
The Regulation of NF-κB Subunits by Phosphorylation


Abstract
:1. Introduction
1.1. NF-κB
1.2. How Phosphorylation Regulates NF-κB Activity and Function
2. Phosphorylation of the NF-κB Subunits
2.1. Processing of p105 and p100
2.2. p105
2.3. p50
2.4. p100 and p52
2.5. RelB
2.6. p65
2.6.1. Phosphorylation of the Rel Homology Domain
S276
2.6.2. Other Rel Homology Domain Phosphorylations
2.6.3. Phosphorylation of the Transactivation Domain
2.6.3.1. S536
2.6.3.2. T435
2.6.3.3. S468
2.6.3.4. T505
2.6.3.5. S529
2.7. c-Rel
3. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| TAD | Transactivation domain |
| RHD | Rel homology domain |
| IKK | Inhibitor of kappaB kinase |
| MAPK | Mitogen activated protein kinase |
| PKA | Protein kinase A |
| NIK | NF-kappaB inducing kinase |
| TLR | Toll-like receptor |
| CDK | Cyclin dependnet kinase |
| CK | Casein kinase |
| GSK | Glycogen synthase kinase |
References
- Ghosh, S.; Hayden, M.S. Celebrating 25 Years of Nf-Kappab Research. Immunol. Rev. 2012, 246, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Nf-Kappab, the First Quarter-Century: Remarkable Progress and Outstanding Questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, C.; Feldmann, M. Nf-Kappab as a Target for Modulating Inflammatory Responses. Curr. Pharm. Design 2012, 18, 5735–5745. [Google Scholar] [CrossRef]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional Regulation Via the Nf-Kappab Signaling Module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [PubMed]
- Www.Nf-Kb.Org. Available Online: Http://Www.Bu.Edu/Nf-Kb/ (accessed on 25 February 2016).
- Siggers, T.; Chang, A.B.; Teixeira, A.; Wong, D.; Williams, K.J.; Ahmed, B.; Ragoussis, J.; Udalova, I.A.; Smale, S.T.; Bulyk, M.L. Principles of Dimer-Specific Gene Regulation Revealed by a Comprehensive Characterization of Nf-Kappab Family DNA Binding. Nat. Immunol. 2012, 13, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Reichardt, A.D.; Cheng, G. Non-Canonical Nf-Kappab Signaling Activation and Regulation: Principles and Perspectives. Immunol. Rev. 2011, 244, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Colleran, A.; Collins, P.E.; O'Carroll, C.; Ahmed, A.; Mao, X.; McManus, B.; Kiely, P.A.; Burstein, E.; Carmody, R.J. Deubiquitination of Nf-Kappab by Ubiquitin-Specific Protease-7 Promotes Transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation Meets Ubiquitination: The Control of Nf-[Kappa]B Activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Orian, A.; Gonen, H.; Bercovich, B.; Fajerman, I.; Eytan, E.; Israel, A.; Mercurio, F.; Iwai, K.; Schwartz, A.L.; Ciechanover, A. Scf(Beta)(-Trcp) Ubiquitin Ligase-Mediated Processing of Nf-Kappab P105 Requires Phosphorylation of Its C-Terminus by Ikappab Kinase. EMBO J. 2000, 19, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; DeMartino, G.N.; Greene, W.C. Cotranslational Biogenesis of Nf-Kappab P50 by the 26s Proteasome. Cell 1998, 92, 819–828. [Google Scholar] [CrossRef]
- Fan, C.M.; Maniatis, T. Generation of P50 Subunit of Nf-Kappa B by Processing of P105 through an Atp-Dependent Pathway. Nature 1991, 354, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Yasuda, H.; Sato, Y.; Yamamoto, K. A Role for Phosphorylation in the Proteolytic Processing of the Human Nf-Kappa B1 Precursor. Gene 1995, 165, 183–189. [Google Scholar] [CrossRef]
- Heissmeyer, V.; Krappmann, D.; Wulczyn, F.G.; Scheidereit, C. Nf-Kappab P105 Is a Target of Ikappab Kinases and Controls Signal Induction of Bcl-3-P50 Complexes. EMBO J. 1999, 18, 4766–4778. [Google Scholar] [CrossRef] [PubMed]
- Heissmeyer, V.; Krappmann, D.; Hatada, E.N.; Scheidereit, C. Shared Pathways of Ikappab Kinase-Induced Scf(Betatrcp)-Mediated Ubiquitination and Degradation for the Nf-Kappab Precursor P105 and Ikappabalpha. Mol. Cell Biol. 2001, 21, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Salmeron, A.; Janzen, J.; Soneji, Y.; Bump, N.; Kamens, J.; Allen, H.; Ley, S.C. Direct Phosphorylation of Nf-Kappab1 P105 by the Ikappab Kinase Complex on Serine 927 Is Essential for Signal-Induced P105 Proteolysis. J. Biol. Chem. 2001, 276, 22215–22222. [Google Scholar] [CrossRef] [PubMed]
