
Hypoxia Induced NF-κB



{kind=link}
Abstract
:1. Introduction
2. Pathways for NF-κB Activation, Canonical, Non-Canonical and Atypical
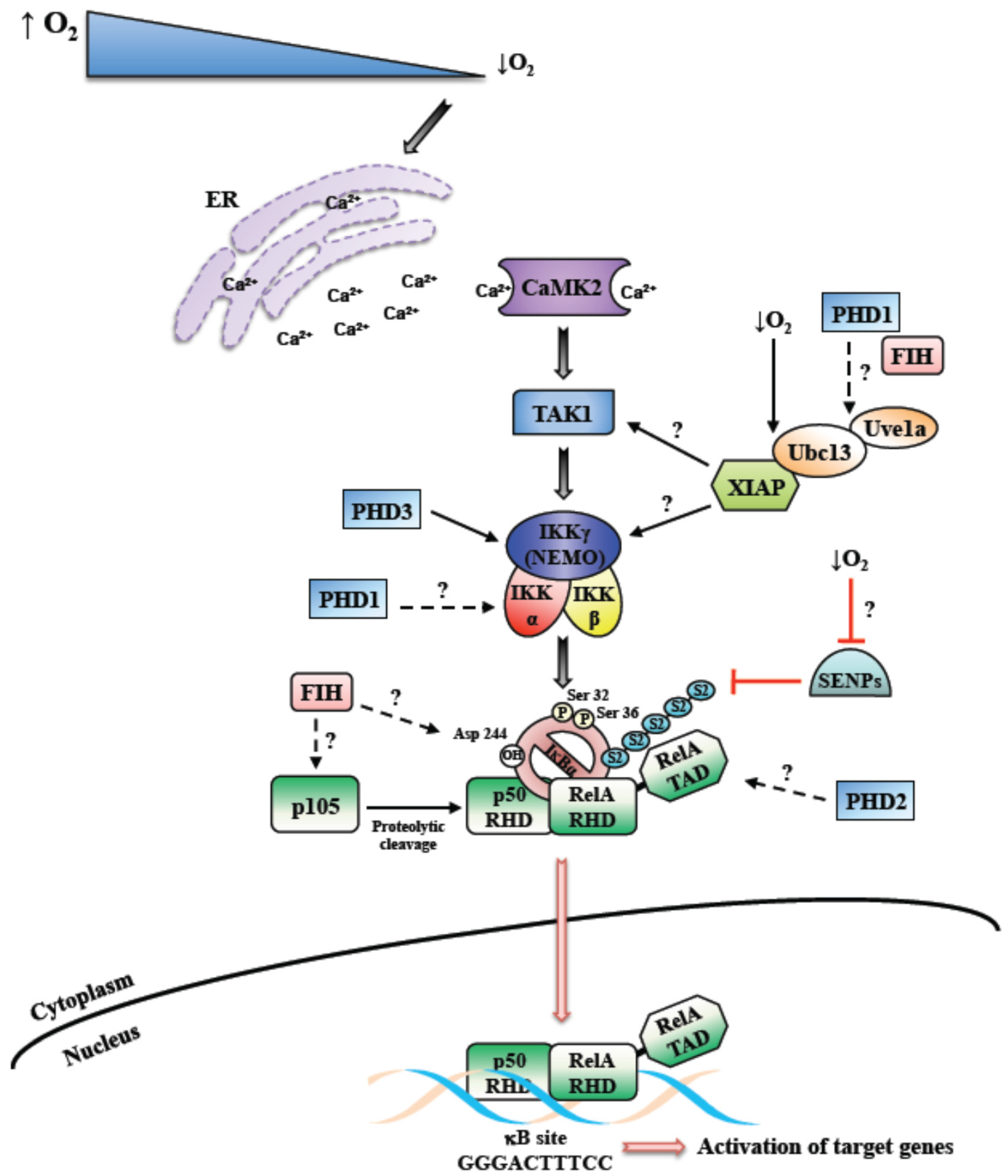
3. Activation of NF-κB by Hypoxia, Role of PHDs and FIH
4. Activation of NF-κB by Hypoxia, Role of TAK and IKK
5. Activation of NF-κB by Hypoxia, Modulation of IκB
6. Role of NF-κB in the Cellular Response to Hypoxia
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Prabhakar, N.R.; Semenza, G.L. Oxygen sensing and homeostasis. Physiology (Bethesda) 2015, 30, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Kenneth, N.S.; Rocha, S. Regulation of gene expression by hypoxia. Biochem. J. 2008, 414, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Moniz, S.; Biddlestone, J.; Rocha, S. Grow(2): The HIF system, energy homeostasis and the cell cycle. Histol. Histopathol. 2014, 29, 589–600. [Google Scholar] [PubMed]
- Myllyharju, J. Prolyl 4-hydroxylases, master regulators of the hypoxia response. Acta Physiol. (Oxf.) 2013, 208, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Lisy, K.; Peet, D.J. Turn me on: Regulating HIF transcriptional activity. Cell Death Differ. 2008, 15, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef] [PubMed]
- Bandarra, D.; Rocha, S. A tale of two transcription factors: NF-kappab and hif crosstalk. OA Mol. Cell Biol. 2013, 1, 1–7. [Google Scholar] [CrossRef]
- Biddlestone, J.; Bandarra, D.; Rocha, S. The role of hypoxia in inflammatory disease (review). Int. J. Mol. Med. 2015, 35, 859–869. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-kappaB and HIF crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [PubMed]
- Hinz, M.; Scheidereit, C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and ikk function. Nat. Rev. Mol. Cell. Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K. Diverse roles of the ubiquitin system in NF-kappaB activation. Biochim Biophys. Acta 2014, 1843, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G. Tumor suppressor cyld: Negative regulation of NF-kappaB signaling and more. Cell Mol. Life Sci. 2008, 65, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F. Linear ubiquitination-mediated NF-kappaB regulation and its related disorders. J. Biochem. 2013, 154, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Napetschnig, J.; Wu, H. Molecular basis of NF-kappaB signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S. Nuclear initiated NF-kappaB signaling: Nemo and atm take center stage. Cell. Res. 2011, 21, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Fandrey, J.; Gorr, T.A.; Gassmann, M. Regulating cellular oxygen sensing by hydroxylation. Cardiovasc. Res. 2006, 71, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Xiao, H.; Ding, F.; Zhong, H.; Zhu, J.; Ma, N.; Mei, J. Over-expression of prolyl hydroxylase-1 blocks NF-kappaB-mediated cyclin D1 expression and proliferation in lung carcinoma cells. Cancer Genet. 2014, 207, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Taubman, M.B. Egln3 inhibition of NF-kappaB is mediated by prolyl hydroxylase-independent inhibition of IkappaB kinase gamma ubiquitination. Mol. Cell Biol. 2013, 33, 3050–3061. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Cavadas, M.A.; Tambuwala, M.M.; Hams, E.; Rodriguez, J.; von Kriegsheim, A.; Cotter, P.; Bruning, U.; Fallon, P.G.; Cheong, A.; et al. Regulation of IL-1beta-induced NF-kappaB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 18490–18495. [Google Scholar] [CrossRef] [PubMed]
