
PACAP and Maxadilan (PAC1 Agonist) Influence Plaque Progression, Migratory Ability, and Mitochondrial Morphology and Dynamics in Vascular Smooth Muscle Cells


Highlights
- PACAP deficiency in vivo increased lumen stenosis but reduced plaque burden in atherosclerotic mice, while in vitro PACAP enhanced the viability of oxLDL-treated human coronary artery smooth muscle cells (HCASMCs).
- Maxadilan, a PAC1 agonist, improved migration in oxLDL-impaired HCASMCs, and both oxLDL and PACAP influenced mitochondrial morphology.
- The findings highlight the complex role of the PACAP/PAC1 system in vascular pathology, suggesting that its targeted modulation could offer novel strategies for stabilizing atherosclerotic plaques.
- Specifically, activating PAC1 or supplementing PACAP may help balance plaque formation and vascular function, warranting further investigation for therapeutic applications.
Abstract
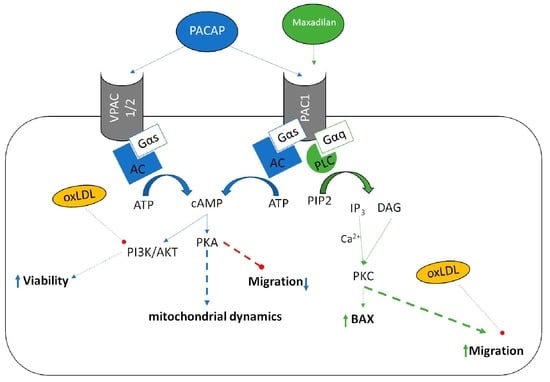
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Genotyping
2.3. Dissection and Tissue Harvesting
2.4. OilRedO Staining
2.5. Immunohistology
2.6. Cell Culture
2.7. LDL Oxidation
2.8. PrestoBlue Viability Assay
2.9. Propidium-Iodide-Staining
2.10. Determination of Lipid Droplets (LDs) by BODIPYTM 493/503
2.11. Scratch Assay
2.12. Fluorescence Labeling of Mitochondria
2.13. SDS-PAGE and Western Blot
2.14. ELISA
2.15. Immunocytofluorescence Confocal Scanning Microscopy
2.16. Statistical Analyses
3. Results
3.1. PACAP Deficiency Affects Aortic Burden, Lumen Stenosis, and Body Weight in Mice
3.2. Cell Migration of HCASMCs
3.3. Viability and Lipid Uptake in HCASMCs
3.4. Apoptosis Analysis in HCASMCs
3.5. Mitochondrial Morphology of HCASMCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTA2 | Alpha-actin-2 |
| BID | BH3-interacting domain death agonist |
| BMI | Body mass index |
| BT | Brachiocephalic trunk |
| cAMP | Cyclic adenosine monophosphate |
| CED | Cholesterol-enriched diet |
| CVDs | Cardiovascular diseases |
| ECM | Extracellular matrix |
| GPCR | G-protein-coupled receptor |
| HCASMCs | Human coronary artery smooth muscle cells |
| HE | Hematoxylin–eosin |
| Klf4 | Krüppel-like factor 4 |
| LD | Lipid droplet |
| LDL | Low-density lipoprotein |
| LOX-1 | Lectin-like oxLDL receptor-1 |
| MՓs | Macrophages |
| MCC | Manders’ Colocalization Coefficient |
| MiNA | Mitochondrial Network Analysis |
| nLDL | Native LDL |
| Oct4 | Octamer-binding protein 4 |
| ORO | OilRedO |
| oxLDL | Oxidized LDL |
| PACAP | Pituitary adenylate cyclase-activating polypeptide |
| PI | Propidium odide |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PCNA | Proliferating Cell Nuclear Antigen |
| PCR | Polymerase chain reaction |
| REM | Relative electrophoretic mobility |
| RT | Room temperature |
| SC | Standard chow |
| SIMH | Stress-induced mitochondrial hyperfusion |
| SMCs | Smooth muscle cells |
| TAGs | Triglycerides |
| TNBSA | 2,4,6-trinitrobenzene sulfonic acid |
| VSMC | Vascular SMC |
References