- Lang, V.; Janzen, J.; Fischer, G.Z.; Soneji, Y.; Beinke, S.; Salmeron, A.; Allen, H.; Hay, R.T.; Ben-Neriah, Y.; Ley, S.C. Betatrcp-Mediated Proteolysis of Nf-Kappab1 P105 Requires Phosphorylation of P105 Serines 927 and 932. Mol. Cell Biol. 2003, 23, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Achbert-Weiner, H.; Ciechanover, A. Dual Effects of Ikappab Kinase Beta-Mediated Phosphorylation on P105 Fate: Scf(Beta-Trcp)-Dependent Degradation and Scf(Beta-Trcp)-Independent Processing. Mol. Cell Biol. 2004, 24, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Demarchi, F.; Bertoli, C.; Sandy, P.; Schneider, C. Glycogen Synthase Kinase-3 Beta Regulates Nf-Kappa B1/P105 Stability. J. Biol. Chem. 2003, 278, 39583–39590. [Google Scholar] [CrossRef] [PubMed]
- Belich, M.P.; Salmeron, A.; Johnston, L.H.; Ley, S.C. Tpl-2 Kinase Regulates the Proteolysis of the Nf-Kappab-Inhibitory Protein Nf-Kappab1 P105. Nature 1999, 397, 363–368. [Google Scholar] [PubMed]
- Beinke, S.; Deka, J.; Lang, V.; Belich, M.P.; Walker, P.A.; Howell, S.; Smerdon, S.J.; Gamblin, S.J.; Ley, S.C. Nf-Kappab1 P105 Negatively Regulates Tpl-2 Mek Kinase Activity. Mol. Cell Biol. 2003, 23, 4739–4752. [Google Scholar] [CrossRef] [PubMed]
- Waterfield, M.R.; Zhang, M.; Norman, L.P.; Sun, S.C. Nf-Kappab1/P105 Regulates Lipopolysaccharide-Stimulated Map Kinase Signaling by Governing the Stability and Function of the Tpl2 Kinase. Mol. Cell 2003, 11, 685–694. [Google Scholar] [CrossRef]
- Hou, S.; Guan, H.; Ricciardi, R.P. Phosphorylation of Serine 337 of Nf-Kappab P50 Is Critical for DNA Binding. J. Biol. Chem. 2003, 278, 45994–45998. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Hou, S.; Ricciardi, R.P. DNA Binding of Repressor Nuclear Factor-Kappab P50/P50 Depends on Phosphorylation of Ser337 by the Protein Kinase a Catalytic Subunit. J. Biol. Chem. 2005, 280, 9957–9962. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Naura, A.S.; Errami, Y.; Zerfaoui, M.; Kim, H.; Kim, J.G.; Abd Elmageed, Z.Y.; Abdel-Mageed, A.B.; Giardina, C.; Beg, A.A.; et al. Phosphorylation of P50 Nf-Kappab at a Single Serine Residue by DNA-Dependent Protein Kinase Is Critical for Vcam-1 Expression Upon Tnf Treatment. J. Biol. Chem. 2010, 285, 41152–41160. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Crawley, C.D.; Kang, S.; Raleigh, D.R.; Yu, X.; Wahlstrom, J.S.; Voce, D.J.; Darga, T.E.; Weichselbaum, R.R.; Yamini, B. P50 (Nf-Kappab1) Is an Effector Protein in the Cytotoxic Response to DNA Methylation Damage. Mol. Cell 2011, 44, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Crawley, C.D.; Raleigh, D.R.; Kang, S.; Voce, D.J.; Schmitt, A.M.; Weichselbaum, R.R.; Yamini, B. DNA Damage-Induced Cytotoxicity Is Mediated by the Cooperative Interaction of Phospho-Nf-Kappab P50 and a Single Nucleotide in the Kappab-Site. Nucleic Acids Res. 2013, 41, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Crawley, C.D.; Kang, S.; Bernal, G.M.; Wahlstrom, J.S.; Voce, D.J.; Cahill, K.E.; Garofalo, A.; Raleigh, D.R.; Weichselbaum, R.R.; Yamini, B. S-Phase-Dependent P50/Nf-Small Ka, Cyrillicb1 Phosphorylation in Response to Atr and Replication Stress Acts to Maintain Genomic Stability. Cell Cycle 2015, 14, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.J.; Ruan, Q.; Palmer, S.; Hilliard, B.; Chen, Y.H. Negative Regulation of Toll-Like Receptor Signaling by Nf-Kappab P50 Ubiquitination Blockade. Science 2007, 317, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Kiely, P.A.; Carmody, R.J. Inhibition of Transcription by B Cell Leukemia 3 (Bcl-3) Protein Requires Interaction with Nuclear Factor Kappab (Nf-Kappab) P50. J. Biol. Chem. 2014, 289, 7059–7067. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.L.; Jurk, D.; Fullard, N.; Banks, P.; Page, A.; Luli, S.; Elsharkawy, A.M.; Gieling, R.G.; Chakraborty, J.B.; Fox, C.; et al. Nfkappab1 Is a Suppressor of Neutrophil-Driven Hepatocellular Carcinoma. Nat. Commun. 2015, 6, 6818. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Fong, A.; Sun, S.C. Induction of P100 Processing by Nf-Kappab-Inducing Kinase Involves Docking Ikappab Kinase Alpha (Ikkalpha) to P100 and Ikkalpha-Mediated Phosphorylation. J. Biol. Chem. 2004, 279, 30099–30105. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. Nf-Kappab-Inducing Kinase Regulates the Processing of Nf-Kappab2 P100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, M.; Sun, S.C. Beta-Trcp Binding and Processing of Nf-Kappab2/P100 Involve Its Phosphorylation at Serines 866 and 870. Cell Signal. 