
B-Cells and Plasmablasts as Architects of Autoimmune Disease: From Molecular Footprints to Precision Therapeutics

Highlights
- B-cell populations in systemic autoimmune diseases can be classified into distinct immunological endotypes—extrafollicular/IFN-high, germinal center/plasma cell-anchored, BAFF-dependent, and tissue-conditioned/fibro-inflammatory—each characterized by specific biomarker signatures, autoantibody profiles, and biological vulnerabilities.
- IgG subclass distribution (IgG1/IgG3 versus IgG4 predominance) critically determines therapeutic response patterns, with IgG4-mediated diseases showing rapid responses to B-cell depletion due to short-lived plasmablast dependence, while IgG1/IgG3-dominant diseases often resist anti-CD20 therapy due to long-lived plasma cell persistence.
- Endotype-based patient stratification enables mechanism-aligned therapeutic selection—directing JAK inhibitors and anti-CD19 therapies toward extrafollicular-dominant patients, proteasome inhibitors or CAR-T towards plasma cell-anchored disease, and BAFF inhibitors toward BAFF-dependent phenotypes—potentially improving response rates and reducing treatment failures.
- The convergence of B-cell endotyping frameworks across organ-specific and systemic autoimmune diseases suggests that shared immunological architectures, rather than traditional diagnostic boundaries, may better guide precision medicine approaches in clinical practice.
Abstract
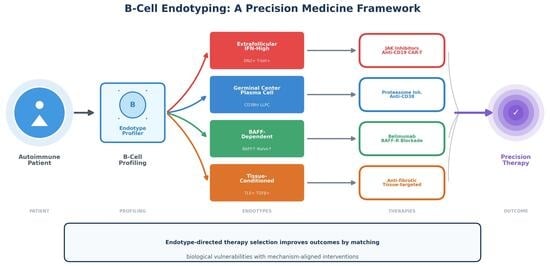
1. Introduction: The B-Cell Renaissance in Autoimmune Disease
2. B-Cell Biology Revisited: From Subsets to High-Dimensional Ecosystems
2.1. Determinants of B-Cell Fate
2.2. Germinal Center and Extrafollicular Circuits
2.3. T-Bet+ and Double-Negative B-Cells as Pathogenic Intermediates
2.4. From Cellular Taxonomy to Systems Immunology
3. B-Cell Ecosystems in Human Autoimmunity
3.1. Human Single-Cell Mapping of B-Cell States
3.2. Tissue Niches and Tertiary Lymphoid Structures
3.3. Clonal Architecture and Immune Repertoire
3.4. Extrafollicular Circuits as Shared Autoimmune Modules
3.5. From Ecosystems to Endotypes
4. Plasmablasts as Dynamic Biomarkers of Immune Activation
5. Autoantibodies as Molecular Footprints of B-Cell Programs
6. Translational Scenarios: How B-Cell Endotypes Shape Disease Architecture
6.1. Scenario A: Interferon High, Extrafollicular Dominant Architecture
6.2. Scenario B: Tissue Anchored, Stromal Conditioned and Plasma Cell Supported Architecture
7. B-Cell-Directed Therapies as Endotype-Dependent Biological Interventions
7.1. Anti-CD20 Monoclonal Antibodies
7.2. BAFF and BAFF-R Inhibition
7.3. Interference with Interferon-Dependent Circuitry
7.4. Plasma-Cell-Directed Vulnerabilities
7.5. Immune Reset with CAR-T Therapy
7.6. From Treatment Classes to Biologically Defined Vulnerability
7.7. IgG Subclass-Dependent Therapeutic Outcomes
8. Beyond B-Cell Depletion: Complementary Immunomodulatory Strategies
8.1. Modulation of T-Cell Help and Immune Regulation
8.2. FcRn-Mediated IgG Clearance: Targeting Autoantibody Persistence
8.3. Antigen-Specific Tolerization: Reprogramming Immune Memory
9. Practical Challenges for Clinical Implementation of B-Cell Endotyping
9.1. Accessibility of Advanced Immunophenotyping Technologies
9.2. Standardization and Inter-Laboratory Reproducibility
9.3. Integration of Multidimensional Data and Clinical Interpretation
10. Disease-Specific B-Cell Signatures
11. Future Perspectives: Actionable Implementation Steps
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | Age-associated B-cell |
| AAV | ANCA-associated vasculitis |
| AI | Artificial intelligence |
| APRIL | A proliferation-inducing ligand |
| BAFF | B-cell activating factor |
| BAFF-R | BAFF receptor |
| BCMA | B-cell maturation antigen |
| BCR | B-cell receptor |
| BM | Bone marrow |
| CAR-T | Chimeric antigen receptor T-cell |
| CCL | Chemokine (C–C motif) ligand |
| CD | Cluster of differentiation |
