


Targeting Glioma Stem Cells: Therapeutic Opportunities and Challenges


Abstract
1. Introduction
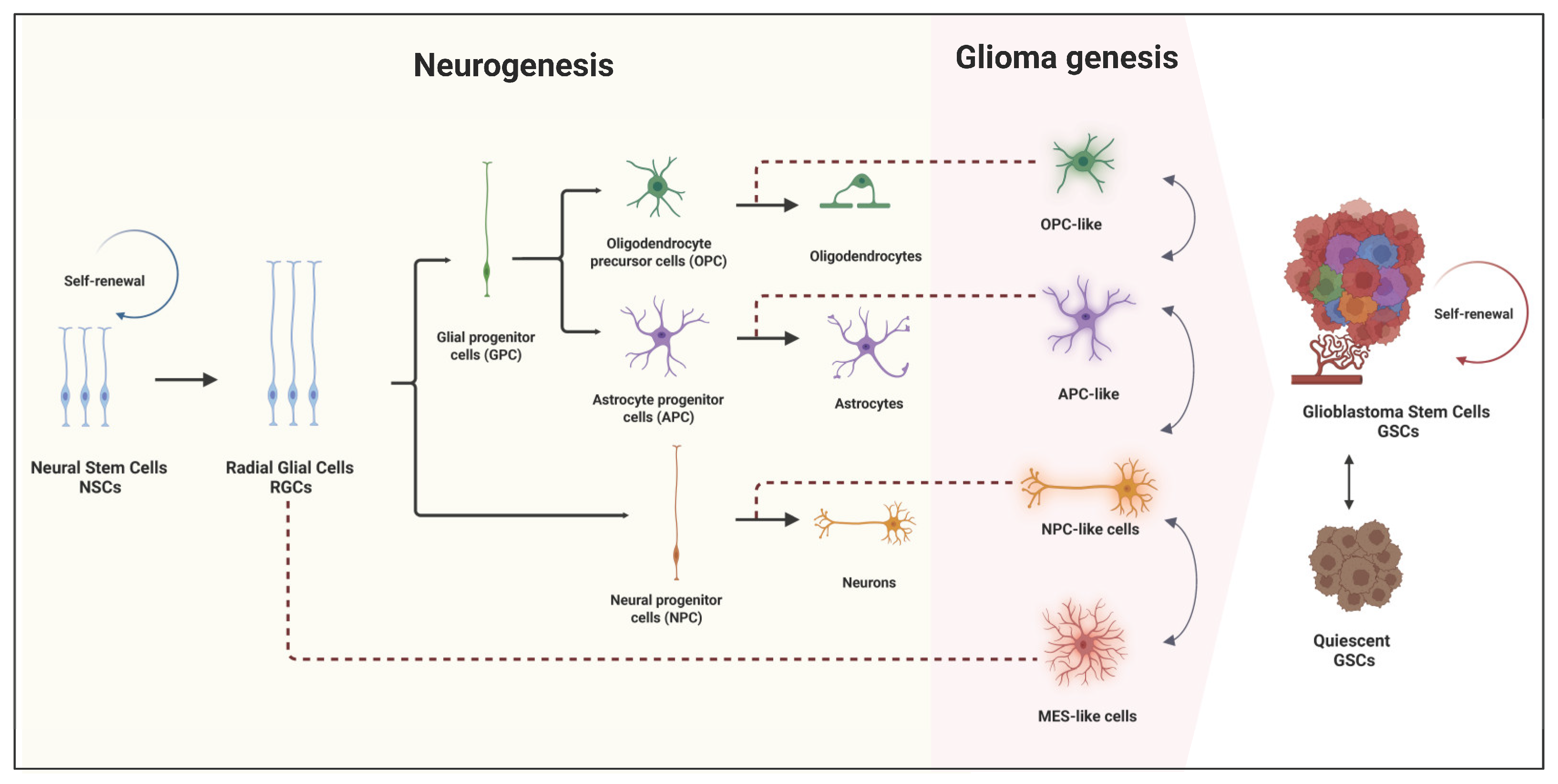
2. Origin and Role of GSCs
3. Therapeutic Opportunities Targeting GSCs
3.1. Targeting GSCs Through Genetic Mechanisms
3.2. Targeting GSCs Through Epigenetic Mechanisms
4. Why Is GBM So Difficult to Defeat?
4.1. Heterogeneity
4.2. Plasticity
4.2.1. Epigenetic Plasticity
4.2.2. Transcriptomic Plasticity
4.3. Quiescence
4.4. Resistance to Therapy
4.4.1. Resistance to Chemotherapy
4.4.2. Resistance to Radiotherapy
5. Perspectives and Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy; Exon Publications: Brisbane, Australia, 2017; pp. 197–241. [Google Scholar]
- Orringer, D.; Lau, D.; Khatri, S.; Zamora-Berridi, G.J.; Zhang, K.; Wu, C.; Chaudhary, N.; Sagher, O. Extent of resection in patients with glioblastoma: Limiting factors, perception of resectability, and effect on survival. J. Neurosurg. 2012, 117, 851–859. [Google Scholar] [CrossRef]
- Murat, A.; Migliavacca, E.; Gorlia, T.; Lambiv, W.L.; Shay, T.; Hamou, M.-F.; De Tribolet, N.; Regli, L.; Wick, W.; Kouwenhoven, M.C. Stem cell–related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J. Clin. Oncol. 2008, 26, 3015–3024. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.L.V.; Gomes, I.N.; Carloni, A.C.; Rosa, M.N.; Da Silva, L.S.; Evangelista, A.F.; Reis, R.M.; Silva, V.A.O. Role of glioblastoma stem cells in cancer therapeutic resistance: A perspective on antineoplastic agents from natural sources and chemical derivatives. Stem Cell Res. Ther. 2021, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.; Pfister, S.M.; Reifenberger, G. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef]
- Clarke, M.F. Clinical and therapeutic implications of cancer stem cells. N. Engl. J. Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Dalerba, P.; Dylla, S.J.; Park, I.-K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef]
- Sanai, N.; Tramontin, A.D.; Quinones-Hinojosa, A.; Barbaro, N.M.; Gupta, N.; Kunwar, S.; Lawton, M.T.; McDermott, M.W.; Parsa, A.T.; Manuel-García Verdugo, J. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 2004, 427, 740–744. [Google Scholar] [CrossRef]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Jackson, E.L.; Garcia-Verdugo, J.M.; Gil-Perotin, S.; Roy, M.; Quinones-Hinojosa, A.; VandenBerg, S.; Alvarez-Buylla, A. PDGFRα-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron 2006, 51, 187–199. [Google Scholar] [CrossRef] [PubMed]
- de Almeida Sassi, F.; Lunardi Brunetto, A.; Schwartsmann, G.; Roesler, R.; Abujamra, A.L. Glioma revisited: From neurogenesis and cancer stem cells to the epigenetic regulation of the niche. J. Oncol. 2012, 2012, 537861. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, R.; Xiong, Y.; Zhou, L.; Yan, X.; Wang, M.; Li, F.; Xie, C.; Zhang, Y.; Huang, Z. Sequential fate-switches in stem-like cells drive the tumorigenic trajectory from human neural stem cells to malignant glioma. Cell Res. 2021, 31, 684–702. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, A.; Di Lullo, E.; Jung, D.; Müller, S.; Crouch, E.E.; Espinosa, C.S.; Ozawa, T.; Alvarado, B.; Spatazza, J.; Cadwell, C.R. Outer radial glia-like cancer stem cells contribute to heterogeneity of glioblastoma. Cell Stem Cell 2020, 26, 48–63.e46. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Zheng, H.; Tomasek, G.J.; Zhang, P.; McKeever, P.E.; Eva, Y.-H.L.; Zhu, Y. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell 2009, 15, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-C.; Kao, C.-Y.; Chung, Y.-F.; Lee, D.-C.; Liu, J.-W.; Chiu, M. Activation of Aurora A kinase through the FGF1/FGFR signaling axis sustains the stem cell characteristics of glioblastoma cells. Exp. Cell Res. 2016, 344, 153–166. [Google Scholar] [CrossRef]
- Joshi, K.; Banasavadi-Siddegowda, Y.; Mo, X.; Kim, S.-H.; Mao, P.; Kig, C.; Nardini, D.; Sobol, R.W.; Chow, L.M.; Kornblum, H.I. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells 2013, 31, 1051–1063. [Google Scholar] [CrossRef]
- Lau, E.O.-C.; Damiani, D.; Chehade, G.; Ruiz-Reig, N.; Saade, R.; Jossin, Y.; Aittaleb, M.; Schakman, O.; Tajeddine, N.; Gailly, P. DIAPH3 deficiency links microtubules to mitotic errors, defective neurogenesis, and brain dysfunction. Elife 2021, 10, e61974. [Google Scholar] [CrossRef]
