L.S. The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell. Rep. 2013, 5, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G.; et al. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, G.; Reyna, S.M.; Dunlap, M.; van Der Kant, R.; Callender, J.A.; Young, J.E.; Roberts, E.A.; Goldstein, L.S. Defective transcytosis of app and lipoproteins in human iPSC-derived neurons with familial Alzheimer’s disease mutations. Cell Rep. 2016, 17, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Lane-Donovan, C.; Philips, G.T.; Herz, J. More than cholesterol transporters: Lipoprotein receptors in CNS function and neurodegeneration. Neuron 2014, 83, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Pierrot, N.; Tyteca, D.; D'Auria, L.; Dewachter, I.; Gailly, P.; Hendrickx, A.; Tasiaux, B.; Haylani, L.E.; Muls, N.; N'Kuli, F.; et al. Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol. Med. 2013, 5, 608–625. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Ye, L.; Chang, J.C.; Beyer, A.I.; Wang, J.; Muench, M.O.; Kan, Y.W. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014, 24, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Fan, Y.; He, W.; Zhu, D.; Niu, X.; Wang, D.; Ou, Z.; Luo, M.; Sun, X. Improved hematopoietic differentiation efficiency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev. 2015, 24, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Kim, D.H.; Son, J.S.; Sung, J.J.; Lee, J.; Bae, S.; Kim, J.H.; Kim, D.W.; Kim, J.S. Functional correction of large factor viii gene chromosomal inversions in hemophilia a patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell 2015, 17, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Horii, T.; Tamura, D.; Morita, S.; Kimura, M.; Hatada, I. Generation of an icf syndrome model by efficient genome editing of human induced pluripotent stem cells using the CRISPR system. Int. J. Mol. Sci. 2013, 14, 19774–19781. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, Y.; Yan, W.; Smith, C.; Ye, Z.; Wang, J.; Gao, Y.; Mendelsohn, L.; Cheng, L. Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells 2015, 33, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Lai, Y.S.; Westin, E.; Khodadadi-Jamayran, A.; Pawlik, K.M.; Lamb, L.S., Jr.; Goldman, F.D.; Townes, T.M. Modeling human severe combined immunodeficiency and correction by CRISPR/Cas9-enhanced gene targeting. Cell. Rep. 2015, 12, 1668–1677. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H.; et al. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 2015, 12, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Howden, S.E.; Maufort, J.P.; Duffin, B.M.; Elefanty, A.G.; Stanley, E.G.; Thomson, J.A. Simultaneous reprogramming and gene correction of patient fibroblasts. Stem Cell Rep. 2015, 5, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rashid, S.T.; Strick-Marchand, H.; Varela, I.; Liu, P.Q.; Paschon, D.E.; Miranda, E.; Ordonez, A.; Hannan, N.R.; Rouhani, F.J.; et al. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature 2011, 478, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of hiv by CCR5 delta32/delta32 stem-cell transplantation. N Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y. CCR5-targeted hematopoietic stem cell gene approaches for hiv disease: Current progress and future prospects. Curr. Stem Cell. Res. Ther. 2012, 7, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5delta32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Daer, R.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The impact of chromatin dynamics on Cas9-mediated genome editing in human cells. ACS Synth Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.N.; Westenskow, P.D.; Todorova, D.; Hu, Z.; Lin, T.; Rong, Z.; Kim, J.; He, J.; Wang, M.; et al. Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell 2015, 17, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yu, M.; Shen, C.; Chen, X.; Feng, T.; Yao, Y.; Li, J.; Li, H.; Tu, W. Negligible immunogenicity of induced pluripotent stem cells derived from human skin fibroblasts. PLoS ONE 2014, 9, e114949. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, F.; Lei, J.; Fang, Y.; Tong, H.; Wu, W.; Liu, C. Pyrosequencing revealed highly microbial phylogenetic diversity in ferromanganese nodules from farmland. Environ. Sci. Process. Impacts 2015, 17, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Miyagawa, S.; Fukushima, S.; Maeda, A.; Kashiyama, N.; Kawamura, A.; Miki, K.; Okita, K.; Yoshida, Y.; Shiina, T.; et al. Cardiomyocytes derived from mhc-homozygous induced pluripotent stem cells exhibit reduced allogeneic immunogenicity in mhc-matched