2006, 18, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Arabi, A.; Ullah, K.; Branca, R.M.; Johansson, J.; Bandarra, D.; Haneklaus, M.; Fu, J.; Aries, I.; Nilsson, P.; Den Boer, M.L.; et al. Proteomic Screen Reveals Fbw7 as a Modulator of the Nf-Kappab Pathway. Nat. Commun. 2012, 3, 976. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.C.; Nabel, G.J. Differential Regulation of Nf-Kappab2(P100) Processing and Control by Amino-Terminal Sequences. Mol. Cell Biol. 1996, 16, 6363–6371. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Xiao, G. Essential Role of Ikappab Kinase Alpha in the Constitutive Processing of Nf-Kappab2 P100. J. Biol. Chem. 2005, 280, 9765–9768. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.; Bergsagel, P.L.; Karin, M. Nonredundant and Complementary Functions of Traf2 and Traf3 in a Ubiquitination Cascade That Activates Nik-Dependent Alternative Nf-Kappab Signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.; Yamazaki, S.; He, J.Q.; Cheng, G. Control of Canonical Nf-Kappab Activation through the Nik-Ikk Complex Pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 3503–3508. [Google Scholar] [CrossRef] [PubMed]
- Fong, A.; Sun, S.C. Genetic Evidence for the Essential Role of Beta-Transducin Repeat-Containing Protein in the Inducible Processing of Nf-Kappa B2/P100. J. Biol. Chem. 2002, 277, 22111–22114. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Haecker, H.; Karin, M.; Ciechanover, A. Mechanism of Processing of the Nf-Kappa B2 P100 Precursor: Identification of the Specific Polyubiquitin Chain-Anchoring Lysine Residue and Analysis of the Role of Nedd8-Modification on the Scf(Beta-Trcp) Ubiquitin Ligase. Oncogene 2004, 23, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Fusco, A.J.; Savinova, O.V.; Talwar, R.; Kearns, J.D.; Hoffmann, A.; Ghosh, G. Stabilization of Relb Requires Multidomain Interactions with P100/P52. J. Biol. Chem. 2008, 283, 12324–12332. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Marienfeld, R.; Wirth, T.; Baumann, B. Critical Role of Relb Serine 368 for Dimerization and P100 Stabilization. J. Biol. Chem. 2003, 278, 39242–39250. [Google Scholar] [CrossRef] [PubMed]
- Marienfeld, R.; Berberich-Siebelt, F.; Berberich, I.; Denk, A.; Serfling, E.; Neumann, M. Signal-Specific and Phosphorylation-Dependent Relb Degradation: A Potential Mechanism of Nf-Kappab Control. Oncogene 2001, 20, 8142–8147. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Klar, S.; Wilisch-Neumann, A.; Hollenbach, E.; Kavuri, S.; Leverkus, M.; Kandolf, R.; Brunner-Weinzierl, M.C.; Klingel, K. Glycogen Synthase Kinase-3beta Is a Crucial Mediator of Signal-Induced Relb Degradation. Oncogene 2011, 30, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Authier, H.; Billot, K.; Derudder, E.; Bordereaux, D.; Riviere, P.; Rodrigues-Ferreira, S.; Nahmias, C.; Baud, V. Ikk Phosphorylates Relb to Modulate Its Promoter Specificity and Promote Fibroblast Migration Downstream of Tnf Receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 14794–14799. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.H.; Chiang, W.C.; Shih, H.M.; Wu, K.J. Stimulation of C-Rel Transcriptional Activity by Pka Catalytic Subunit Beta. J. Mol. Med. (Berl.) 2004, 82, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Mosialos, G.; Hamer, P.; Capobianco, A.J.; Laursen, R.A.; Gilmore, T.D. A Protein Kinase-a Recognition Sequence Is Structurally Linked to Transformation by P59v-Rel and Cytoplasmic Retention of P68c-Rel. Mol. Cell Biol. 1991, 11, 5867–5877. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Valdepeñas, C.; Martín, A.G.; Ramakrishnan, P.; Wallach, D.; Fresno, M. Nf-Κb-Inducing Kinase Is Involved in the Activation of the Cd28 Responsive Element through Phosphorylation of C-Rel and Regulation of Its Transactivating Activity. J. Immunol. 2006, 176, 4666–4674. [Google Scholar] [CrossRef] [PubMed]
- Starczynowski, D.T.; Reynolds, J.G.; Gilmore, T.D. Mutations of Tumor Necrosis Factor Alpha-Responsive Serine Residues within the C-Terminal Transactivation Domain of Human Transcription Factor Rel Enhance Its in Vitro Transforming Ability. Oncogene 2005, 24, 7355–7368. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.G.; San-Antonio, B.; Fresno, M. Regulation of Nuclear Factor Κb Transactivation: Implication of Phosphatidylinositol 3-Kinase and Protein Kinase C Ζ in C-Rel Activation by Tumor Necrosis Factor Α. J. Biol. Chem. 2001, 276, 15840–15849. [Google Scholar] [CrossRef] [PubMed]