- Damiani, D.; Goffinet, A.M.; Alberts, A.; Tissir, F. Lack of Diaph3 relaxes the spindle checkpoint causing the loss of neural progenitors. Nat. Commun. 2016, 7, 13509. [Google Scholar] [CrossRef] [PubMed]
- Chehade, G.; El Hajj, N.; Aittaleb, M.; Alkailani, M.I.; Bejaoui, Y.; Mahdi, A.; Aldaalis, A.A.; Verbiest, M.; Lelotte, J.; Ruiz-Reig, N. DIAPH3 predicts survival of patients with MGMT-methylated glioblastoma. Front. Oncol. 2024, 14, 1359652. [Google Scholar] [CrossRef]
- Alkailani, M.I.; Aittaleb, M.; Tissir, F. WNT signaling at the intersection between neurogenesis and brain tumorigenesis. Front. Mol. Neurosci. 2022, 15, 1017568. [Google Scholar] [CrossRef]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Rajakulendran, N.; Rowland, K.J.; Selvadurai, H.J.; Ahmadi, M.; Park, N.I.; Naumenko, S.; Dolma, S.; Ward, R.J.; So, M.; Lee, L. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes. Dev. 2019, 33, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, H.; Mu, X.; Cui, J.; Peng, Z. Dysregulation of Fra1 expression by Wnt/β-catenin signalling promotes glioma aggressiveness through epithelial–mesenchymal transition. Biosci. Rep. 2017, 37, BSR20160643. [Google Scholar] [CrossRef]
- Harwood, D.S.L.; Pedersen, V.; Bager, N.S.; Schmidt, A.Y.; Stannius, T.O.; Areškevičiūtė, A.; Josefsen, K.; Nørøxe, D.S.; Scheie, D.; Rostalski, H. Glioblastoma cells increase expression of notch signaling and synaptic genes within infiltrated brain tissue. Nat. Commun. 2024, 15, 7857. [Google Scholar] [CrossRef]
- Boucherie, C.; Boutin, C.; Jossin, Y.; Schakman, O.; Goffinet, A.M.; Ris, L.; Gailly, P.; Tissir, F. Neural progenitor fate decision defects, cortical hypoplasia and behavioral impairment in Celsr1-deficient mice. Mol. Psychiatry 2018, 23, 723–734. [Google Scholar] [CrossRef]
- Frankel, S.A.; German, W.J. Glioblastoma multiforme: Review of 219 cases with regard to natural history, pathology, diagnostic methods, and treatment. J. Neurosurg. 1958, 15, 489–503. [Google Scholar] [CrossRef]
- Newlands, E.; Stevens, M.; Wedge, S.; Wheelhouse, R.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma-a comprehensive review. Cancer Drug Resist. 2021, 4, 17. [Google Scholar] [CrossRef]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E.; Dowell, J.M.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Wagner, M. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin. Cancer Res. 2007, 13, 1253–1259. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Miyake, K.; Tabei, Y.; Ohara, K.; Sampetrean, O.; Kono, M.; Mizutani, K.; Yamamoto, Y.; Murayama, Y. Histopathological investigation of glioblastomas resected under bevacizumab treatment. Oncotarget 2016, 7, 52423. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Mol, C.D.; Dougan, D.R.; Schneider, T.R.; Skene, R.J.; Kraus, M.L.; Scheibe, D.N.; Snell, G.P.; Zou, H.; Sang, B.-C.; Wilson, K.P. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J. Biol. Chem. 2004, 279, 31655–31663. [Google Scholar] [CrossRef]
- Östman, A.; Böhmer, F.-D. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. 2001, 11, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Yang, K.; Wu, B.; Zhang, Z.; Peng, G.; Huang, J.; Hong, X.; Ding, Q.; Shi, L.; Wang, X.; Zhao, H. Anlotinib for patients with newly diagnosed glioblastoma with unmethylated MGMT promoter: An open-label, single-center, phase 2 clinical trial. J. Clin. Oncol. 2023, 41, e14039. [Google Scholar] [CrossRef]
- Wen, P.Y.; Drappatz, J.; de Groot, J.; Prados, M.D.; Reardon, D.A.; Schiff, D.; Chamberlain, M.; Mikkelsen, T.; Desjardins, A.; Holland, J.; et al. Phase II study of cabozantinib in patients with progressive glioblastoma: Subset analysis of patients naive to antiangiogenic therapy. Neuro Oncol. 2018, 20, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Pugh, S.L.; Gilbert, M.R.; Aldape, K.D.; Geinoz, S.; Beumer, J.H.; Christner, S.M.; Komaki, R.; DeAngelis, L.M.; Gaur, R.; et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro Oncol. 2015, 17, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Anderson, S.K.; Twohy, E.L.; Carrero, X.W.; Dixon, J.G.; Tran, D.D.; Jeyapalan, S.A.; Anderson, D.M.; Kaufmann, T.J.; Feathers, R.W.; et al. A phase 1 and randomized, placebo-controlled phase 2 trial of bevacizumab plus dasatinib in patients with recurrent glioblastoma: Alliance/North Central Cancer Treatment Group N0872. Cancer 2019, 125, 3790–3800. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Schiff, D.; Ahluwalia, M.S.; Lesser, G.J.; Nayak, L.; Lee, E.Q.; Rinne, M.L.; Muzikansky, A.; Dietrich, J.; Purow, B.; et al. Phase II trial of triple tyrosine kinase receptor inhibitor nintedanib in recurrent high-grade gliomas. J. Neurooncol. 2015, 121, 297–302. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Lamborn, K.R.; Robins, H.I.; Mehta, M.P.; Chang, S.M.; Butowski, N.A.; Deangelis, L.M.; Abrey, L.E.; Zhang, W.T.; Prados, M.D.; et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro Oncol. 2010, 12, 855–861. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016, 18, 557–564. [Google Scholar] [CrossRef]
- Mendez, J.S.; Cohen, A.L.; Eckenstein, M.; Jensen, R.L.; Burt, L.M.; Salzman, K.L.; Chamberlain, M.; Hsu, H.H.; Hutchinson, M.; Iwamoto, F.; et al. Phase 1b/2 study of orally administered pexidartinib in combination with radiation therapy and temozolomide in patients with newly diagnosed glioblastoma. Neurooncol. Adv. 2024, 6, vdae202. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Muzikansky, A.; Duda, D.G.; Gaffey, S.; Dietrich, J.; Nayak, L.; Chukwueke, U.N.; Beroukhim, R.; Doherty, L.; Laub, C.K.; et al. Phase II trial of ponatinib in patients with bevacizumab-refractory glioblastoma. Cancer Med. 2019, 8, 5988–5994. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Grisanti, S.; Ferrari, V.D.; Buglione, M.; Agazzi, G.M.; Liserre, R.; Poliani, L.; Buttolo, L.; Gipponi, S.; Pedersini, R.; Consoli, F.; et al. Second line treatment of recurrent glioblastoma with sunitinib: Results of a phase II study and systematic review of literature. J. Neurosurg. Sci. 2019, 63, 458–467. [Google Scholar] [CrossRef]
- Lee, E.Q.; Kaley, T.J.; Duda, D.G.; Schiff, D.; Lassman, A.B.; Wong, E.T.; Mikkelsen, T.; Purow, B.W.; Muzikansky, A.; Ancukiewicz, M.; et al. A Multicenter, Phase II, Randomized, Noncomparative Clinical Trial of Radiation and Temozolomide with or without Vandetanib in Newly Diagnosed Glioblastoma Patients. Clin. Cancer Res. 2015, 21, 3610–3618. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Vredenburgh, J.J.; Desjardins, A.; Peters, K.; Gururangan, S.; Sampson, J.H.; Marcello, J.; Herndon, J.E., 2nd; McLendon, R.E.; Janney, D.; et al. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: Results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J. Neurooncol. 2011, 101, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Schiff, D.; Jaeckle, K.A.; Anderson, S.K.; Galanis, E.; Giannini, C.; Buckner, J.C.; Stella, P.; Flynn, P.J.; Erickson, B.J.; Schwerkoske, J.F. Phase 1/2 trial of temsirolimus and sorafenib in the treatment of patients with recurrent glioblastoma: North Central Cancer Treatment Group Study/Alliance N0572. Cancer 2018, 124, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Kuhn, J.; Lamborn, K.R.; Abrey, L.; DeAngelis, L.M.; Lieberman, F.; Robins, H.I.; Chang, S.M.; Yung, W.K.; Drappatz, J.; et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro Oncol. 2012, 14, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): A north central cancer treatment group trial. Clin. Cancer Res. 2013, 19, 4816–4823. [Google Scholar] [CrossRef]
- Lassman, A.B.; Pugh, S.L.; Wang, T.J.C.; Aldape, K.; Gan, H.K.; Preusser, M.; Vogelbaum, M.A.; Sulman, E.P.; Won, M.; Zhang, P.; et al. Depatuxizumab mafodotin in EGFR-amplified newly diagnosed glioblastoma: A phase III randomized clinical trial. Neuro Oncol. 2023, 25, 339–350. [Google Scholar] [CrossRef]
- Cabezas-Camarero, S.; Pérez-Alfayate, R.; Polidura, C.; Gómez-Ruiz, M.N.; Gil-Martínez, L.; Casado-Fariñas, I.; Bartolomé, J.; Pérez-Segura, P. Durable benefit and slowdown in tumor growth dynamics with erdafitinib in a FGFR3-TACC3 fusion-positive IDH-wild type glioblastoma. Neurooncol. Adv. 2024, 6, vdae139. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Wang, M.; Robins, H.I.; Lautenschlaeger, T.; Curran, W.J.; Brachman, D.G.; Schultz, C.J.; Choucair, A.; Dolled-Filhart, M.; Christiansen, J.; et al. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1206–1211. [Google Scholar] [CrossRef]