| CDC | Complement-dependent cytotoxicity |
| CRS | Cytokine release syndrome |
| DN2 | Double-negative 2 B-cell |
| DsDNA | Double-stranded DNA |
| ECM | Extracellular matrix |
| EF | Extrafollicular |
| ELISA | Enzyme-linked immunosorbent assay |
| FcRn | Neonatal Fc receptor |
| FDC | Follicular dendritic cell |
| GC | Germinal center |
| GOF | Gain of function |
| HSCT | Hematopoietic stem cell transplantation |
| IIM | Idiopathic inflammatory myopathy |
| IFN | Interferon |
| ISG | Interferon-stimulated gene |
| JAK | Janus kinase |
| LLPC | Long-lived plasma cell |
| MCTD | Mixed connective tissue disease |
| MHC | Major histocompatibility complex |
| MSA | Myositis-specific autoantibodies |
| mTOR | Mechanistic target of rapamycin |
| NLE | Neonatal lupus erythematosus |
| NK | Natural killer cell |
| PAH | Pulmonary arterial hypertension |
| PB | Plasmablast |
| PC | Plasma cell |
| pSjD | Primary Sjögren’s disease |
| RA | Rheumatoid arthritis |
| RNA-seq | RNA sequencing |
| RP-ILD | Rapidly progressive interstitial lung disease |
| RTX | Rituximab |
| ScRNA-seq | Single-cell RNA sequencing |
| SDC | Syndecan 1 (CD138) |
| SHM | Somatic hypermutation |
| SLE | Systemic lupus erythematosus |
| SSc | Systemic sclerosis |
| STAT | Signal transducer and activator of transcription |
| TCR | T-cell receptor |
| Tfh | T follicular helper cell |
| TGF-β | Transforming growth factor beta |
| TLS | Tertiary lymphoid structure |
| TLR | Toll-like receptor |
| Treg | Regulatory T-cell |
| V(D)J | Variable, diversity and joining |
| VTE | Venous thromboembolism |
References
- Shlomchik, M.J. Sites and stages of autoreactive B cell activation and regulation. Immunity 2008, 28, 18–28. [Google Scholar] [CrossRef]
- Rawlings, D.J.; Metzler, G.; Wray-Dutra, M.; Jackson, S.W. Altered B cell signalling in autoimmunity. Nat. Rev. Immunol. 2017, 17, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Dorner, T.; Jacobi, A.M.; Lipsky, P.E. B cells in autoimmunity. Arthritis Res. Ther. 2009, 11, 247. [Google Scholar]
- Corsiero, E.; Nerviani, A.; Bombardieri, M.; Pitzalis, C. Ectopic Lymphoid Structures: Powerhouse of Autoimmunity. Front. Immunol. 2016, 7, 430. [Google Scholar] [CrossRef] [PubMed]
- Asam, S.; Nayar, S.; Gardner, D.; Barone, F. Stromal cells in tertiary lymphoid structures: Architects of autoimmunity. Immunol. Rev. 2021, 302, 184–195. [Google Scholar] [CrossRef]
- Cyster, J.G.; Allen, C.D.C. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 2019, 177, 524–540. [Google Scholar] [CrossRef]
- Elsner, R.A.; Shlomchik, M.J. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 2020, 53, 1136–1150. [Google Scholar] [CrossRef] [PubMed]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739.e6. [Google Scholar] [CrossRef]
- Victora, G.D.; Nussenzweig, M.C. Germinal centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef]
- Mesin, L.; Ersching, J.; Victora, G.D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. [Google Scholar] [CrossRef]
- Bannard, O.; Cyster, J.G. Germinal centers: Programmed for affinity maturation and antibody diversification. Curr. Opin. Immunol. 2017, 45, 21–30. [Google Scholar] [CrossRef]
- Eisenbarth, S.C.; Batista, F.; Cyster, J.; Elsner, R.; Kelsoe, G.; Lund, F.E.; Shlomchik, M.J.; Sweet, R.A.; Vinuesa, C.G.; Bhattacharya, D.; et al. A roadmap for defining “extrafollicular” B cell responses. Immunity 2025, 58, 2627–2645. [Google Scholar] [CrossRef]
- Pipi, E.; Nayar, S.; Gardner, D.H.; Colafrancesco, S.; Smith, C.; Barone, F. Tertiary Lymphoid Structures: Autoimmunity Goes Local. Front. Immunol. 2018, 9, 1952. [Google Scholar] [CrossRef] [PubMed]
- Rivellese, F.; Mauro, D.; Nerviani, A.; Pagani, S.; Fossati-Jimack, L.; Messemaker, T.C.; Clark, E.; Rosber, M.K.; Lewis, M.; Bornstein, S.; et al. Mast cells in early rheumatoid arthritis associate with disease severity and support B cell autoantibody production. Ann. Rheum. Dis. 2018, 77, 1773–1781. [Google Scholar] [CrossRef]