- Starczynowski, D.T.; Trautmann, H.; Pott, C.; Harder, L.; Arnold, N.; Africa, J.A.; Leeman, J.R.; Siebert, R.; Gilmore, T.D. Mutation of an Ikk Phosphorylation Site within the Transactivation Domain of Rel in Two Patients with B-Cell Lymphoma Enhances Rel's in Vitro Transforming Activity. Oncogene 2007, 26, 2685–2694. [Google Scholar] [CrossRef] [PubMed]
- Garbati, M.R.; Gilmore, T.D. Ser484 and Ser494 in Rel Are the Major Sites of Ikk Phosphorylation in Vitro: Evidence That Ikk Does Not Directly Enhance Gal4-Rel Transactivation. Gene Exp. 2008, 14, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Kracht, M.; Schmitz, M.L. The Cytokine-Induced Conformational Switch of Nuclear Factor Kappab P65 Is Mediated by P65 Phosphorylation. Biochem. J. 2014, 457, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; SuYang, H.; Erdjument-Bromage, H.; Tempst, P.; Ghosh, S. The Transcriptional Activity of Nf-Κb Is Regulated by the Iκb-Associated Pkac Subunit through a Cyclic Amp–Independent Mechanism. Cell 1997, 89, 413–424. [Google Scholar] [CrossRef]
- Ishinaga, H.; Jono, H.; Lim, J.H.; Kweon, S.M.; Xu, H.; Ha, U.H.; Xu, H.; Koga, T.; Yan, C.; Feng, X.H.; et al. Tgf-Β Induces P65 Acetylation to Enhance Bacteria-Induced Nf-Κb Activation. EMBO J. 2007, 26, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Hibi, Y.; Cueno, M.; Asamitsu, K.; Okamoto, T. A-Kinase-Interacting Protein 1 (Akip1) Acts as a Molecular Determinant of Pka in Nf-Kappa B Signaling. J. Biol. Chem. 2010, 285, 28097–28104. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Voll, R.E.; Ghosh, S. Phosphorylation of Nf-Kappa B P65 by Pka Stimulates Transcriptional Activity by Promoting a Novel Bivalent Interaction with the Coactivator Cbp/P300. Mol. Cell. 1998, 1, 661–671. [Google Scholar] [CrossRef]
- Doucas, V.; Shi, Y.; Miyamoto, S.; West, A.; Verma, I.; Evans, R.M. Cytoplasmic Catalytic Subunit of Protein Kinase a Mediates Cross-Repression by Nf-Κb and the Glucocorticoid Receptor. Proc. Natl. Acad. Sci. 2000, 97, 11893–11898. [Google Scholar] [CrossRef] [PubMed]
- Tago, K.; Funakoshi-Tago, M.; Sakinawa, M.; Mizuno, N.; Itoh, H. Kappab-Ras Is a Nuclear-Cytoplasmic Small Gtpase That Inhibits Nf-Kappab Activation through the Suppression of Transcriptional Activation of P65/Rela. J. Biol. Chem. 2010, 285, 30622–30633. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.E.; Tian, B.; Jamaluddin, M.; Boldogh, I.; Vergara, L.A.; Choudhary, S.; Brasier, A.R. Rela Ser276 Phosphorylation Is Required for Activation of a Subset of Nf-Kappab-Dependent Genes by Recruiting Cyclin-Dependent Kinase 9/Cyclin T1 Complexes. Mol. Cell Biol. 2008, 28, 3623–3638. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Jimi, E.; Zhong, H.; Hayden, M.S.; Ghosh, S. Repression of Gene Expression by Unphosphorylated Nf-Kappab P65 through Epigenetic Mechanisms. Genes Dev. 2008, 22, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mayo, M.W.; Nagji, A.S.; Smith, P.W.; Ramsey, C.S.; Li, D.; Jones, D.R. Phosphorylation of Rela/P65 Promotes Dnmt-1 Recruitment to Chromatin and Represses Transcription of the Tumor Metastasis Suppressor Gene Brms1. Oncogene 2012, 31, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; den Dunnen, J.; Litjens, M.; van het Hof, B.; van Kooyk, Y.; Geijtenbeek, T.B.H. C-Type Lectin Dc-Sign Modulates Toll-Like Receptor Signaling Via Raf-1 Kinase-Dependent Acetylation of Transcription Factor Nf-Κb. Immunity 2007, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; van der Vlist, M.; van den Berg, L.M.; den Dunnen, J.; Litjens, M.; Geijtenbeek, T.B.H. Hiv-1 Exploits Innate Signaling by Tlr8 and Dc-Sign for Productive Infection of Dendritic Cells. Nat. Immunol. 2010, 11, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Wilde, G.; Van Damme, P.; Vanden Berghe, W.; Haegeman, G. Transcriptional Activation of the Nf-Kappab P65 Subunit by Mitogen- and Stress-Activated Protein Kinase-1 (Msk1). EMBO J. 2003, 22, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Reber, L.; Vermeulen, L.; Haegeman, G.; Frossard, N. Ser276 Phosphorylation of Nf-Kb P65 by Msk1 Controls Scf Expression in Inflammation. PLoS ONE 2009, 4, e4393. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.; Tian, B.; Boldogh, I.; Garofalo, R.P.; Brasier, A.R. Respiratory Syncytial Virus Infection Induces a Reactive Oxygen Species-Msk1-Phospho-Ser-276 Rela Pathway Required for Cytokine Expression. J. Virol. 2009, 83, 10605–10615. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Haegeman, G.; Frossard, N.; Reber, L. Ser276 Phosphorylation of Nf-Kb P65 by Msk1 Controls Scf Expression in Inflammation. Figshare 2009, 1. [Google Scholar]