- Uhm, J.H.; Ballman, K.V.; Wu, W.; Giannini, C.; Krauss, J.C.; Buckner, J.C.; James, C.D.; Scheithauer, B.W.; Behrens, R.J.; Flynn, P.J.; et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 347–353. [Google Scholar] [CrossRef]
- Liang, J.; Zubovitz, J.; Petrocelli, T.; Kotchetkov, R.; Connor, M.K.; Han, K.; Lee, J.H.; Ciarallo, S.; Catzavelos, C.; Beniston, R.; et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 2002, 8, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; Cavallo, G.; Lonardi, S.; Magrini, E.; Tosoni, A.; Grosso, D.; Scopece, L.; Blatt, V.; Urbini, B.; Pession, A.; et al. Gefitinib in patients with progressive high-grade gliomas: A multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br. J. Cancer 2007, 96, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Lassman, A.B.; Mischel, P.S.; Rosen, N.; Scher, H.I.; Teruya-Feldstein, J.; Shaffer, D.; Lis, E.; Abrey, L.E. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM). J. Neurooncol. 2009, 92, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Endersby, R.; Whitehouse, J.; Hii, H.; Greenall, S.A.; Johns, T.G.; Gottardo, N.G. A Pre-Clinical Assessment of the Pan-ERBB Inhibitor Dacomitinib in Pediatric and Adult Brain Tumors. Neoplasia 2018, 20, 432–442. [Google Scholar] [CrossRef]
- Sepúlveda-Sánchez, J.M.; Vaz, M.; Balañá, C.; Gil-Gil, M.; Reynés, G.; Gallego, Ó.; Martínez-García, M.; Vicente, E.; Quindós, M.; Luque, R.; et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol. 2017, 19, 1522–1531. [Google Scholar] [CrossRef]
- Chi, A.S.; Cahill, D.P.; Reardon, D.A.; Wen, P.Y.; Mikkelsen, T.; Peereboom, D.M.; Wong, E.T.; Gerstner, E.R.; Dietrich, J.; Plotkin, S.R.; et al. Exploring Predictors of Response to Dacomitinib in EGFR-Amplified Recurrent Glioblastoma. JCO Precis. Oncol. 2020, 4, 593–613. [Google Scholar] [CrossRef]
- Thiessen, B.; Stewart, C.; Tsao, M.; Kamel-Reid, S.; Schaiquevich, P.; Mason, W.; Easaw, J.; Belanger, K.; Forsyth, P.; McIntosh, L.; et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010, 65, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Chagoya, G.; Kwatra, S.G.; Nanni, C.W.; Roberts, C.M.; Phillips, S.M.; Nullmeyergh, S.; Gilmore, S.P.; Spasojevic, I.; Corcoran, D.L.; Young, C.C.; et al. Efficacy of osimertinib against EGFRvIII+ glioblastoma. Oncotarget 2020, 11, 2074–2082. [Google Scholar] [CrossRef]
- Makhlin, I.; Salinas, R.D.; Zhang, D.; Jacob, F.; Ming, G.L.; Song, H.; Saxena, D.; Dorsey, J.F.; Nasrallah, M.P.; Morrissette, J.J.; et al. Clinical activity of the EGFR tyrosine kinase inhibitor osimertinib in EGFR-mutant glioblastoma. CNS Oncol. 2019, 8, Cns43. [Google Scholar] [CrossRef]
- Cardona, A.F.; Jaramillo-Velásquez, D.; Ruiz-Patiño, A.; Polo, C.; Jiménez, E.; Hakim, F.; Gómez, D.; Ramón, J.F.; Cifuentes, H.; Mejía, J.A.; et al. Efficacy of osimertinib plus bevacizumab in glioblastoma patients with simultaneous EGFR amplification and EGFRvIII mutation. J. Neurooncol. 2021, 154, 353–364. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Chen, Y.-H.; Chang, C.-H.; Liang, H.-K.T. Recurrent fibroblast growth factor receptor3 fusion glioblastoma treated with pemigatinib: A case report and review of the literature. Neuro-Oncol. Adv. 2024, 6, vdae072. [Google Scholar] [CrossRef] [PubMed]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J.; et al. AZD4547: An Orally Bioavailable, Potent, and Selective Inhibitor of the Fibroblast Growth Factor Receptor Tyrosine Kinase Family. Cancer Res. 2012, 72, 2045–2056. [Google Scholar] [CrossRef]
- Komla-Ebri, D.; Dambroise, E.; Kramer, I.; Benoist-Lasselin, C.; Kaci, N.; Le Gall, C.; Martin, L.; Busca, P.; Barbault, F.; Graus-Porta, D.; et al. Tyrosine kinase inhibitor NVP-BGJ398 functionally improves FGFR3-related dwarfism in mouse model. J. Clin. Investig. 2016, 126, 1871–1884. [Google Scholar] [CrossRef] [PubMed]
- Borad, M.J.; Bridgewater, J.A.; Morizane, C.; Shroff, R.T.; Oh, D.-Y.; Moehler, M.H.; Furuse, J.; Benhadji, K.A.; He, H.; Valle, J.W. A phase III study of futibatinib (TAS-120) versus gemcitabine-cisplatin (gem-cis) chemotherapy as first-line (1L) treatment for patients (pts) with advanced (adv) cholangiocarcinoma (CCA) harboring fibroblast growth factor receptor 2 (FGFR2) gene rearrangements (FOENIX-CCA3). J. Clin. Oncol. 2020, 38, TPS600. [Google Scholar]