- Mueller, S.N.; Germain, R.N. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat. Rev. Immunol. 2009, 9, 618–629. [Google Scholar] [CrossRef]
- Arvidsson, G.; Czarnewski, P.; Johansson, A.; Raine, A.; Imgenberg-Kreuz, J.; Nordlund, J.; Petri, A.; Svenungsson, E.; Cavalli, G.; Nordmark, G.; et al. Multimodal Single-Cell Sequencing of B Cells in Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2024, 76, 255–267. [Google Scholar] [CrossRef]
- Su, C.; Wang, W.; Cheng, F.; Zhao, F.; Zheng, S.G. The role of B cells in Sjogren’s syndrome and their impact on the nervous system. Autoimmun. Rev. 2025, 24, 103852. [Google Scholar] [CrossRef]
- Wu, C.; Jiang, S.; Chen, Z.; Li, T.; Gu, X.; Dai, M.; Chen, X.; Zhang, L.; Tang, H.; Zhou, J.; et al. Single-cell transcriptomics reveal potent extrafollicular B cell response linked with granzyme K(+) CD8 T cell activation in lupus kidney. Ann. Rheum. Dis. 2024, 84, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bai, X.; Zhou, C.; Ouyang, Q.; Zhang, Y.; Zhang, X.; Wang, H.; Li, Z.; Chen, M.; Liu, Y.; et al. Cxcl9(high) macrophages recruit circulating Cxcr3+ plasmablasts into kidneys to promote pathogenesis of lupus nephritis mice. Commun. Biol. 2025, 8, 1446. [Google Scholar] [CrossRef] [PubMed]
- Jenks, S.A.; Cashman, K.S.; Woodruff, M.C.; Lee, F.E.; Sanz, I. Extrafollicular responses in humans and SLE. Immunol. Rev. 2019, 288, 136–148. [Google Scholar] [CrossRef]
- Brown, G.J.; Canete, P.F.; Wang, H.; Medhavy, A.; Bones, J.; Roco, J.A.; He, Y.; Lawber, K.E.; Burgio, G.; Almeida-Santos, J.; et al. TLR7 gain-of-function genetic variation causes human lupus. Nature 2022, 605, 349–356. [Google Scholar] [CrossRef]
- Mackay, F.; Figgett, W.A.; Saulep, D.; Lepage, M.; Hibbs, M.L. B-cell stage and context-dependent requirements for survival signals from BAFF and the B-cell receptor. Immunol. Rev. 2010, 237, 205–225. [Google Scholar] [CrossRef]
- Meffre, E.; O’Connor, K.C. Impaired B-cell tolerance checkpoints promote the development of autoimmune diseases and pathogenic autoantibodies. Immunol. Rev. 2019, 292, 90–101. [Google Scholar] [CrossRef]
- Li, J.; Zhao, M.; Luo, W.; Huang, J.; Zhao, B.; Zhou, Z. B cell metabolism in autoimmune diseases: Signaling pathways and interventions. Front. Immunol. 2023, 14, 1232820. [Google Scholar] [CrossRef] [PubMed]
- Boothby, M.; Rickert, R.C. Metabolic Regulation of the Immune Humoral Response. Immunity 2017, 46, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Nussenzweig, M.C. Germinal Centers. Annu. Rev. Immunol. 2022, 40, 413–442. [Google Scholar] [CrossRef]
- Radbruch, A.; Muehlinghaus, G.; Luger, E.O.; Inamine, A.; Smith, K.G.; Dorner, T.; Heer, A.K.; Berek, C. Competence and competition: The challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 2006, 6, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Moser, K.; Tokoyoda, K.; Radbruch, A.; MacLennan, I.; Manz, R.A. Stromal niches, plasma cell differentiation and survival. Curr. Opin. Immunol. 2006, 18, 265–270. [Google Scholar] [CrossRef]
- Pyzik, M.; Kozicky, L.K.; Gandhi, A.K.; Blumberg, R.S. The therapeutic age of the neonatal Fc receptor. Nat. Rev. Immunol. 2023, 23, 415–432. [Google Scholar] [CrossRef]
- Xiang, N.; Xu, H.; Zhou, Z.; Wang, J.; Cai, P.; Wang, L.; Tang, J.; Wang, X.; Liu, C.; Chen, S.; et al. Single-cell transcriptome profiling reveals immune and stromal cell heterogeneity in primary Sjogren’s syndrome. iScience 2023, 26, 107943. [Google Scholar] [CrossRef]
- Huang, J.; Tang, J.; Zhang, C.; Liu, T.; Deng, Z.; Liu, L. Single-cell transcriptomic analysis uncovers heterogeneity in the labial gland microenvironment of primary Sjogren’s syndrome. J. Transl. Autoimmun. 2024, 9, 100248. [Google Scholar] [CrossRef]
- Inamo, J.; Takeshita, M.; Suzuki, K.; Tsunoda, K.; Usuda, S.; Kuramoto, J.; Moody, J.; Hon, C.-C.; Ando, Y.; Sasaki, T.; et al. Comparative single-cell and spatial profiling of anti-SSA-positive and anti-centromere-positive Sjogren’s disease reveals common and distinct immune activation and fibroblast-mediated inflammation. Nat. Commun. 2025, 16, 8299. [Google Scholar] [CrossRef]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef]