- Culver, C.; Sundqvist, A.; Mudie, S.; Melvin, A.; Xirodimas, D.; Rocha, S. Mechanism of hypoxia-induced NF-kappaB. Mol. Cell Biol. 2010, 30, 4901–4921. [Google Scholar] [CrossRef] [PubMed]
- Melvin, A.; Mudie, S.; Rocha, S. Further insights into the mechanism of hypoxia-induced NFkappaB. [corrected]. Cell Cycle 2011, 10, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, W.; Jiang, S.; Ye, W.; Yang, H.; Shapiro, I.M.; Risbud, M.V. Prolyl-4-hydroxylase domain protein 2 controls NF-kappaB/p65 transactivation and enhances the catabolic effects of inflammatory cytokines on cells of the nucleus pulposus. J. Biol. Chem. 2015, 290, 7195–7207. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Lancaster, D.E.; Stolze, I.P.; Hewitson, K.S.; McDonough, M.A.; Coleman, M.L.; Coles, C.H.; Yu, X.; Hay, R.T.; Ley, S.C.; et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc. Natl. Acad. Sci. USA 2006, 103, 14767–14772. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Rodriguez, J.; Pickel, C.; Burr, S.; Fabrizio, J.A.; Nolan, K.A.; Spielmann, P.; Cavadas, M.A.; Crifo, B.; Halligan, D.N.; et al. Fih regulates cellular metabolism through hydroxylation of the deubiquitinase otub1. PLoS Biol. 2016, 14, e1002347. [Google Scholar] [CrossRef] [PubMed]
- Devries, I.L.; Hampton-Smith, R.J.; Mulvihill, M.M.; Alverdi, V.; Peet, D.J.; Komives, E.A. Consequences of IkappaB alpha hydroxylation by the factor inhibiting HIF (FIH). FEBS Lett. 2010, 584, 4725–4730. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Gilmore, T.D. Good cop, bad cop: The different faces of NF-kappaB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Ho, W.; Banerjee, A. Unravelling the complexities of the NF-kappaB signalling pathway using mouse knockout and transgenic models. Oncogene 2006, 25, 6781–6799. [Google Scholar] [CrossRef] [PubMed]
- Koong, A.C.; Chen, E.Y.; Giaccia, A.J. Hypoxia causes the activation of nuclear factor kappa B through the phosphorylation of I kappa b alpha on tyrosine residues. Cancer Res. 1994, 54, 1425–1430. [Google Scholar] [PubMed]
- Fitzpatrick, S.F.; Tambuwala, M.M.; Bruning, U.; Schaible, B.; Scholz, C.C.; Byrne, A.; O’Connor, A.; Gallagher, W.M.; Lenihan, C.R.; Garvey, J.F.; et al. An intact canonical NF-kappaB pathway is required for inflammatory gene expression in response to hypoxia. J. Immunol. 2011, 186, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Bandarra, D.; Biddlestone, J.; Mudie, S.; Muller, H.A.; Rocha, S. Hypoxia activates IKK-NF-kappaB and the immune response in drosophila melanogaster. Biosci. Rep. 2014, 34, e00127. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; Xu, M.; Chen, Z.J. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 2007, 26, 3214–3226. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Wong, E.T.; Shi, Y.; Niu, J.; Chen, Z.; Miyamoto, S.; Tergaonkar, V. Atm- and nemo-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol. Cell 2010, 40, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israel, A.; Wallach, D.; Courtois, G. The tumour suppressor cyld negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Mo, D.; Liu, H.; Veena, M.S.; Srivatsan, E.S.; Massoumi, R.; Rettig, M.B. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell 2008, 14, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappab family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. Sumo-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef]