- Chiba, Y.; Sudo, K.; Kojima, Y.; Okuma, H.; Kohsaka, S.; Machida, R.; Ichimura, M.; Anjo, K.; Kurishita, K.; Okita, N.; et al. A multicenter investigator-initiated Phase 2 trial of E7090 in patients with advanced or recurrent solid tumor with fibroblast growth factor receptor (FGFR) gene alteration: FORTUNE trial. BMC Cancer 2022, 22, 869. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Schuler, M.; Kang, Y.-K.; Yen, C.-J.; Edeline, J.; Choo, S.P.; Lin, C.-C.; Okusaka, T.; Weiss, K.-H.; Macarulla, T.; et al. A first-in-human phase 1/2 study of FGF401 and combination of FGF401 with spartalizumab in patients with hepatocellular carcinoma or biomarker-selected solid tumors. J. Exp. Clin. Cancer Res. 2022, 41, 189. [Google Scholar] [CrossRef] [PubMed]
- Joshi, J.J.; Coffey, H.; Corcoran, E.; Tsai, J.; Huang, C.-L.; Ichikawa, K.; Prajapati, S.; Hao, M.-H.; Bailey, S.; Wu, J.; et al. H3B-6527 Is a Potent and Selective Inhibitor of FGFR4 in FGF19-Driven Hepatocellular Carcinoma. Cancer Res. 2017, 77, 6999–7013. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Han, R.; Wang, Z.; Yang, Q.; Huang, Y.; Yan, Y. BLU-554, A selective inhibitor of FGFR4, exhibits anti-tumour activity against gastric cancer in vitro. Biochem. Biophys. Res. Commun. 2022, 595, 22–27. [Google Scholar] [CrossRef]
- Michael, M.; Bang, Y.J.; Park, Y.S.; Kang, Y.K.; Kim, T.M.; Hamid, O.; Thornton, D.; Tate, S.C.; Raddad, E.; Tie, J. A Phase 1 Study of LY2874455, an Oral Selective pan-FGFR Inhibitor, in Patients with Advanced Cancer. Target. Oncol. 2017, 12, 463–474. [Google Scholar] [CrossRef]
- Zhou, W.; Hur, W.; McDermott, U.; Dutt, A.; Xian, W.; Ficarro, S.B.; Zhang, J.; Sharma, S.V.; Brugge, J.; Meyerson, M.; et al. A structure-guided approach to creating covalent FGFR inhibitors. Chem. Biol. 2010, 17, 285–295. [Google Scholar] [CrossRef]
- Nguyen, P.T.; Tsunematsu, T.; Yanagisawa, S.; Kudo, Y.; Miyauchi, M.; Kamata, N.; Takata, T. The FGFR1 inhibitor PD173074 induces mesenchymal-epithelial transition through the transcription factor AP-1. Br. J. Cancer 2013, 109, 2248–2258. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.K.; Németh, G.; Ng, Y.R.; Pang, E.; Szántai-Kis, C.; Zsákai, L.; Breza, N.; Greff, Z.; Horváth, Z.; Pató, J.; et al. Developing FGFR4 inhibitors as potential anti-cancer agents via in silico design, supported by in vitro and cell-based testing. Curr. Med. Chem. 2013, 20, 1203–1217. [Google Scholar] [CrossRef]
- Hagel, M.; Miduturu, C.; Sheets, M.; Rubin, N.; Weng, W.; Stransky, N.; Bifulco, N.; Kim, J.L.; Hodous, B.; Brooijmans, N.; et al. First Selective Small Molecule Inhibitor of FGFR4 for the Treatment of Hepatocellular Carcinomas with an Activated FGFR4 Signaling Pathway. Cancer Discov. 2015, 5, 424–437. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.; Azaro, A.; De Vos, F.; Sepulveda, J.; Yung, W.K.A.; Wen, P.Y.; Lassman, A.B.; Joerger, M.; Tabatabai, G.; Rodon, J.; et al. A Phase Ib/II, open-label, multicenter study of INC280 (capmatinib) alone and in combination with buparlisib (BKM120) in adult patients with recurrent glioblastoma. J. Neurooncol. 2020, 146, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Martínez-García, M.; Velasco, G.; Pineda, E.; Gil-Gil, M.; Alameda, F.; Capellades, J.; Martín-Soberón, M.C.; López-Valero, I.; Tovar Ambel, E.; Foro, P.; et al. Safety and Efficacy of Crizotinib in Combination with Temozolomide and Radiotherapy in Patients with Newly Diagnosed Glioblastoma: Phase Ib GEINO 1402 Trial. Cancers 2022, 14, 2393. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.R.; Rath, P.; Oyinlade, O.; Lopez, H.; Mughal, S.; Xia, S.; Li, Y.; Kaur, H.; Zhou, X.; Ahmed, A.K.; et al. Crizotinib and erlotinib inhibits growth of c-Met(+)/EGFRvIII(+) primary human glioblastoma xenografts. Clin. Neurol. Neurosurg. 2018, 171, 26–33. [Google Scholar] [CrossRef]
- Shi, F.; Zhang, J.; Liu, H.; Wu, L.; Jiang, H.; Wu, Q.; Liu, T.; Lou, M.; Wu, H. The dual PI3K/mTOR inhibitor dactolisib elicits anti-tumor activity in vitro and in vivo. Oncotarget 2018, 9, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Netland, I.A.; Førde, H.E.; Sleire, L.; Leiss, L.; Rahman, M.A.; Skeie, B.S.; Gjerde, C.H.; Enger, P.; Goplen, D. Dactolisib (NVP-BEZ235) toxicity in murine brain tumour models. BMC Cancer 2016, 16, 657. [Google Scholar] [CrossRef]