- Jacks, K.A.; Koch, C.A. Differential Regulation of Mitogen- and Stress-Activated Protein Kinase-1 and -2 (Msk1 and Msk2) by Ck2 Following Uv Radiation. J. Biol. Chem. 2010, 285, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Moreau, F.; Hirota, C.L.; MacNaughton, W.K. Proteinase-Activated Receptors Induce Interleukin-8 Expression by Intestinal Epithelial Cells through Erk/Rsk90 Activation and Histone Acetylation. FASEB J. 2010, 24, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mo, X.; Piper, M.G.; Wang, H.; Parinandi, N.L.; Guttridge, D.; Marsh, C.B. M-Csf Induces Monocyte Survival by Activating Nf-Κb P65 Phosphorylation at Ser276 Via Protein Kinase C. PLoS ONE 2011, 6, e28081. [Google Scholar] [CrossRef] [PubMed]
- Hochrainer, K.; Racchumi, G.; Anrather, J. Site-Specific Phosphorylation of the P65 Protein Subunit Mediates Selective Gene Expression by Differential Nf-Kappab and Rna Polymerase Ii Promoter Recruitment. J. Biol. Chem. 2013, 288, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Williams, S.A.; Mu, Y.; Nakano, H.; Duerr, J.M.; Buckbinder, L.; Greene, W.C. Nf-Kappab Rela Phosphorylation Regulates Rela Acetylation. Mol. Cell Biol. 2005, 25, 7966–7975. [Google Scholar] [CrossRef] [PubMed]
- Nihira, K.; Ando, Y.; Yamaguchi, T.; Kagami, Y.; Miki, Y.; Yoshida, K. Pim-1 Controls Nf-Kappab Signalling by Stabilizing Rela/P65. Cell. Death Differ. 2010, 17, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Racchumi, G.; Iadecola, C. Cis-Acting, Element-Specific Transcriptional Activity of Differentially Phosphorylated Nuclear Factor-Kappa B. J. Biol. Chem. 2005, 280, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Gong, K.; Feng, W.; Nozell, S.E.; Chen, Y.-F.; Chatham, J.C.; Oparil, S. O-Glcnac Modification of Nfκb P65 Inhibits Tnf-Α-Induced Inflammatory Mediator Expression in Rat Aortic Smooth Muscle Cells. PLoS ONE 2011, 6, e24021. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of Nf-Kappab Signaling by Pin1-Dependent Prolyl Isomerization and Ubiquitin-Mediated Proteolysis of P65/Rela. Mol. Cell. 2003, 12, 1413–1426. [Google Scholar] [CrossRef]
- Duran, A.; Diaz-Meco, M.T.; Moscat, J. Essential Role of Rela Ser311 Phosphorylation by Zetapkc in Nf-Kappab Transcriptional Activation. EMBO J. 2003, 22, 3910–3918. [Google Scholar] [CrossRef] [PubMed]
- Clavijo, P.E.; Frauwirth, K.A. Anergic Cd8+ T Lymphocytes Have Impaired Nf-Kappab Activation with Defects in P65 Phosphorylation and Acetylation. J. Immunol. 2012, 188, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Kuo, A.J.; Chang, Y.; Schaefer, U.; Kitson, C.; Cheung, P.; Espejo, A.; Zee, B.M.; Liu, C.L.; Tangsombatvisit, S.; et al. Lysine Methylation of the Nf-[Kappa]B Subunit Rela by Setd6 Couples Activity of the Histone Methyltransferase Glp at Chromatin to Tonic Repression of Nf-[Kappa]B Signaling. Nat. Immunol. 2011, 12, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Levy, D.; Horton, J.R.; Peng, J.; Zhang, X.; Gozani, O.; Cheng, X. Structural Basis of Setd6-Mediated Regulation of the Nf-Kb Network Via Methyl-Lysine Signaling. Nucleic Acids Res. 2011, 39, 6380–6389. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wei, H.; Prabhu, L.; Zhao, W.; Martin, M.; Hartley, A.V.; Lu, T. Role of Novel Serine 316 Phosphorylation of the P65 Subunit of Nf-Kappab in Differential Gene Regulation. J. Biol. Chem. 2015, 290, 20336–20347. [Google Scholar] [CrossRef] [PubMed]
- Buss, H.; Dörrie, A.; Schmitz, M.L.; Hoffmann, E.; Resch, K.; Kracht, M. Constitutive and Interleukin-1-Inducible Phosphorylation of P65 Nf-Κb at Serine 536 Is Mediated by Multiple Protein Kinases Including Iκb Kinase (Ikk)-Α, Ikkβ, Ikkϵ, Traf Family Member-Associated (Tank)-Binding Kinase 1 (Tbk1), and an Unknown Kinase and Couples P65 to Tata-Binding Protein-Associated Factor Ii31-Mediated Interleukin-8 Transcription. J. Biol. Chem. 2004, 279, 55633–55643. [Google Scholar] [PubMed]