- Dong, Y.; Wang, T.; Wu, H. Tertiary lymphoid structures in autoimmune diseases. Front. Immunol. 2023, 14, 1322035. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, S.M.; Beccaria, C.G.; Iannacone, M.; Linterman, M.A. Tertiary Lymphoid Structures Across Organs: Context, Composition, and Clinical Levers. Immunol. Rev. 2025, 335, e70063. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Qian, D.; Luo, R.; Cheng, Y.; Xu, G.; Ge, S. Identifying potential mechanism and targets for treatment of tertiary lymphoid structure in lupus nephritis based on bioinformatics analysis. Int. Immunopharmacol. 2025, 148, 114084. [Google Scholar] [CrossRef]
- Wang, Q.; Feng, D.; Jia, S.; Lu, Q.; Zhao, M. B-Cell Receptor Repertoire: Recent Advances in Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2024, 66, 76–98. [Google Scholar] [CrossRef]
- Deguine, J.; Xavier, R.J. B cell tolerance and autoimmunity: Lessons from repertoires. J. Exp. Med. 2024, 221, e20231314. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Nakano, M.; Nagafuchi, Y.; Kobayashi, S.; Hatano, H.; Yoshida, R.; Akutsu, Y.; Itamiya, T.; Ban, N.; Tsuchida, Y.; et al. Multimodal repertoire analysis unveils B cell biology in immune-mediated diseases. Ann. Rheum. Dis. 2023, 82, 1455–1463. [Google Scholar] [CrossRef]
- Wilbrink, R.; van der Weele, L.; Spoorenberg, A.J.P.L.; de Vries, N.; Niewold, I.T.G.; Verstappen, G.M.; Kroese, F.G.M. B Cell Receptor Repertoire Analysis of the CD21(lo) B Cell Compartment in Healthy Individuals, Patients With Sjogren’s Disease, and Patients With Radiographic Axial Spondyloarthritis. Eur. J. Immunol. 2025, 55, e202451398. [Google Scholar] [CrossRef]
- Zaslavsky, M.E.; Craig, E.; Michuda, J.K.; Sehgal, N.; Ram-Mohan, N.; Lee, J.Y.; Nguyen, K.D.; Hoh, R.A.; Pham, T.D.; Röltgen, K.; et al. Disease diagnostics using machine learning of B cell and T cell receptor sequences. Science 2025, 387, eadp2407. [Google Scholar] [CrossRef]
- Voss, L.F.; Howarth, A.J.; Wittenborn, T.R.; Hummelgaard, S.; Juul-Madsen, K.; Kastberg, K.S.; Pedersen, M.K.; Jensen, L.; Papanastasiou, A.D.; Vorup-Jensen, T.; et al. The extrafollicular response is sufficient to drive initiation of autoimmunity and early disease hallmarks of lupus. Front. Immunol. 2022, 13, 1021370. [Google Scholar] [CrossRef]
- Al-Aubodah, T.A.; Aoudjit, L.; Pascale, G.; Perinpanayagam, M.A.; Langlais, D.; Bhargava, R.; Bhalla, N.; Lee, S.J.; Liu, Q.; Bhalla, A.; et al. The extrafollicular B cell response is a hallmark of childhood idiopathic nephrotic syndrome. Nat. Commun. 2023, 14, 7682. [Google Scholar] [CrossRef]
- Zhu, D.Y.; Maurer, D.P.; Castrillon, C.; Deng, Y.; Mohamed, F.A.N.; Ma, M.; Schuck, P.; Bhargava, R.; Li, X.; He, L.; et al. CD21 primes extrafollicular differentiation of autoreactive B cells in a TLR7-driven lupus model. Sci. Immunol. 2025, 10, eads8226. [Google Scholar] [CrossRef]
- Horisberger, A.; Griffith, A.; Keegan, J.; Arazi, A.; Pulford, J.; Murzin, E.; Cordoba, S.; Hacohen, N.; Diamond, B.; Bhalla, A.; et al. Blood immunophenotyping identifies distinct kidney histopathology and outcomes in patients with lupus nephritis. J. Clin. Investig. 2025, 135, e181034. [Google Scholar] [CrossRef]
- Wan, L.; Guo, J.; Sun, A.; Chen, H.; Hu, B.; Liu, C. The application value of peripheral plasmablasts in the assessment of disease activity and treatment response in systemic lupus erythematosus. Clin. Exp. Rheumatol. 2025. Online ahead of print. [Google Scholar]