- World Health Organization. Global Action Plan for the Prevention and Control of Noncommunicable Diseases, 2013–2020; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Jarad, S.; Gill, G.; Amadi, P.; Gu, H.-M.; Zhang, D.-W. VSMCs in Atherosclerosis: Implications on the Role of Inflammation and Extracellular Matrix Remodelling. Pharmacol. Res. 2025, 218, 107833. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Weissberg, P.L. Smooth muscle cell heterogeneity: Patterns of gene expression in vascular smooth muscle cells in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Sobue, K.; Hayashi, K.; Nishida, W. Molecular mechanism of phenotypic modulation of smooth muscle cells. Horm. Res. 1998, 50, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jørgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Alencar, G.F.; Owsiany, K.M.; Karnewar, S.; Sukhavasi, K.; Mocci, G.; Nguyen, A.T.; Williams, C.M.; Shamsuzzaman, S.; Mokry, M.; Henderson, C.A.; et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef] [PubMed]
- Kansakar, U.; Jankauskas, S.S.; Gambardella, J.; Santulli, G. Targeting the phenotypic switch of vascular smooth muscle cells to tackle atherosclerosis. Atherosclerosis 2021, 324, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Miyata, A.; Arimura, A.; Dahl, R.R.; Minamino, N.; Uehara, A.; Jiang, L.; Culler, M.D.; Coy, D.H. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem. Biophys. Res. Commun. 1989, 164, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Vaudry, D.; Gonzalez, B.J.; Basille, M.; Yon, L.; Fournier, A.; Vaudry, H. Pituitary adenylate cyclase-activating polypeptide and its receptors: From structure to functions. Pharmacol. Rev. 2000, 52, 269–324. [Google Scholar] [CrossRef]
- Laburthe, M.; Couvineau, A. Molecular pharmacology and structure of VPAC Receptors for VIP and PACAP. Regul. Pept. 2002, 108, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Harmar, A.J.; Fahrenkrug, J.; Gozes, I.; Laburthe, M.; May, V.; Pisegna, J.R.; Vaudry, D.; Vaudry, H.; Waschek, J.A.; Said, S.I. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br. J. Pharmacol. 2012, 166, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Couvineau, A.; Rouyer-Fessard, C.; Darmoul, D.; Maoret, J.J.; Carrero, I.; Ogier-Denis, E.; Laburthe, M. Human intestinal VIP receptor: Cloning and functional expression of two cDNA encoding proteins with different N-terminal domains. Biochem. Biophys. Res. Commun. 1994, 200, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Rasbach, E.; Splitthoff, P.; Bonaterra, G.A.; Schwarz, A.; Mey, L.; Schwarzbach, H.; Eiden, L.E.; Weihe, E.; Kinscherf, R. PACAP deficiency aggravates atherosclerosis in ApoE deficient mice. Immunobiology 2019, 224, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Witzel, R.; Block, A.; Pollmann, S.; Oetzel, L.; Fleck, F.; Bonaterra, G.A.; Kinscherf, R.; Schwarz, A. PACAP regulates VPAC1 expression, inflammatory processes and lipid homeostasis in M1- and M2-macrophages. Front. Cardiovasc. Med. 2023, 10, 1264901. [Google Scholar] [CrossRef] [PubMed]
- Splitthoff, P.; Rasbach, E.; Neudert, P.; Bonaterra, G.A.; Schwarz, A.; Mey, L.; Schwarzbach, H.; Eiden, L.E.; Weihe, E.; Kinscherf, R. PAC1 deficiency attenuates progression of atherosclerosis in ApoE deficient mice under cholesterol-enriched diet. Immunobiology 2020, 225, 151930. [Google Scholar] [CrossRef] [PubMed]