- Lawrence, T.; Bebien, M.; Liu, G.Y.; Nizet, V.; Karin, M. Ikk[Alpha] Limits Macrophage Nf-[Kappa]B Activation and Contributes to the Resolution of Inflammation. Nature 2005, 434, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Chiba, H.; Miyoshi, H.; Sugita, T.; Toriumi, W. Iκb Kinases Phosphorylate Nf-Κb P65 Subunit on Serine 536 in the Transactivation Domain. J. Biol. Chem. 1999, 274, 30353–30356. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, N.; Lerner, N.; Dombrowski, N.; Sakurai, H.; Stark, G.R. Distinct Roles of the Ikappa B Kinase Alpha and Beta Subunits in Liberating Nuclear Factor Kappa B (Nf-Kappa B) from Ikappa B and in Phosphorylating the P65 Subunit of Nf-Kappa B. J. Biol. Chem. 2002, 277, 3863–3869. [Google Scholar] [CrossRef] [PubMed]
- Haller, D.; Russo, M.P.; Sartor, R.B.; Jobin, C. Ikkβ and Phosphatidylinositol 3-Kinase/Akt Participate in Non-Pathogenic Gram-Negative Enteric Bacteria-Induced Rela Phosphorylation and Nf-Κb Activation in Both Primary and Intestinal Epithelial Cell Lines. J. Biol. Chem. 2002, 277, 38168–38178. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, I.; Sebald, A.; Bucher, C.; Charles, R.P.; Nakano, H.; Doi, T.; Kracht, M.; Schmitz, M.L. Transient and Selective Nf-Kappa B P65 Serine 536 Phosphorylation Induced by T Cell Costimulation Is Mediated by I Kappa B Kinase Beta and Controls the Kinetics of P65 Nuclear Import. J. Immunol. 2004, 172, 6336–6344. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lu, Q.; Bottero, V.; Estepa, G.; Morrison, L.; Mercurio, F.; Verma, I.M. Enhanced Nf-Kappab Activation and Cellular Function in Macrophages Lacking Ikappab Kinase 1 (Ikk1). Proc. Natl. Acad. Sci. USA 2005, 102, 12425–12430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, L.; Tong, X.; Ye, X. Casein Kinase 1gamma1 Inhibits the Rig-I/Tlr Signaling Pathway through Phosphorylating P65 and Promoting Its Degradation. J. Immunol. 2014, 192, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.E.; Ihekwaba, A.E.C.; Elliott, M.; Johnson, J.R.; Gibney, C.A.; Foreman, B.E.; Nelson, G.; See, V.; Horton, C.A.; Spiller, D.G.; et al. Oscillations in Nf-Κb Signaling Control the Dynamics of Gene Expression. Science 2004, 306, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.C.; Ramaswami, S.; Juvekar, A.; Vu, H.Y.; Galdieri, L.; Davidson, D.; Vancurova, I. Gene-Specific Repression of Proinflammatory Cytokines in Stimulated Human Macrophages by Nuclear Ikappabalpha. J. Immunol. 2010, 185, 3685–3693. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.Y.; Barberi, T.J.; Ghosh, P.; Longo, D.L. Phosphorylation of Rela/P65 on Serine 536 Defines an Iκbα-Independent Nf-Κb Pathway. J. Biol. Chem. 2005, 280, 34538–34547. [Google Scholar] [CrossRef] [PubMed]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory Syncytial Virus-Mediated Nf-Kappa B P65 Phosphorylation at Serine 536 Is Dependent on Rig-I, Traf6, and Ikk Beta. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Indukuri, H.; Liu, T.; Liao, S.L.; Tian, B.; Brasier, A.R.; Garofalo, R.P.; Casola, A. Ikkepsilon Modulates Rsv-Induced Nf-Kappab-Dependent Gene Transcription. Virology 2010, 408, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Moreno, R.; Sobotzik, J.M.; Schultz, C.; Schmitz, M.L. Specification of the Nf-Kappab Transcriptional Response by P65 Phosphorylation and Tnf-Induced Nuclear Translocation of Ikk Epsilon. Nucleic Acids Res. 2010, 38, 6029–6044. [Google Scholar] [CrossRef] [PubMed]
- Bohuslav, J.; Chen, L.-f.; Kwon, H.; Mu, Y.; Greene, W.C. P53 Induces Nf-Κb Activation by an Iκb Kinase-Independent Mechanism Involving Phosphorylation of P65 by Ribosomal S6 Kinase 1. J. Biol. Chem. 2004, 279, 26115–26125. [Google Scholar] [CrossRef] [PubMed]
- Pringle, L.M.; Young, R.; Quick, L.; Riquelme, D.N.; Oliveira, A.M.; May, M.J.; Chou, M.M. Atypical Mechanism of Nf-Kappab Activation by Tre17/Ubiquitin-Specific Protease 6 (Usp6) Oncogene and Its Requirement in Tumorigenesis. Oncogene 2012, 31, 3525–3535. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, J.N.; Poghosyan, S.; Hontelez, S.; Louche, P.; Looman, M.W.G.; Ansems, M.; Adema, G.J. Dc-Script Regulates Il-10 Production in Human Dendritic Cells by Modulating Nf-Κbp65 Activation. J. Immunol. 2015, 195, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- O'Shea, J.M.; Perkins, N.D. Thr435 Phosphorylation Regulates Rela (P65) Nf-Kappab Subunit Transactivation. Biochem. J. 2010, 426, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.Y.; Yeh, K.H.; Chuang, S.E.; Song, Y.C.; Cheng, A.L. Suppression of Mek/Erk Signaling Pathway Enhances Cisplatin-Induced Nf-Kappab Activation by Protein Phosphatase 4-Mediated Nf-Kappab P65 Thr Dephosphorylation. J. Biol. Chem. 2004, 279, 26143–26148. [Google Scholar] [CrossRef] [PubMed]