- Jacobi, A.M.; Odendahl, M.; Reiter, K.; Bruns, A.; Burmester, G.R.; Radbruch, A.; Voll, R.E.; Dorner, T. Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Guo, F.; Liao, S.; Liao, H.; Xiao, H.; Yang, L.; Li, X.; Wang, X.; Zhang, J.; Chen, L.; et al. Altered frequency of peripheral B-cell subsets and their correlation with disease activity in patients with systemic lupus erythematosus: A comprehensive analysis. J. Cell. Mol. Med. 2020, 24, 12044–12053. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Chen, X.; Gao, Y.; Yang, M.; Zhou, S.; Lu, L.; Tian, R.; Wang, X.; Li, Y.; Zhang, Z.; et al. Age-Associated B Cells in Autoimmune Diseases: Pathogenesis and Clinical Implications. Clin. Rev. Allergy Immunol. 2025, 68, 18. [Google Scholar] [CrossRef]
- Huang, W.; Quach, T.D.; Dascalu, C.; Liu, Z.; Leung, T.; Byrne-Steele, M.; Bhargava, A.; Bhalla, R.; Bhalla, A.; Han, S.; et al. Belimumab promotes negative selection of activated autoreactive B cells in systemic lupus erythematosus patients. JCI Insight 2018, 3, e122525. [Google Scholar] [CrossRef]
- Malkiel, S.; Barlev, A.N.; Atisha-Fregoso, Y.; Suurmond, J.; Diamond, B. Plasma Cell Differentiation Pathways in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Suurmond, J.; Diamond, B. Autoantibodies in systemic autoimmune diseases: Specificity and pathogenicity. J. Clin. Investig. 2015, 125, 2194–2202. [Google Scholar] [CrossRef]
- Hiepe, F.; Radbruch, A. Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat. Rev. Nephrol. 2016, 12, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Dorner, T.; Lipsky, P.E. Beyond pan-B-cell-directed therapy—New avenues and insights into the pathogenesis of SLE. Nat. Rev. Rheumatol. 2016, 12, 645–657. [Google Scholar] [CrossRef]
- Edwards, J.C.; Szczepanski, L.; Szechinski, J.; Filipowicz-Sosnowska, A.; Emery, P.; Close, D.R.; Stevens, R.M.; Shaw, T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N. Engl. J. Med. 2004, 350, 2572–2581. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Hiepe, F.; Dorner, T.; Hauser, A.E.; Hoyer, B.F.; Mei, H.; Radbruch, A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat. Rev. Rheumatol. 2011, 7, 170–178. [Google Scholar] [CrossRef]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Browning, J.L. BAFF: A fundamental survival factor for B cells. Nat. Rev. Immunol. 2002, 2, 465–475. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzman, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.; Thomas, M.; Kim, H.Y.; Leon, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Dorner, T.; Kinnman, N.; Tak, P.P. Targeting B cells in immune-mediated inflammatory disease: A comprehensive review of mechanisms of action and identification of biomarkers. Pharmacol. Ther. 2010, 125, 464–475. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- Zhu, S.; Li, S.; Shi, L.; Chu, T.; Huang, Z.; Lu, X.; Wang, Y.; Zhang, L.; Chen, H.; Li, M.; et al. Baricitinib could improve the prognosis of anti-MDA5 antibody positive dermatomyositis associated interstitial lung disease. Arthritis Res. Ther. 2025, 27, 214. [Google Scholar] [CrossRef]
- Yanagihara, T.; Mirza, R.D.; Kolb, M.R.J. Tofacitinib in anti-MDA5-positive dermatomyositis-associated interstitial lung disease: A new standard of care emerges. Eur. Respir. J. 2025, 65, 2500458. [Google Scholar] [CrossRef]
- Alexander, T.; Sarfert, R.; Klotsche, J.; Kuhl, A.A.; Rubbert-Roth, A.; Lorenz, H.M.; Rech, J.; Schleenvoigt, B.T.; Arnold, R.; Blaschek, B.; et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann. Rheum. Dis. 2015, 74, 1474–1478. [Google Scholar] [CrossRef]
- Manz, R.A.; Hauser, A.E.; Hiepe, F.; Radbruch, A. Maintenance of serum antibody levels. Annu. Rev. Immunol. 2005, 23, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.W.; Cavacini, L., Jr. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Mackensen, A.; Muller, F.; Mougiakakos, D.; Boltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.; Taubmann, J.; Bucci, L.; Wilhelm, A.; Bergmann, C.; Volkl, S.; Aigner, M.; Rothe, T.; Minopoulou, I.; Tur, C.; et al. CD19 CAR T-Cell Therapy in Autoimmune Disease—A Case Series with Follow-up. N. Engl. J. Med. 2024, 390, 687–700. [Google Scholar] [CrossRef]