- Moro, O.; Lerner, E.A. Maxadilan, the vasodilator from sand flies, is a specific pituitary adenylate cyclase activating peptide type I receptor agonist. J. Biol. Chem. 1997, 272, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Mey, L.; Bonaterra, G.A.; Hoffmann, J.; Schwarzbach, H.; Schwarz, A.; Eiden, L.E.; Weihe, E.; Kinscherf, R. PAC1 Agonist Maxadilan Reduces Atherosclerotic Lesions in Hypercholesterolemic ApoE-Deficient Mice. Int. J. Mol. Sci. 2024, 25, 13245. [Google Scholar] [CrossRef] [PubMed]
- Juhász, T.; Matta, C.; Katona, É.; Somogyi, C.; Takács, R.; Gergely, P.; Csernoch, L.; Panyi, G.; Tóth, G.; Reglődi, D.; et al. Pituitary adenylate cyclase activating polypeptide (PACAP) signalling exerts chondrogenesis promoting and protecting effects: Implication of calcineurin as a downstream target. PLoS ONE 2014, 9, e91541. [Google Scholar] [CrossRef] [PubMed]
- Juhász, T.; Matta, C.; Katona, É.; Somogyi, C.; Takács, R.; Hajdú, T.; Helgadottir, S.L.; Fodor, J.; Csernoch, L.; Tóth, G.; et al. Pituitary adenylate cyclase-activating polypeptide (PACAP) signalling enhances osteogenesis in UMR-106 cell line. J. Mol. Neurosci. 2014, 54, 555–573. [Google Scholar] [CrossRef] [PubMed]
- Blümm, C.; Bonaterra, G.A.; Schwarzbach, H.; Eiden, L.E.; Weihe, E.; Kinscherf, R. PAC1 deficiency reduces chondrogenesis in atherosclerotic lesions of hypercholesterolemic ApoE-deficient mice. BMC Cardiovasc. Disord. 2023, 23, 566. [Google Scholar] [CrossRef] [PubMed]
- Hamelink, C.; Tjurmina, O.; Damadzic, R.; Young, W.S.; Weihe, E.; Lee, H.-W.; Eiden, L.E. Pituitary adenylate cyclase-activating polypeptide is a sympathoadrenal neurotransmitter involved in catecholamine regulation and glucohomeostasis. Proc. Natl. Acad. Sci. USA 2002, 99, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, S.; Gramanzini, M.; Megna, R.; Greco, A.; Albanese, S.; Manfredi, C.; Brunetti, A. Evaluation of growth patterns and body composition in C57Bl/6J mice using dual energy X-ray absorptiometry. BioMed Res. Int. 2014, 2014, 253067. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-Y.; Qin, L.; Simons, M. Imaging and Analysis of Oil Red O-Stained Whole Aorta Lesions in an Aneurysm Hyperlipidemia Mouse Model. J. Vis. Exp. 2022, 183, e61277. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Kinscherf, R.; Deigner, H.P.; Usinger, C.; Pill, J.; Wagner, M.; Kamencic, H.; Hou, D.; Chen, M.; Schmiedt, W.; Schrader, M.; et al. Induction of mitochondrial manganese superoxide dismutase in macrophages by oxidized LDL: Its relevance in atherosclerosis of humans and heritable hyperlipidemic rabbits. FASEB J. 1997, 11, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Galle, J.; Wanner, C. Oxidized LDL and Lp(a). Preparation, modification, and analysis. Methods Mol. Biol. 1998, 108, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, U.P. Oxidation of human low density lipoprotein results in derivatization of lysine residues of apolipoprotein B by lipid peroxide decomposition products. J. Biol. Chem. 1987, 262, 3603–3608. [Google Scholar] [CrossRef]
- Foxx, K.K.; Roberts, R.L.; Waxdal, M.J. Effect of Copper Ion Concentration on the Oxidation of Human LDL; Kalen Biomedical, LLC.: Gaithersburg, MD, USA, 2008. [Google Scholar]
- Lougheed, M.; Steinbrecher, U.P. Mechanism of uptake of copper-oxidized low density lipoprotein in macrophages is dependent on its extent of oxidation. J. Biol. Chem. 1996, 271, 11798–11805. [Google Scholar] [CrossRef] [PubMed]