- Buss, H.; Dörrie, A.; Schmitz, M.L.; Frank, R.; Livingstone, M.; Resch, K.; Kracht, M. Phosphorylation of Serine 468 by Gsk-3β Negatively Regulates Basal P65 Nf-Κb Activity. J. Biol. Chem. 2004, 279, 49571–49574. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, I.; Geng, H.; Sebald, A.; Hodel, M.; Bucher, C.; Kracht, M.; Schmitz, M.L. Inducible Phosphorylation of Nf-Κb P65 at Serine 468 by T Cell Costimulation Is Mediated by Ikkϵ. J. Biol. Chem. 2006, 281, 6175–6183. [Google Scholar] [CrossRef] [PubMed]
- Renner, F.; Moreno, R.; Schmitz, M.L. Sumoylation-Dependent Localization of Ikkepsilon in Pml Nuclear Bodies Is Essential for Protection against DNA-Damage-Triggered Cell Death. Mol. Cell 2010, 37, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Sakurai, H. Ikkbeta Phosphorylates P65 at S468 in Transactivaton Domain 2. FASEB J. 2005, 19, 1758–1760. [Google Scholar] [PubMed]
- Mao, X.; Gluck, N.; Li, D.; Maine, G.N.; Li, H.; Zaidi, I.W.; Repaka, A.; Mayo, M.W.; Burstein, E. Gcn5 Is a Required Cofactor for a Ubiquitin Ligase That Targets Nf-Κb/Rela. Gene. Dev. 2009, 23, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Wittwer, T.; Dittrich-Breiholz, O.; Kracht, M.; Schmitz, M.L. Phosphorylation of Nf-Κb P65 at Ser468 Controls Its Commd1-Dependent Ubiquitination and Target Gene-Specific Proteasomal Elimination. EMBO Rep. 2009, 10, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Witty, J.M.; Rocha, S.; Perkins, N.D. Cisplatin Mimics Arf Tumor Suppressor Regulation of Rela (P65) Nuclear Factor-Kappab Transactivation. Cancer Res. 2006, 66, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Msaki, A.; Sanchez, A.M.; Koh, L.F.; Barre, B.; Rocha, S.; Perkins, N.D.; Johnson, R.F. The Role of Rela (P65) Threonine 505 Phosphorylation in the Regulation of Cell Growth, Survival, and Migration. Mol. Biol. Cell 2011, 22, 3032–3040. [Google Scholar] [CrossRef] [PubMed]
- Moles, A.; Butterworth, J.A.; Sanchez, A.; Hunter, J.E.; Leslie, J.; Sellier, H.; Tiniakos, D.; Cockell, S.J.; Mann, D.A.; Oakley, F.; et al. A Rela(P65) Thr505 Phospho-Site Mutation Reveals an Important Mechanism Regulating Nf-Kappab-Dependent Liver Regeneration and Cancer. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.A.; Schooley, K.; Dower, S.K.; Hagen, H.; Virca, G.D. Activation of Nuclear Transcription Factor Nf-Kappab by Interleukin-1 Is Accompanied by Casein Kinase Ii-Mediated Phosphorylation of the P65 Subunit. J. Biol. Chem. 1997, 272, 32606–32612. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S., Jr. Tumor Necrosis Factor Alpha-Induced Phosphorylation of Rela/P65 on Ser529 Is Controlled by Casein Kinase Ii. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Baldwin, A.S. Activation of Nuclear Factor-Κb-Dependent Transcription by Tumor Necrosis Factor-Α Is Mediated through Phosphorylation of Rela/P65 on Serine 529. J. Biol. Chem. 1998, 273, 29411–29416. [Google Scholar] [CrossRef] [PubMed]
- Bristow, C.L.; Wolkowicz, R.; Trucy, M.; Franklin, A.; Di Meo, F.; Kozlowski, M.T.; Winston, R.; Arnold, R.R. Nf-Kappab Signaling, Elastase Localization, and Phagocytosis Differ in Hiv-1 Permissive and Nonpermissive U937 Clones. J. Immunol. 2008, 180, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Bijli, K.M.; Fazal, F.; Minhajuddin, M.; Rahman, A. Activation of Syk by Protein Kinase C-Δ Regulates Thrombin-Induced Intercellular Adhesion Molecule-1 Expression in Endothelial Cells Via Tyrosine Phosphorylation of Rela/P65. J. Biol. Chem. 2008, 283, 14674–14684. [Google Scholar] [CrossRef] [PubMed]
- Bijli, K.M.; Minhajuddin, M.; Fazal, F.; O'Reilly, M.A.; Platanias, L.C.; Rahman, A. C-Src Interacts with and Phosphorylates Rela/P65 to Promote Thrombin-Induced Icam-1 Expression in Endothelial Cells. Am. J. Physiol.-Lung C. 2007, 292, L396–L404. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Gerondakis, S. The C-Rel Transcription Factor in Development and Disease. Gene Cancer 2011, 2, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Gapuzan, M.-E.R.; Pitoc, G.A.; Gilmore, T.D. Mutations within a Conserved Protein Kinase a Recognition Sequence Confer Temperature-Sensitive and Partially Defective Activities onto Mouse C-Rel. Biochem. Biophys. Res. Commun. 2003, 307, 92–99. [Google Scholar] [CrossRef]