- Wen, P.Y.; Omuro, A.; Ahluwalia, M.S.; Fathallah-Shaykh, H.M.; Mohile, N.; Lager, J.J.; Laird, A.D.; Tang, J.; Jiang, J.; Egile, C.; et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015, 17, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Salphati, L.; Alicke, B.; Heffron, T.P.; Shahidi-Latham, S.; Nishimura, M.; Cao, T.; Carano, R.A.; Cheong, J.; Greve, J.; Koeppen, H.; et al. Brain Distribution and Efficacy of the Brain Penetrant PI3K Inhibitor GDC-0084 in Orthotopic Mouse Models of Human Glioblastoma. Drug Metab. Dispos. 2016, 44, 1881–1889. [Google Scholar] [CrossRef]
- Wen, P.Y.; De Groot, J.F.; Battiste, J.; Goldlust, S.A.; Garner, J.S.; Friend, J.; Simpson, J.A.; Damek, D.; Olivero, A.; Cloughesy, T.F. Paxalisib in patients with newly diagnosed glioblastoma with unmethylated MGMT promoter status: Final phase 2 study results. J. Clin. Oncol. 2022, 40, 2047. [Google Scholar] [CrossRef]
- Wen, P.Y.; Cloughesy, T.F.; Olivero, A.G.; Morrissey, K.M.; Wilson, T.R.; Lu, X.; Mueller, L.U.; Coimbra, A.F.; Ellingson, B.M.; Gerstner, E.; et al. First-in-Human Phase I Study to Evaluate the Brain-Penetrant PI3K/mTOR Inhibitor GDC-0084 in Patients with Progressive or Recurrent High-Grade Glioma. Clin. Cancer Res. 2020, 26, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.R.; Duchatel, R.J.; Staudt, D.E.; Persson, M.L.; Mannan, A.; Yadavilli, S.; Parackal, S.; Game, S.; Chong, W.C.; Jayasekara, W.S.N.; et al. ONC201 in Combination with Paxalisib for the Treatment of H3K27-Altered Diffuse Midline Glioma. Cancer Res. 2023, 83, 2421–2437. [Google Scholar] [CrossRef]
- Wen, P.Y.; Rodon, J.A.; Mason, W.; Beck, J.T.; DeGroot, J.; Donnet, V.; Mills, D.; El-Hashimy, M.; Rosenthal, M. Phase I, open-label, multicentre study of buparlisib in combination with temozolomide or with concomitant radiation therapy and temozolomide in patients with newly diagnosed glioblastoma. ESMO Open 2020, 5, e000673. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Becker, K.P.; Mekhail, T.; Chowdhary, S.A.; Eakle, J.F.; Wright, D.; Langdon, R.M.; Yost, K.J.; Padula, G.D.A.; West-Osterfield, K.; et al. Phase I/II study of bevacizumab with BKM120, an oral PI3K inhibitor, in patients with refractory solid tumors (phase I) and relapsed/refractory glioblastoma (phase II). J. Neurooncol. 2019, 144, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-f.; Wang, J.; Shao, W.; Wu, C.-p.; Chen, Z.-p.; To, S.-s.T.; Li, W.-p. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Pitz, M.W.; Eisenhauer, E.A.; MacNeil, M.V.; Thiessen, B.; Easaw, J.C.; Macdonald, D.R.; Eisenstat, D.D.; Kakumanu, A.S.; Salim, M.; Chalchal, H.; et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro Oncol. 2015, 17, 1270–1274. [Google Scholar] [CrossRef]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro Oncol. 2015, 17, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; van den Bent, M.J.; Taphoorn, M.J.; Steuve, J.; Brandes, A.A.; Hamou, M.F.; Wick, A.; et al. Phase II Study of Radiotherapy and Temsirolimus versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation (EORTC 26082). Clin. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef]
- Galanis, E.; Buckner, J.C.; Maurer, M.J.; Kreisberg, J.I.; Ballman, K.; Boni, J.; Peralba, J.M.; Jenkins, R.B.; Dakhil, S.R.; Morton, R.F.; et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: A North Central Cancer Treatment Group Study. J. Clin. Oncol. 2005, 23, 5294–5304. [Google Scholar] [CrossRef]
- Turcan, S.; Fabius, A.W.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Rei, M.; Woodhouse, I.; Ferris, K.; Kirschner, S.; Chandran, A.; Gileadi, U.; Chen, J.L.; Pereira Pinho, M.; Ariosa-Morejon, Y.; et al. Decitabine increases neoantigen and cancer testis antigen expression to enhance T-cell-mediated toxicity against glioblastoma. Neuro Oncol. 2022, 24, 2093–2106. [Google Scholar] [CrossRef]
- Kratzsch, T.; Kuhn, S.A.; Joedicke, A.; Hanisch, U.K.; Vajkoczy, P.; Hoffmann, J.; Fichtner, I. Treatment with 5-azacitidine delay growth of glioblastoma xenografts: A potential new treatment approach for glioblastomas. J. Cancer Res. Clin. Oncol. 2018, 144, 809–819. [Google Scholar] [CrossRef]