- Boncler, M.; Różalski, M.; Krajewska, U.; Podsędek, A.; Watala, C. Comparison of PrestoBlue and MTT assays of cellular viability in the assessment of anti-proliferative effects of plant extracts on human endothelial cells. J. Pharmacol. Toxicol. Methods 2014, 69, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Farese, R.V. Lipid Droplets and Cellular Lipid Metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Bakare, A.B.; Daniel, J.; Stabach, J.; Rojas, A.; Bell, A.; Henry, B.; Iyer, S. Quantifying Mitochondrial Dynamics in Patient Fibroblasts with Multiple Developmental Defects and Mitochondrial Disorders. Int. J. Mol. Sci. 2021, 22, 6263. [Google Scholar] [CrossRef] [PubMed]
- Kiryu-Seo, S.; Tamada, H.; Kato, Y.; Yasuda, K.; Ishihara, N.; Nomura, M.; Mihara, K.; Kiyama, H. Mitochondrial fission is an acute and adaptive response in injured motor neurons. Sci. Rep. 2016, 6, 28331. [Google Scholar] [CrossRef] [PubMed]
- Zemirli, N.; Morel, E.; Molino, D. Mitochondrial Dynamics in Basal and Stressful Conditions. Int. J. Mol. Sci. 2018, 19, 564. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q. Experimental study of the effects of pituitary adenylate cyclase-activating polypeptide (PACAP) and its mechanism on the vascular cell components—The possible relationship between PACAP and atherosclerosis. Sheng Li Ke Xue Jin Zhan 1997, 28, 132–135. [Google Scholar] [PubMed]
- Gasz, B.; Rácz, B.; Roth, E.; Borsiczky, B.; Ferencz, A.; Tamás, A.; Cserepes, B.; Lubics, A.; Gallyas, F.; Tóth, G.; et al. Pituitary adenylate cyclase activating polypeptide protects cardiomyocytes against oxidative stress-induced apoptosis. Peptides 2006, 27, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Merriam, L.A.; Baran, C.N.; Girard, B.M.; Hardwick, J.C.; May, V.; Parsons, R.L. Pituitary adenylate cyclase 1 receptor internalization and endosomal signaling mediate the pituitary adenylate cyclase activating polypeptide-induced increase in guinea pig cardiac neuron excitability. J. Neurosci. 2013, 33, 4614–4622. [Google Scholar] [CrossRef] [PubMed]
- Schoenhagen, P.; Nissen, S.E.; Tuzcu, E.M. Coronary arterial remodeling: From bench to bedside. Curr. Atheroscler. Rep. 2003, 5, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Glagov, S.; Weisenberg, E.; Zarins, C.K.; Stankunavicius, R.; Kolettis, G.J. Compensatory enlargement of human atherosclerotic coronary arteries. N. Engl. J. Med. 1987, 316, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Hein, L.; Brede, M.; Jahns, R.; Engelhardt, S.; Gröne, H.-J.; Schütz, G. Pulmonary hypertension and right heart failure in pituitary adenylate cyclase-activating polypeptide type I receptor-deficient mice. Circulation 2004, 110, 3245–3251. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Kolodgie, F.D.; Farb, A.; Weber, D.; Virmani, R. Morphological predictors of arterial remodeling in coronary atherosclerosis. Circulation 2002, 105, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Pant, R.; Marok, R.; Klein, L.W. Pathophysiology of coronary vascular remodeling: Relationship with traditional risk factors for coronary artery disease. Cardiol. Rev. 2014, 22, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.C.; Kwiecinski, J.; Doris, M.; McElhinney, P.; D’Souza, M.S.; Cadet, S.; Adamson, P.D.; Moss, A.J.; Alam, S.; Hunter, A.; et