- Harris, J.; Oliere, S.; Sharma, S.; Sun, Q.; Lin, R.; Hiscott, J.; Grandvaux, N. Nuclear Accumulation of Crel Following C-Terminal Phosphorylation by Tbk1/Ikk Epsilon. J. Immunol. 2006, 177, 2527–2535. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Neumann, M.; Okuda, K.; Franza, B.R.; Griffin, J.D. Rel Is Rapidly Tyrosine-Phosphorylated Following Granulocyte-Colony-Stimulating Factor Treatment of Human Neutrophils. J. Biol. Chem. 1994, 269, 5387–5390. [Google Scholar] [PubMed]
- Neumann, M.; Tsapos, K.; Scheppler, J.A.; Ross, J.; Franza, B.R., Jr. Identification of Complex Formation between Two Intracellular Tyrosine Kinase Substrates: Human C-Rel and the P105 Precursor of P50 Nf-Kappa B. Oncogene 1992, 7, 2095–2104. [Google Scholar] [PubMed]
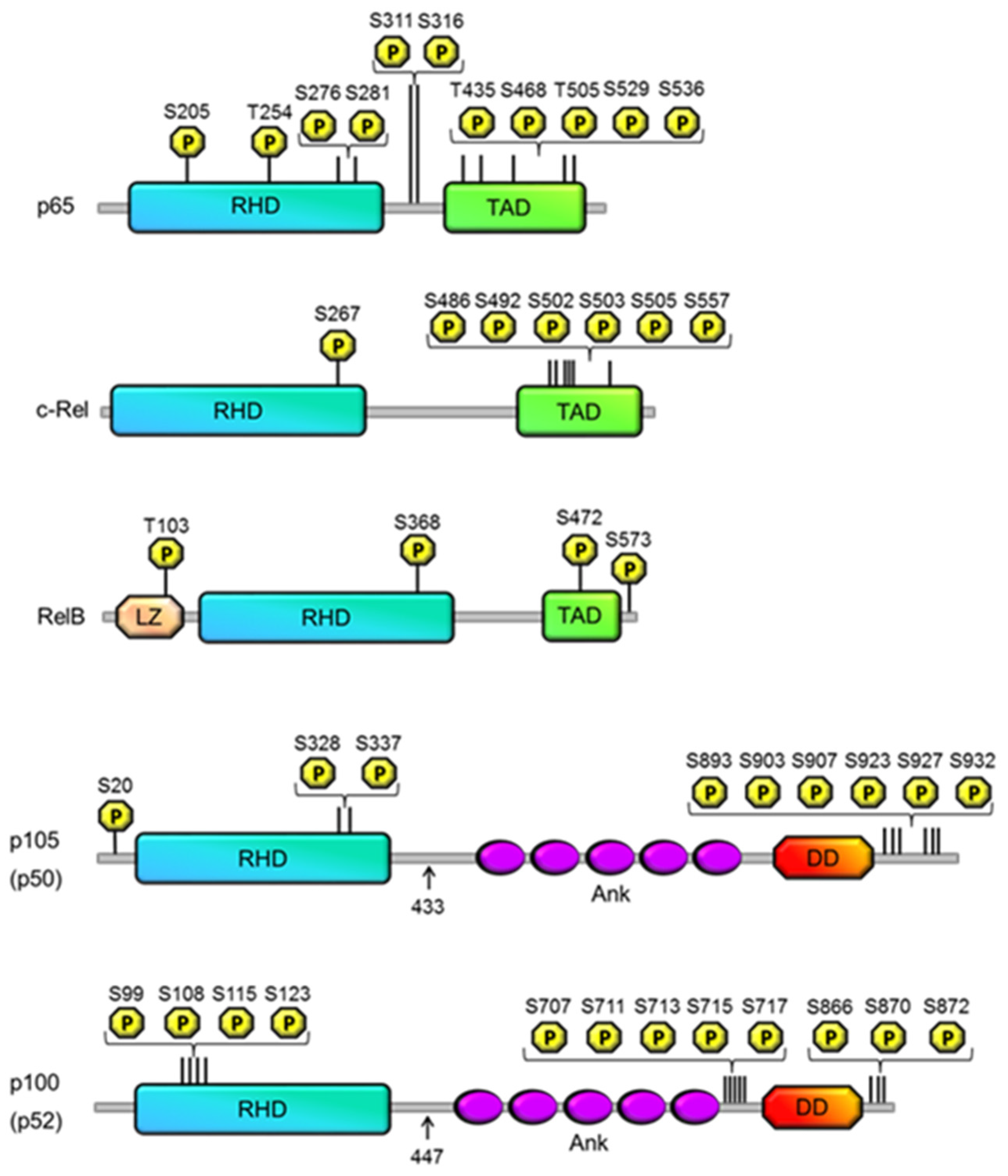

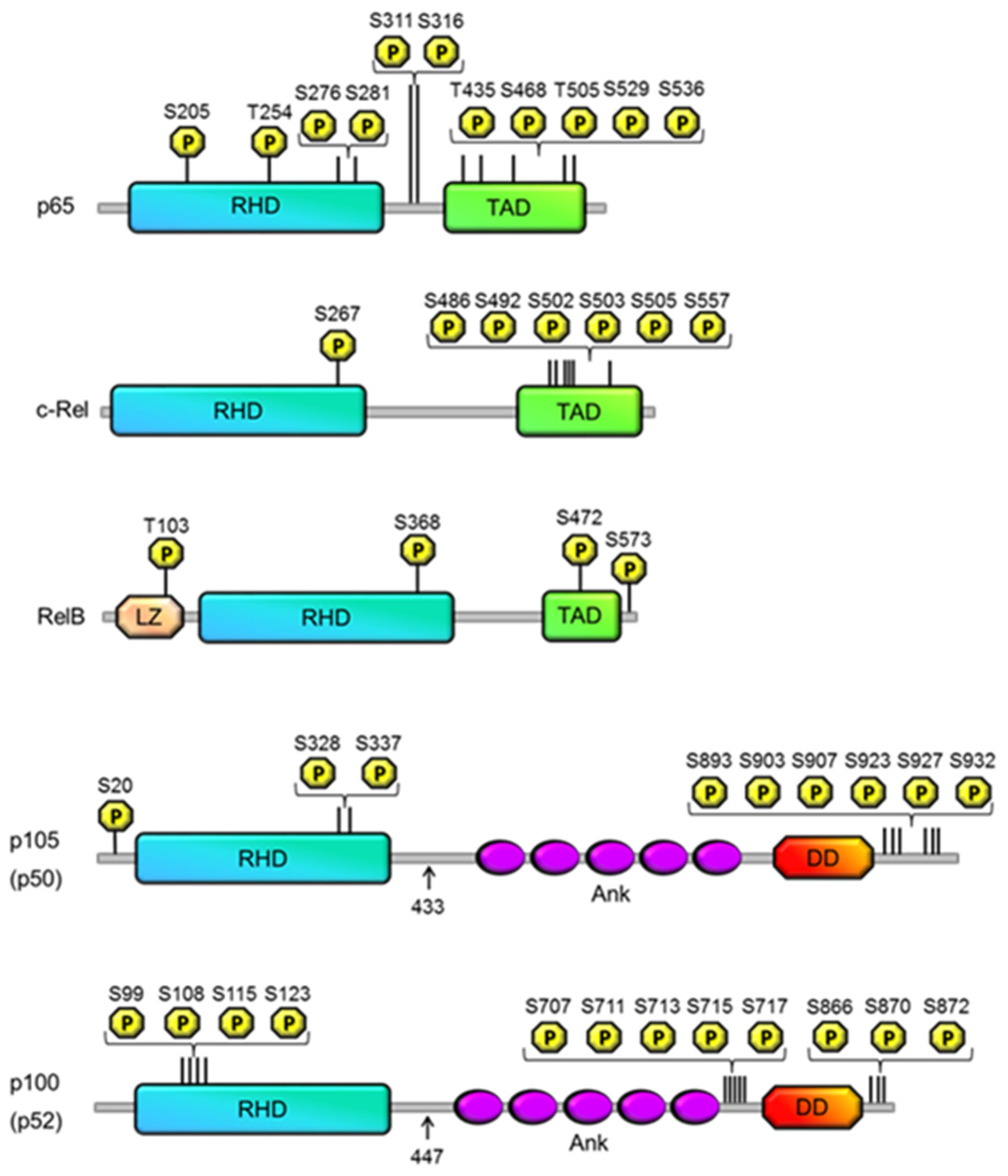{kind=link}
| Site | Kinase | Effect | Reference |
|---|---|---|---|
| p50 | |||
| S20 | DNA-dependent PKA | DNA binding specificity | [25] |
| S328 | Chk1 | DNA binding specificity | [26,27,28] |
| S337 | Protein kinase A | Enhanced binding affinity | [23,24] |
| p105 | |||
| S893 | Unknown | p105 processing | [13] |
| S903 | GSK3β | Resting state: Stabilization TNF-α induced state: ubiquitination | [19] |
| S907 | GSK3β | Resting state: Stabilization TNF-α induced state: ubiquitination | [13,19] |
| S923 | IKKβ | Ubiquitination | [14,15] |
| S927 | IKKβ | Ubiquitination | [17] |
| S932 | IKKβ | Ubiquitination | [14,17] |
| p100 | |||
| S99 | IKKα | Ubiquitination | [32] |
| S108 | IKKα | Ubiquitination | [32] |
| S115 | IKKα | Ubiquitination | [32] |
| S870 | Unknown | IKKα recruitment | [32,33,34] |
| S872 | IKKα | Ubiquitination | [32] |
| S707 | GSK3β | Ubiquitination | [35] |
| S866 | Unknown | IKKα recruitment | [32,33,34] |
| S711 | GSK3β | Ubiquitination | [35] |
| S123 | IKKα | Ubiquitination | [32] |
| S713 | unknown | Ubiquitination | [36] |
| S715 | unknown | Ubiquitination | [36] |
| S717 | unknown | Ubiquitination | [36] |
| Site | Kinase | Effect | Reference |
|---|---|---|---|
| RelB | |||
| T103 | Unknown | Degradation | [44] |
| S390 | Unknown | Dimerization | [43] |
| S573 | GSK-3β | Degradation | [44,45] |
| S472 | IKK complex | DNA binding specificity | [46] |
| c-Rel | |||
| S267 | PKA (in vitro only) | stimulates transcriptional activity, nuclear localisation | [47,48] |
| S486, S492, S502, S503, S505 | NIK? | TNFα-induced transactivation activity | [49,50] |
| S503 | PKCζ, NIK? | TNFα-induced transactivation activity | [51] |
| S557 | IKKα/β (in vitro only) | transactivation activity | [52,53] |
| Site | Kinase | Effect | Reference |
|---|---|---|---|
| S205 | unknown | transactivation | [76] |
| T254 | unknown | prolyl isomerisation, transactivation | [77,78] |
| S276 | PKA-C, | transactivation, K310 acetylation | [55,56,58] |
| MSK1, | [66,67,68,69] | ||
| MSK2, | [70] | ||
| Pim-1, | [75] | ||
| RSK p90, | [71] | ||
| PKCα | [72] | ||
| S281 | unknown | transactivation | [73,76] |
| S311 | PKCζ | K310 acetylation, transactivation | [79,80,81,82] |
| S316 | unknown | transactivation | [83] |
| T435 | unknown (CK2, PLK1?) | transactivation | [101,102] |
| S468 | GSK3β | Inhibition | [103] |
| IKKε | transactivation | [104] | |
| IKKβ | slight inhibition | [106] | |
| T505 | Chk1 | inhibition | [105,109,110] |
| S529 | CK2 | transactivation | [112,113,114,115] |
| S536 | IKKβ, | transactivation, K310 acetylation | [84,86,87,88,95] |
| RSK1, | [84] | ||
| IKKα, | [84,85,86,87] | ||
| IKKε, | [84,96] | ||
| NAK/TBK1 | [84] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. https://doi.org/10.3390/cells5010012
Christian F, Smith EL, Carmody RJ. The Regulation of NF-κB Subunits by Phosphorylation. Cells. 2016; 5(1):12. https://doi.org/10.3390/cells5010012
Chicago/Turabian StyleChristian, Frank, Emma L. Smith, and Ruaidhrí J. Carmody. 2016. "The Regulation of NF-κB Subunits by Phosphorylation" Cells 5, no. 1: 12. https://doi.org/10.3390/cells5010012
APA StyleChristian, F., Smith, E. L., & Carmody, R. J. (2016). The Regulation of NF-κB Subunits by Phosphorylation. Cells, 5(1), 12. https://doi.org/10.3390/cells5010012