- Chung, J.B.; Brudno, J.N.; Borie, D.; Kochenderfer, J.N. Chimeric antigen receptor T cell therapy for autoimmune disease. Nat. Rev. Immunol. 2024, 24, 830–845. [Google Scholar] [CrossRef]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghazi, M.; Soto, H.; Bhargava, A.; Bhalla, A.; et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci. Transl. Med. 2019, 11, eaav1648. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Rispens, T.; Huijbers, M.G. The unique properties of IgG4 and its roles in health and disease. Nat. Rev. Immunol. 2023, 23, 763–778. [Google Scholar] [CrossRef]
- Khosroshahi, A.; Carruthers, M.N.; Deshpande, V.; Unizony, S.; Bloch, D.B.; Stone, J.H. Rituximab for the treatment of IgG4-related disease: Lessons from 10 consecutive patients. Medicine 2012, 91, 57–66. [Google Scholar] [CrossRef]
- Huijbers, M.G.; Plomp, J.J.; van der Maarel, S.M.; Verschuuren, J.J. IgG4-mediated autoimmune diseases: A niche of antibody-mediated disorders. Ann. N. Y. Acad. Sci. 2018, 1413, 92–103. [Google Scholar] [CrossRef]
- Zhang, H.; Li, P.; Wu, D.; Xu, D.; Hou, Y.; Wang, Q.; Li, M.; Li, Y.; Zeng, X.; Zhang, F.; et al. Serum IgG subclasses in autoimmune diseases. Medicine 2015, 94, e387. [Google Scholar] [CrossRef]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Churlaud, G.; Mallone, R.; Six, A.; Derian, N.; Chaara, W.; Lorenzon, R.; Long, S.A.; Buckner, J.H.; Afonso, G.; et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J. Autoimmun. 2015, 58, 48–58. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Zhu, L.N.; Hou, H.M.; Wang, S.; Zhang, S.; Wang, G.G.; Guo, Z.Y.; Wang, X.; Li, Y.; Chen, Z.; Zhang, J.; et al. FcRn inhibitors: A novel option for the treatment of myasthenia gravis. Neural Regen. Res. 2023, 18, 1637–1644. [Google Scholar]
- LaMothe, R.A.; Kolte, P.N.; Vo, T.; Ferrari, J.D.; Gelsinger, T.C.; Wong, J.; Chan, V.T.; Ahmed, S.; Srinivasan, A.; Deitemeyer, P.; et al. Tolerogenic Nanoparticles Induce Antigen-Specific Regulatory T Cells and Provide Therapeutic Efficacy and Transferrable Tolerance against Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2018, 9, 281. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Buckner, J.H.; Herold, K.C. Immunotherapy: Building a bridge to a cure for type 1 diabetes. Science 2021, 373, 510–516. [Google Scholar] [CrossRef]
- Kenison, J.E.; Jhaveri, A.; Li, Z.; Khadse, N.; Tjon, E.; Tezza, S.; Mayo, L.; Bhalla, A.; Bhargava, R.; Quintana, F.J.; et al. Tolerogenic nanoparticles suppress central nervous system inflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 32017–32028. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- See, P.; Lum, J.; Chen, J.; Ginhoux, F. A Single-Cell Sequencing Guide for Immunologists. Front. Immunol. 2018, 9, 2425. [Google Scholar] [CrossRef]
- van Dongen, J.J.M.; Lhermitte, L.; Bottcher, S.; Almeida, J.; van der Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lecrevisse, Q.; Lucio, P.; et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef]
- Solly, F.; Angelot-Delettre, F.; Ticchioni, M.; Genevieve, F.; Rambaud, H.; Baseggio, L.; Plesa, A.; Debliquis, A.; Garnache-Ottou, F.; Roggy, A.; et al. Standardization of Flow Cytometric Immunophenotyping for Hematological Malignancies: The FranceFlow Group Experience. Cytom. Part A 2019, 95, 1008–1018. [Google Scholar] [CrossRef]
- Kelleher, P.; Greathead, L.; Whitby, L.; Brando, B.; UK NEQAS Leucocyte Immunophenotyping Steering Committee; Barnett, D.; Bloxham, D.; Detute, R.; Dunlop, A.; Farren, T.; et al. European flow cytometry quality assurance guidelines for the diagnosis of primary immune deficiencies and assessment of immune reconstitution following B cell depletion therapies and transplantation. Cytom. Part B Clin. Cytom. 2024, 106, 424–436. [Google Scholar] [CrossRef]
- Greiff, V.; Menzel, U.; Haessler, U.; Cook, S.C.; Friedensohn, S.; Khan, T.A.; Pogson, M.; Hellmann, I.; Reddy, S.T. Quantitative assessment of the robustness of next-generation sequencing of antibody variable gene repertoires from immunized mice. BMC Immunol. 2014, 15, 40. [Google Scholar] [CrossRef]