- Liu, Q.; Li, J.; Khoury, J.; Colgan, S.P.; Ibla, J.C. Adenosine signaling mediates SUMO-1 modification of IkappaBalpha during hypoxia and reoxygenation. J. Biol. Chem. 2009, 284, 13686–13695. [Google Scholar] [CrossRef] [PubMed]
- Aillet, F.; Lopitz-Otsoa, F.; Egana, I.; Hjerpe, R.; Fraser, P.; Hay, R.T.; Rodriguez, M.S.; Lang, V. Heterologous SUMO-2/3-ubiquitin chains optimize IkappaBalpha degradation and NF-kappaB activity. PLoS ONE 2012, 7, e51672. [Google Scholar] [CrossRef] [PubMed]
- Mulero, M.C.; Ferres-Marco, D.; Islam, A.; Margalef, P.; Pecoraro, M.; Toll, A.; Drechsel, N.; Charneco, C.; Davis, S.; Bellora, N.; et al. Chromatin-bound IkappaBalpha regulates a subset of polycomb target genes in differentiation and cancer. Cancer Cell 2013, 24, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Vegran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter mct1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.J.; Gallagher, R.; Seaton, A.; Wilson, C.; Scullin, P.; Pettigrew, J.; Stratford, I.J.; Williams, K.J.; Johnston, P.G.; Waugh, D.J. HIF-1 and NF-kappaB-mediated upregulation of CXCR1 and CXCR2 expression promotes cell survival in hypoxic prostate cancer cells. Oncogene 2007, 26, 7333–7345. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of nf-kappab in the heart: To be or not to NF-kappaB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, J.N.; Qiu, J.; Rassin, D.; Grafe, M.; Wood, T.; Perez-Pol, J.R. Transcriptional regulation of the BCL-X gene by NF-kappaB is an element of hypoxic responses in the rat brain. Neurochem. Res. 2001, 26, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Sarnico, I.; Lanzillotta, A.; Boroni, F.; Benarese, M.; Alghisi, M.; Schwaninger, M.; Inta, I.; Battistin, L.; Spano, P.; Pizzi, M. NF-kappaB p50/rela and c-Rel-containing dimers: Opposite regulators of neuron vulnerability to ischaemia. J. Neurochem. 2009, 108, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, Y.; Qian, Y.; Zhang, H.; Guo, S.; Sunagawa, M.; Hisamitsu, T.; Liu, Y. Interleukin-17a promotes rheumatoid arthritis synoviocytes migration and invasion under hypoxia by increasing mmp2 and mmp9 expression through NF-kappaB/HIF-1alpha pathway. Mol. Immunol. 2013, 53, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K. Camp-response element-binding protein (creb) and NF-kappaB transcription factors are activated during prolonged hypoxia and cooperatively regulate the induction of matrix metalloproteinase mmp1. J. Biol. Chem. 2013, 288, 22584–22595. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Ma, L.; Zhang, J.; Huang, K.; Yang, Q.; Guo, Y.Y.; Liu, S.B.; Liu, Y.H.; Wu, Y.M. The migration of neural progenitor cell mediated by SDF-1 is NF-kappaB/HIF-1alpha dependent upon hypoxia. CNS Neurosci. Ther. 2013, 19, 145–153. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Ignazio, L.; Rocha, S. Hypoxia Induced NF-κB. Cells 2016, 5, 10. https://doi.org/10.3390/cells5010010
D’Ignazio L, Rocha S. Hypoxia Induced NF-κB. Cells. 2016; 5(1):10. https://doi.org/10.3390/cells5010010
Chicago/Turabian StyleD’Ignazio, Laura, and Sonia Rocha. 2016. "Hypoxia Induced NF-κB" Cells 5, no. 1: 10. https://doi.org/10.3390/cells5010010
APA StyleD’Ignazio, L., & Rocha, S. (2016). Hypoxia Induced NF-κB. Cells, 5(1), 10. https://doi.org/10.3390/cells5010010