al. Low-Attenuation Noncalcified Plaque on Coronary Computed Tomography Angiography Predicts Myocardial Infarction. Circulation 2020, 141, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.B.; Dzaye, O.; Steffensen, F.H.; Bøtker, H.E.; Jensen, J.M.; Rønnow Sand, N.P.; Kragholm, K.H.; Sørensen, H.T.; Leipsic, J.; Mæng, M.; et al. Impact of Plaque Burden Versus Stenosis on Ischemic Events in Patients with Coronary Atherosclerosis. J. Am. Coll. Cardiol. 2020, 76, 2803–2813. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Torres, E.; Yuhara, A.; Jongsma, H.; Otto, C.; Korhonen, L.; Abiru, Y.; Skoglösa, Y.; Schütz, G.; Hatanaka, H.; et al. Pituitary adenylate cyclase-activating polypeptide promotes the survival of basal forebrain cholinergic neurons in vitro and in vivo: Comparison with effects of nerve growth factor. Eur. J. Neurosci. 2000, 12, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Reglodi, D.; Tamas, A.; Bosnyak, I.; Atlasz, T.; Szabo, E.; Li, L.; Horvath, G.; Opper, B.; Kiss, P.; Lucas, L.; et al. Protective Effects of PACAP in Diabetic Complications: Retinopathy, Nephropathy and Neuropathy. Int. J. Mol. Sci. 2025, 26, 9650. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Zhou, X.; Li, Y.; Yang, L. PACAP inhibits sepsis-associated acute lung injury by inhibiting the Sp1/AQP1 pathway. Peptides 2025, 191, 171411. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Guan, Z.; Zhang, J.; Lv, Z.; Duan, Y.; Tian, S. PACAP mitigates traumatic brain injury progression via ELF3/ZO-1 axis-mediated blood-brain barrier repair. Psychoneuroendocrinology 2025, 180, 107573. [Google Scholar] [CrossRef] [PubMed]
- Frontini, M.J.; O’Neil, C.; Sawyez, C.; Chan, B.M.C.; Huff, M.W.; Pickering, J.G. Lipid incorporation inhibits Src-dependent assembly of fibronectin and type I collagen by vascular smooth muscle cells. Circ. Res. 2009, 104, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, E.R.; Pugach, I.M.; Orekhov, A.N. Subendothelial smooth muscle cells of human aorta express macrophage antigen in situ and in vitro. Atherosclerosis 1997, 135, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Benitez, R.; Delgado-Maroto, V.; Caro, M.; Forte-Lago, I.; Duran-Prado, M.; O’Valle, F.; Lichtman, A.H.; Gonzalez-Rey, E.; Delgado, M. Vasoactive Intestinal Peptide Ameliorates Acute Myocarditis and Atherosclerosis by Regulating Inflammatory and Autoimmune Responses. J. Immunol. 2018, 200, 3697–3710. [Google Scholar] [CrossRef] [PubMed]
- Lerner, E.A.; Iuga, A.O.; Reddy, V.B. Maxadilan, a PAC1 receptor agonist from sand flies. Peptides 2007, 28, 1651–1654. [Google Scholar] [CrossRef] [PubMed]
- Uchida, D.; Tatsuno, I.; Tanaka, T.; Hirai, A.; Saito, Y.; Moro, O.; Tajima, M. Maxadilan is a specific agonist and its deleted peptide (M65) is a specific antagonist for PACAP type 1 receptor. Ann. N. Y. Acad. Sci. 1998, 865, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Ammann, K.R.; DeCook, K.J.; Li, M.; Slepian, M.J. Migration versus proliferation as contributor to in vitro wound healing of vascular endothelial and smooth muscle cells. Exp. Cell Res. 2019, 376, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Jeong, J.-Y.; Huh, W.