- Shemesh, O.; Polak, P.; Lundin, K.E.A.; Sollid, L.M.; Yaari, G. Machine Learning Analysis of Naive B-Cell Receptor Repertoires Stratifies Celiac Disease Patients and Controls. Front. Immunol. 2021, 12, 627813. [Google Scholar] [CrossRef]
- Esteva, A.; Robicquet, A.; Ramsundar, B.; Kuleshov, V.; DePristo, M.; Chou, K.; Cui, C.; Corrado, G.; Thrun, S.; Dean, J.; et al. A guide to deep learning in healthcare. Nat. Med. 2019, 25, 24–29. [Google Scholar] [CrossRef]
- Dorner, T.; Lipsky, P.E. B cells: Depletion or functional modulation in rheumatic diseases. Curr. Opin. Rheumatol. 2014, 26, 228–236. [Google Scholar] [CrossRef]
- Fillatreau, S.; Manfroi, B.; Dorner, T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat. Rev. Rheumatol. 2021, 17, 98–108. [Google Scholar] [CrossRef]
- Bombardieri, M.; Lewis, M.; Pitzalis, C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat. Rev. Rheumatol. 2017, 13, 141–154. [Google Scholar] [CrossRef]
- Thoreau, B.; Chaigne, B.; Mouthon, L. Role of B-Cell in the Pathogenesis of Systemic Sclerosis. Front. Immunol. 2022, 13, 933468. [Google Scholar] [CrossRef]
- Reyes-Huerta, R.F.; Mandujano-Lopez, V.; Velasquez-Ortiz, M.G.; Alcala-Carmona, B.; Ostos-Prado, M.J.; Reyna-Juarez, Y.; Garcia-Blanco, A.; Maldonado-Bernal, C.; Meza-Sanchez, D.; Llorente-Garcia, L.; et al. Novel B-cell subsets as potential biomarkers in idiopathic inflammatory myopathies: Insights into disease pathogenesis and disease activity. J. Leukoc. Biol. 2024, 116, 84–94. [Google Scholar] [CrossRef]
- Franco, C.; Gatto, M.; Iaccarino, L.; Ghirardello, A.; Doria, A. Lymphocyte immunophenotyping in inflammatory myositis: A review. Curr. Opin. Rheumatol. 2021, 33, 522–528. [Google Scholar] [CrossRef]
- Pan, Z.; Li, M.; Zhang, P.; Li, T.; Liu, R.; Liu, J.; Wang, Y.; Chen, H.; Zhang, L.; Li, X.; et al. Peripheral Blood Lymphocyte Subsets and Heterogeneity of B Cell Subsets in Patients of Idiopathic Inflammatory Myositis with Different Myositis-specific Autoantibodies. Inflammation 2025, 48, 118–132. [Google Scholar] [CrossRef]
- Li, L.; Liu, S.; Yu, J. Autoimmune thyroid disease and type 1 diabetes mellitus: Same pathogenesis; new perspective? Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820958329. [Google Scholar] [CrossRef]
- Naser, S.S.; Mahdi, B.M. Type 1 diabetes and autoimmune thyroid disease—The genetic link. Front. Endocrinol. 2021, 12, 618213. [Google Scholar] [CrossRef]
- Ragusa, F.; Fallahi, P.; Elia, G.; Gonnella, D.; Paparo, S.R.; Giusti, C.; Churilov, L.P.; Ferrari, S.M.; Antonelli, A. Hashimoto thyroiditis: Epidemiology, pathogenesis, clinic and therapy. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101367. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Verschuuren, J.J. Myasthenia gravis: Subgroup classification and therapeutic strategies. Lancet Neurol. 2015, 14, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Koneczny, I.; Herbst, R. Myasthenia gravis: Pathogenic effects of autoantibodies on neuromuscular architecture. Cells 2019, 8, 671. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autoantibody | Dominant B-Cell Endotype | Typical Clinical Associations | Key Immunobiological Features | Therapeutic Vulnerabilities (Context-Dependent) |
|---|---|---|---|---|
| Anti-MDA5 | IFN-high/Extrafollicular (EF-biased) | Dermatomyositis, RP-ILD | Low SHM, DN2/ABC expansion, high IFN signature, plasmablast bursts | JAK inhibitors, glucocorticoids, RTX (case-series and emerging evidence) |
| Anti-U1RNP | IFN-high/EF-enriched | MCTD, overlap syndromes, vasculopathy | IFN-driven activation, plasmablast bias, Raynaud association | IFN-pathway targeting, JAK inhibitors, immunosuppression |
| Anti-Ro52 (TRIM21) | IFN-high/EF-enriched | pSjD, IIM, SSc-ILD | Type I/II IFN activation, vascular tropism, lung involvement | High-intensity immunosuppression in selected severe phenotypes |