- Xu, K.; Ramesh, K.; Huang, V.; Gurbani, S.S.; Cordova, J.S.; Schreibmann, E.; Weinberg, B.D.; Sengupta, S.; Voloschin, A.D.; Holdhoff, M.; et al. Final Report on Clinical Outcomes and Tumor Recurrence Patterns of a Pilot Study Assessing Efficacy of Belinostat (PXD-101) with Chemoradiation for Newly Diagnosed Glioblastoma. Tomography 2022, 8, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Reardon, D.A.; Schiff, D.; Drappatz, J.; Muzikansky, A.; Grimm, S.A.; Norden, A.D.; Nayak, L.; Beroukhim, R.; Rinne, M.L.; et al. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro Oncol. 2015, 17, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Jaeckle, K.A.; Maurer, M.J.; Reid, J.M.; Ames, M.M.; Hardwick, J.S.; Reilly, J.F.; Loboda, A.; Nebozhyn, M.; Fantin, V.R.; et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: A north central cancer treatment group study. J. Clin. Oncol. 2009, 27, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. Embo J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef]
- Han, W.; Guan, W. Valproic Acid: A Promising Therapeutic Agent in Glioma Treatment. Front. Oncol. 2021, 11, 687362. [Google Scholar] [CrossRef]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Beer, P.A.; Cartwright, C.A.; Haymaker, C.; Vo, H.H.; Kiany, S.; Cecil, A.R.L.; Dow, J.; Haque, K.; Silva, F.A.; et al. Preclinical Development and First-in-Human Study of KA2507, a Selective and Potent Inhibitor of Histone Deacetylase 6, for Patients with Refractory Solid Tumors. Clin. Cancer Res. 2021, 27, 3584–3594. [Google Scholar] [CrossRef]
- Martín, V.; Sanchez-Sanchez, A.M.; Herrera, F.; Gomez-Manzano, C.; Fueyo, J.; Alvarez-Vega, M.A.; Antolín, I.; Rodriguez, C. Melatonin-induced methylation of the ABCG2/BCRP promoter as a novel mechanism to overcome multidrug resistance in brain tumour stem cells. Br. J. Cancer 2013, 108, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhou, L.; Zhao, W.; Yang, H.; Lu, Z.; Zhang, L.; Zhang, Y.; Xie, Y.; Lu, H.; Han, W. The combination of temozolomide and perifosine synergistically inhibit glioblastoma by impeding DNA repair and inducing apoptosis. Cell Death Discov. 2024, 10, 315. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133+ glioma stem cells to temozolomide therapy. Mol. Med. 2011, 17, 103–112. [Google Scholar] [CrossRef]
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.A.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H. RAD51 is a selective DNA repair target to radiosensitize glioma stem cells. Stem Cell Rep. 2017, 8, 125–139. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Lin, B.; Ziebro, J.; Smithberger, E.; Skinner, K.R.; Zhao, E.; Cloughesy, T.F.; Binder, Z.A.; O’Rourke, D.M.; Nathanson, D.A.; Furnari, F.B. EGFR, the Lazarus target for precision oncology in glioblastoma. Neuro-Oncol. 2022, 24, 2035–2062. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.D.; Krishnan, S.; Sarkaria, J.N.; Wu, W.; Jaeckle, K.A.; Uhm, J.H.; Geoffroy, F.J.; Arusell, R.; Kitange, G.; Jenkins, R.B.; et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J. Clin. Oncol. 2008, 26, 5603–5609. [Google Scholar] [CrossRef]
- Prados, M.D.; Chang, S.M.; Butowski, N.; DeBoer, R.; Parvataneni, R.; Carliner, H.; Kabuubi, P.; Ayers-Ringler, J.; Rabbitt, J.; Page, M.; et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J. Clin. Oncol. 2009, 27, 579–584. [Google Scholar] [CrossRef]
- Clarke, J.L.; Molinaro, A.M.; Phillips, J.J.; Butowski, N.A.; Chang, S.M.; Perry, A.; Costello, J.F.; DeSilva, A.A.; Rabbitt, J.E.; Prados, M.D. A single-institution phase II trial of radiation, temozolomide, erlotinib, and bevacizumab for initial treatment of glioblastoma. Neuro Oncol. 2014, 16, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Giglio, P.; Hu, J.; Groves, M.; Merrell, R.; Conrad, C.; Phuphanich, S.; Puduvalli, V.K.; Loghin, M.; Paleologos, N.; et al. A phase II study of bevacizumab and erlotinib after radiation and temozolomide in MGMT unmethylated GBM patients. J. Neurooncol. 2016, 126, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.K.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y.; et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010, 12, 95–103. [Google Scholar] [CrossRef]