-K. Activation of the pituitary adenylate cyclase-activating polypeptide type I receptor promotes neuroblastoma proliferation and migration through distinct G protein pathways. Cell Commun. Signal. 2026, 24, 105. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, O.A.; Pidkovka, N.A.; Sarmento, O.F.; Yoshida, T.; Gan, Q.; Adiguzel, E.; Bendeck, M.P.; Berliner, J.; Leitinger, N.; Owens, G.K. Oxidized phospholipids induce type VIII collagen expression and vascular smooth muscle cell migration. Circ. Res. 2009, 104, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Pidkovka, N.A.; Cherepanova, O.A.; Yoshida, T.; Alexander, M.R.; Deaton, R.A.; Thomas, J.A.; Leitinger, N.; Owens, G.K. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ. Res. 2007, 101, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ren, Y.; Kang, L.; Zhang, L. Oxidized low-density lipoprotein increases the proliferation and migration of human coronary artery smooth muscle cells through the upregulation of osteopontin. Int. J. Mol. Med. 2014, 33, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.W.; Hessler, J.R.; Chisolm, G.M. Low density lipoprotein cytotoxicity induced by free radical peroxidation of lipid. J. Lipid Res. 1983, 24, 1070–1076. [Google Scholar] [CrossRef]
- Jovinge, S.; Crisby, M.; Thyberg, J.; Nilsson, J. DNA fragmentation and ultrastructural changes of degenerating cells in atherosclerotic lesions and smooth muscle cells exposed to oxidized LDL in vitro. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2225–2231. [Google Scholar] [CrossRef] [PubMed]
- Björkerud, B.; Björkerud, S. Contrary effects of lightly and strongly oxidized LDL with potent promotion of growth versus apoptosis on arterial smooth muscle cells, macrophages, and fibroblasts. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Kume, N.; Miyamoto, S.; Minami, M.; Morimoto, M.; Hayashida, K.; Hashimoto, N.; Kita, T. Oxidized LDL modulates Bax/Bcl-2 through the lectinlike Ox-LDL receptor-1 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Okura, Y.; Brink, M.; Itabe, H.; Scheidegger, K.J.; Kalangos, A.; Delafontaine, P. Oxidized low-density lipoprotein is associated with apoptosis of vascular smooth muscle cells in human atherosclerotic plaques. Circulation 2000, 102, 2680–2686. [Google Scholar] [CrossRef] [PubMed]
- Bornfeldt, K.E.; Graves, L.M.; Raines, E.W.; Igarashi, Y.; Wayman, G.; Yamamura, S.; Yatomi, Y.; Sidhu, J.S.; Krebs, E.G.; Hakomori, S. Sphingosine-1-phosphate inhibits PDGF-induced chemotaxis of human arterial smooth muscle cells: Spatial and temporal modulation of PDGF chemotactic signal transduction. J. Cell Biol. 1995, 130, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Bornfeldt, K.E.; Krebs, E.G. Crosstalk between protein kinase A and growth factor receptor signaling pathways in arterial smooth muscle. Cell Signal. 1999, 11, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Langer, T.; Baker, M.J. Proteolytic control of mitochondrial function and morphogenesis. Biochim. Biophys. Acta 2013, 1833, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Trimmer, P.A.; Swerdlow, R.H.; Parks, J.K.; Keeney, P.; Bennett, J.P.; Miller, S.W.; Scott, W.; Davis, R.E.; Parker, W.D. Abnormal Mitochondrial Morphology in Sporadic Parkinson’s and Alzheimer’s Disease Cybrid Cell Lines. Exp. Neurol. 