| Anti-centromere | Germinal Center/Plasma Cell-anchored | Limited SSc, PAH | High SHM, LLPC persistence, BM niching | Supportive care; B-cell targeting under investigation |
| Anti-Ro60/Anti-La (SS-A/B) | GC/LLPC-dominant | pSjD, SLE, NLE | LLPC niches, structured TLS, chronic autoantibody production | BAFF-R targeting (investigational), plasma-cell modulation (experimental) |
| Anti-Scl-70 (Topo-I) | Tissue-conditioned (fibro-inflammatory) | Diffuse SSc, ILD | Stromal imprinting, TLS presence, ECM remodeling | Antifibrotics, IL-6 blockade; B-cell targeting under investigation |
| RF/Anti-CCP | Mixed GC/BAFF-modulated | Seropositive RA | GC activity, BAFF sensitivity, transitional B-cell expansion | B-cell depletion, costimulation blockade; BAFF inhibition in subsets |
| Biological Target | Mechanism | Endotype Association | Biological Limitation |
|---|---|---|---|
| Mature B-cells (CD20) | Depletion of antigen-experienced B-cells | Memory-dominant/GC-associated endotypes | Spares plasma cells; rapid serological rebound |
| BAFF survival axis | Disruption of transitional and naïve B-cell survival | BAFF-high survival-biased architectures | Slow pharmacodynamic effect |
| IFN signaling hubs (JAK–STAT) | Inhibition of inflammatory license | IFN-high/extrafollicular endotypes | Indirect effect on B-cells |
| Plasma cells (CD38/proteasome) | Disruption of antibody secretion machinery | Plasma-cell-anchored architectures | Niche protection limits durability |
| B-cell lineage (CD19) | Immune circuit collapse | Refractory immune configurations | Immune toxicity risk |
| Biomarker | Definition/Panel | Endotype Indication | Clinical Utility | Availability |
|---|---|---|---|---|
| DN2 B-cells | CD19+CD27−IgD−CD11c+ T-bet+ | IFN-high/EF | Flare risk; immune pathway stratification | Specialized flow cytometry |
| Plasmablasts | CD19+CD27++CD38++ CD20−/low | Active immune circuit | Disease activity monitoring | Standard flow cytometry |
| Type I IFN score | ISG expression profile (e.g., IFI27, IFI44, IFIT1, ISG15) | IFN-high endotype | IFN-pathway activity | Commercial immune panels |
| Serum BAFF | ELISA-based quantification | BAFF-driven endotype | BAFF-axis activation | Reference laboratories |
| Free light chains | κ, λ, κ/λ ratio | Plasma-cell-anchored | Plasma-cell activity marker | Routine laboratories |
| Endotype | Key Biological Identifiers | First-Line Therapies | Refractory Options | Avoid/Limited Benefit |
|---|---|---|---|---|
| IFN-high/EF | DN2 expansion, T-bet+ plasmablasts, high IFN score | JAK inhibitors + rituximab | CAR-T, anti-CD19, anifrolumab | Belimumab alone; plasma cell-directed therapy |
| GC/Plasma cell-anchored | High SHM autoantibodies, stable titers | Daratumumab, bortezomib | CAR-T, BCMA-targeting | Rituximab alone |
| BAFF-driven | High BAFF, transitional and naïve B-cell expansion | Belimumab + rituximab | Ianalumab, anti-CD19 | Rituximab monotherapy |
| TGF-β-fibrotic | Anti-Scl-70, fibrosis biomarkers | Nintedanib, tocilizumab | Anti-CD19, CAR-T, HSCT | B-cell depletion alone |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Sarrand, J.; Soyfoo, M. B-Cells and Plasmablasts as Architects of Autoimmune Disease: From Molecular Footprints to Precision Therapeutics. Cells 2026, 15, 119. https://doi.org/10.3390/cells15020119
Sarrand J, Soyfoo M. B-Cells and Plasmablasts as Architects of Autoimmune Disease: From Molecular Footprints to Precision Therapeutics. Cells. 2026; 15(2):119. https://doi.org/10.3390/cells15020119
Chicago/Turabian StyleSarrand, Julie, and Muhammad Soyfoo. 2026. "B-Cells and Plasmablasts as Architects of Autoimmune Disease: From Molecular Footprints to Precision Therapeutics" Cells 15, no. 2: 119. https://doi.org/10.3390/cells15020119
APA StyleSarrand, J., & Soyfoo, M. (2026). B-Cells and Plasmablasts as Architects of Autoimmune Disease: From Molecular Footprints to Precision Therapeutics. Cells, 15(2), 119. https://doi.org/10.3390/cells15020119