non-human primates. Stem Cell Rep. 2016, 6, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Wang, M.; Hu, Z.; Stradner, M.; Zhu, S.; Kong, H.; Yi, H.; Goldrath, A.; Yang, Y.G.; Xu, Y.; et al. An effective approach to prevent immune rejection of human esc-derived allografts. Cell Stem Cell 2014, 14, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Shen, X.Z.; Jiang, F.; Wu, Y.; Han, C. DNA-guided genome editing using the natronobacterium gregoryi argonaute. Nat. Biotechnol. 2016, 34, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Dong, Z.; Shi, Y.; Wang, X.; Qin, Y.; Wang, Y.; Liu, D. NgAgo-based fabp11a gene knockdown causes eye developmental defects in zebrafish. Cell Res. 2016, 26, 1349–1352. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Updated: NgAgo gene-editing controversy escalates in peer-reviewed papers. Nature 2016, 540, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Replications, ridicule and a recluse: The controversy over ngago gene-editing intensifies. Nature 2016, 536, 136–137. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Cheng, L.; Gu, F.; Huang, J.; Huang, Z.; Lin, S.; Li, J.; Li, W.; Qin, W.; Sun, Y.; et al. Questions about ngago. Protein Cell 2016, 7, 913–915. [Google Scholar] [CrossRef] [PubMed]
- Sandoe, J.; Eggan, K. Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nat. Neurosci. 2013, 16, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.E.; Dai, D.F.; Laflamme, M.A. Human pluripotent stem cells: Prospects and challenges as a source of cardiomyocytes for in vitro modeling and cell-based cardiac repair. Adv. Drug Deliv. Rev. 2016, 96, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Ding, S.; Rando, T.A.; Trounson, A. Translational strategies and challenges in regenerative medicine. Nat. Med. 2014, 20, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Trounson, A.; DeWitt, N.D. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol. 2016, 17, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Picanco-Castro, V.; Moreira, L.F.; Kashima, S.; Covas, D.T. Can pluripotent stem cells be used in cell-based therapy? Cell Reprogram 2014, 16, 98–107. [Google Scholar] [CrossRef] [PubMed]
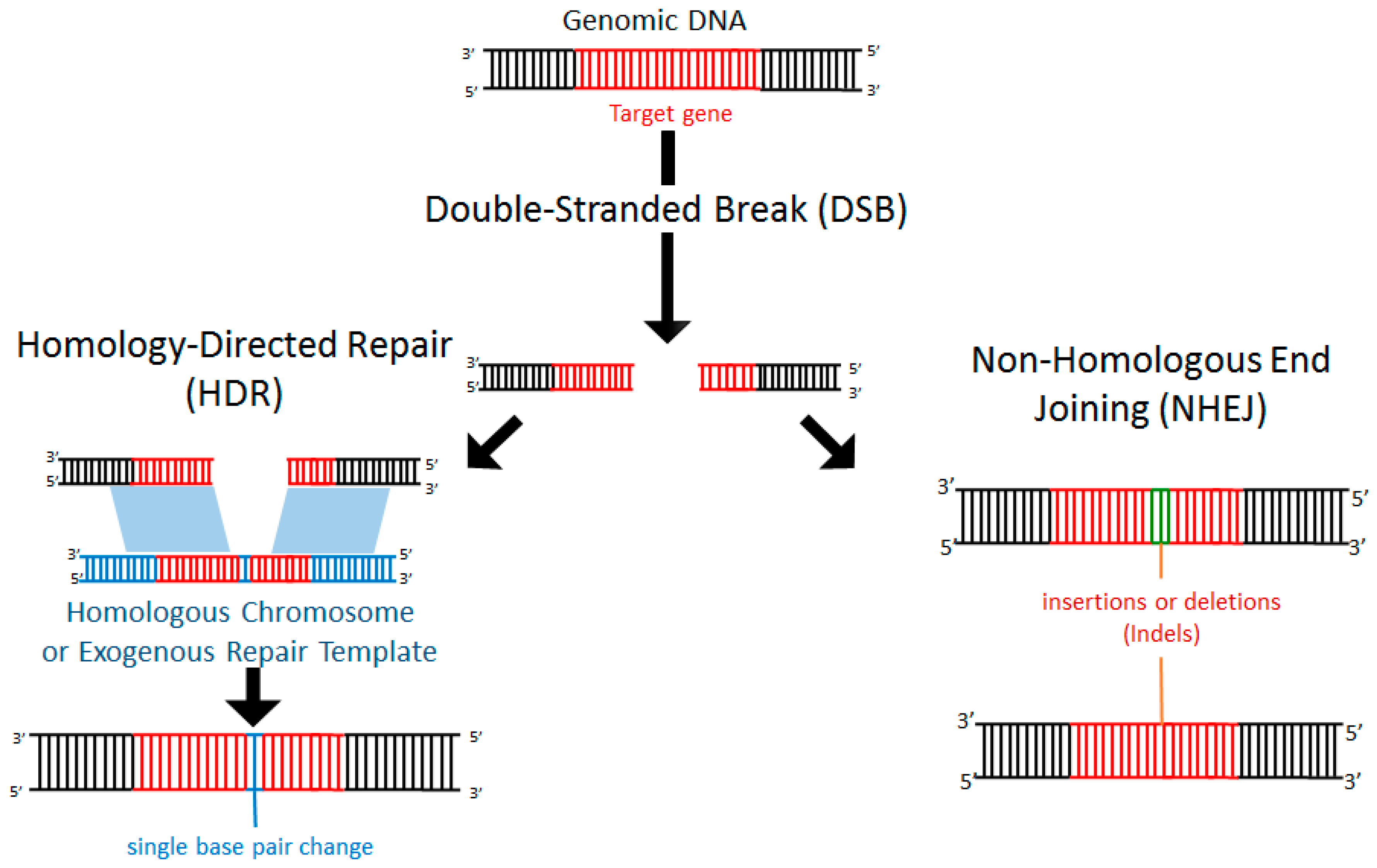
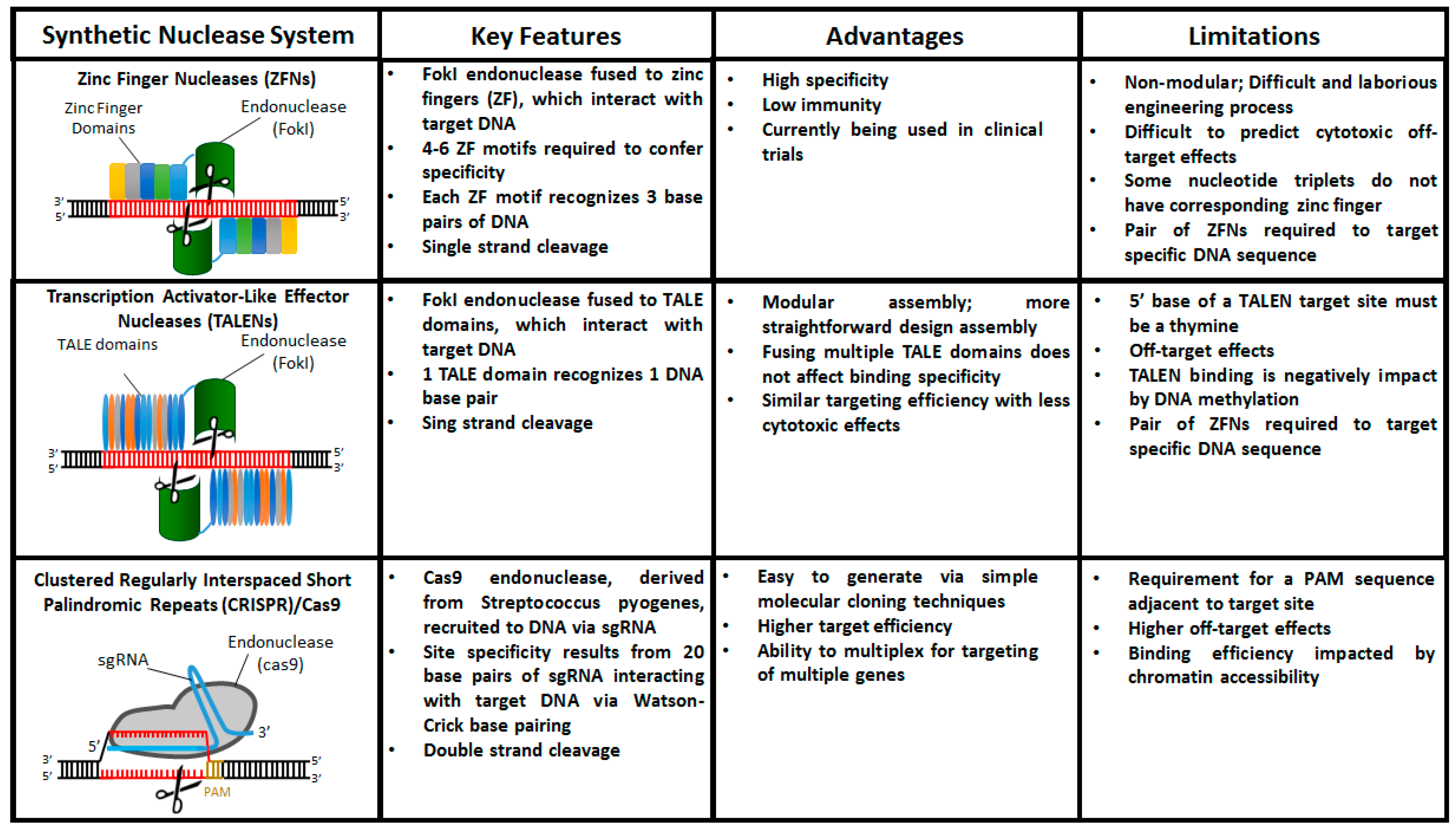
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).