2000, 162, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Hoppel, C.L.; Tandler, B.; Fujioka, H.; Riva, A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int. J. Biochem. Cell Biol. 2009, 41, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Goyal, G.; Fell, B.; Sarin, A.; Youle, R.J.; Sriram, V. Role of mitochondrial remodeling in programmed cell death in Drosophila melanogaster. Dev. Cell 2007, 12, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Jing, D.; Huang, X.; Yang, W.; Shao, Z. Drp1-mediated mitochondrial fission is involved in oxidized low-density lipoprotein-induced AF cella poptosis. J. Orthop. Res. 2021, 39, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhao, Y.; Yao, H.; Zhang, L.; Chen, C.; Zheng, Z.; Jin, S. DRP1 knockdown and atorvastatin alleviate ox-LDL-induced vascular endothelial cells injury: DRP1 is a potential target for preventing atherosclerosis. Exp. Cell Res. 2023, 429, 113688. [Google Scholar] [CrossRef] [PubMed]
- Nakada, K.; Inoue, K.; Ono, T.; Isobe, K.; Ogura, A.; Goto, Y.I.; Nonaka, I.; Hayashi, J.I. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 2001, 7, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Miyatake, T.; Attardi, G. Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol. Cell Biol. 1994, 14, 2699–2712. [Google Scholar] [CrossRef]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Rolland, S.G.; Motori, E.; Memar, N.; Hench, J.; Frank, S.; Winklhofer, K.F.; Conradt, B. Impaired complex IV activity in response to loss of LRPPRC function can be compensated by mitochondrial hyperfusion. Proc. Natl. Acad. Sci. USA 2013, 110, E2967–E2976. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age [Weeks] | Body Weight [g] | Body Height [cm] | BMI [g/cm2] | n | |
|---|---|---|---|---|---|
| ApoE−/− | 31.68 ± 3.47 | 30.08 ± 2.49 | 9.39 ± 0.48 | 3.21 ± 0.29 | 19 |
| PACAP−/−/ApoE−/− | 30.23 ± 1.86 | 27.17 *** ± 1.67 | 9.19 ± 0.67 | 2.97 ** ± 0.22 | 11 |
| p value | 1.00 | ≤0.001 | 0.18 | ≤0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Brauschke, J.; Schütz, L.-M.; Bonaterra, G.A.; Kinscherf, R.; Schwarz, A. PACAP and Maxadilan (PAC1 Agonist) Influence Plaque Progression, Migratory Ability, and Mitochondrial Morphology and Dynamics in Vascular Smooth Muscle Cells. Cells 2026, 15, 1127. https://doi.org/10.3390/cells15121127
Brauschke J, Schütz L-M, Bonaterra GA, Kinscherf R, Schwarz A. PACAP and Maxadilan (PAC1 Agonist) Influence Plaque Progression, Migratory Ability, and Mitochondrial Morphology and Dynamics in Vascular Smooth Muscle Cells. Cells. 2026; 15(12):1127. https://doi.org/10.3390/cells15121127
Chicago/Turabian StyleBrauschke, Julia, Lisa-Marie Schütz, Gabriel A. Bonaterra, Ralf Kinscherf, and Anja Schwarz. 2026. "PACAP and Maxadilan (PAC1 Agonist) Influence Plaque Progression, Migratory Ability, and Mitochondrial Morphology and Dynamics in Vascular Smooth Muscle Cells" Cells 15, no. 12: 1127. https://doi.org/10.3390/cells15121127
APA StyleBrauschke, J., Schütz, L.-M., Bonaterra, G. A., Kinscherf, R., & Schwarz, A. (2026). PACAP and Maxadilan (PAC1 Agonist) Influence Plaque Progression, Migratory Ability, and Mitochondrial Morphology and Dynamics in Vascular Smooth Muscle Cells. Cells, 15(12), 1127. https://doi.org/10.3390/cells15121127