- Yung, W.K.; Vredenburgh, J.J.; Cloughesy, T.F.; Nghiemphu, P.; Klencke, B.; Gilbert, M.R.; Reardon, D.A.; Prados, M.D. Safety and efficacy of erlotinib in first-relapse glioblastoma: A phase II open-label study. Neuro Oncol. 2010, 12, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef]
- de Groot, J.F.; Gilbert, M.R.; Aldape, K.; Hess, K.R.; Hanna, T.A.; Ictech, S.; Groves, M.D.; Conrad, C.; Colman, H.; Puduvalli, V.K.; et al. Phase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastoma. J. Neurooncol. 2008, 90, 89–97. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Ahluwalia, M.S.; Ye, X.; Supko, J.G.; Hilderbrand, S.L.; Phuphanich, S.; Nabors, L.B.; Rosenfeld, M.R.; Mikkelsen, T.; Grossman, S.A. NABTT 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol. 2013, 15, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Gururangan, S.; Friedman, A.H.; Herndon, J.E., 2nd; Marcello, J.; Norfleet, J.A.; McLendon, R.E.; Sampson, J.H.; et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J. Neurooncol. 2010, 96, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Norden, A.D.; Cloughesy, T.F.; Robins, H.I.; Lieberman, F.S.; Gilbert, M.R.; Mehta, M.P.; et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro Oncol. 2014, 16, 567–578. [Google Scholar] [CrossRef]
- Sathornsumetee, S.; Desjardins, A.; Vredenburgh, J.J.; McLendon, R.E.; Marcello, J.; Herndon, J.E.; Mathe, A.; Hamilton, M.; Rich, J.N.; Norfleet, J.A.; et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Oncol. 2010, 12, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandris, Q.G.; Montano, N.; Cenci, T.; Martini, M.; Lauretti, L.; Bianchi, F.; Larocca, L.M.; Maira, G.; Fernandez, E.; Pallini, R. Targeted therapy with bevacizumab and erlotinib tailored to the molecular profile of patients with recurrent glioblastoma. Preliminary experience. Acta Neurochir. 2013, 155, 33–40. [Google Scholar] [CrossRef]
- Zalcman, N.; Gutreiman, M.; Shahar, T.; Weller, M.; Lavon, I. Androgen receptor activation in glioblastoma can be achieved by ligand-independent signaling through EGFR—A potential therapeutic target. Int. J. Mol. Sci. 2021, 22, 10954. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wang, F.; Ahmed, S.; Liu, K.; Zhang, C.; Cathcart, S.J.; DiMaio, D.J.; Punsoni, M.; Guan, B.; Zhou, P. Androgen receptor, although not a specific marker for, is a novel target to suppress glioma stem cells as a therapeutic strategy for glioblastoma. Front. Oncol. 2021, 11, 616625. [Google Scholar] [CrossRef]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C. Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Dono, A.; El Achi, H.; Bundrant, B.E.; Goli, P.S.; Zhu, P.; Ozkizilkaya, H.I.; Esquenazi, Y.; Ballester, L.Y. Infiltrating gliomas with FGFR alterations: Histologic features, genetic alterations, and potential clinical implications. Cancer Biomark. 2023, 36, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Chen, T.; Chen, S.; Song, C.; Shan, D.; Xu, S.; Xu, S. Case report: Identification of multiple TERT and FGFR2 gene fusions in a pineal region glioblastoma case. Front. Oncol. 2021, 11, 739309. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.-M.; Islam, M.Z.; Li, Y.; Traylor, J.; Nanda, A. Novel targetable FGFR2 and FGFR3 alterations in glioblastoma associate with aggressive phenotype and distinct gene expression programs. Acta Neuropathol. Commun. 2021, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Sepúlveda-Sánchez, J.M.; Cloughesy, T.F.; Gil-Gil, M.J.; Puduvalli, V.K.; Raizer, J.J.; De Vos, F.Y.F.; Wen, P.Y.; Butowski, N.A.; Clement, P.M.J.; et al. Infigratinib in Patients with Recurrent Gliomas and FGFR Alterations: A Multicenter Phase II Study. Clin. Cancer Res. 2022, 28, 2270–2277. [Google Scholar] [CrossRef]
- Garnett, J.; Chumbalkar, V.; Vaillant, B.; Gururaj, A.E.; Hill, K.S.; Latha, K.; Yao, J.; Priebe, W.; Colman, H.; Elferink, L.A.; et al. Regulation of HGF expression by ΔEGFR-mediated c-Met activation in glioblastoma cells. Neoplasia 2013, 15, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Saunders, V.C.; Lafitte, M.; Adrados, I.; Quereda, V.; Feurstein, D.; Ling, Y.; Fallahi, M.; Rosenberg, L.H.; Duckett, D.R. Identification of an EGFRvIII-JNK2-HGF/c-Met-Signaling Axis Required for Intercellular Crosstalk and Glioblastoma Multiforme Cell Invasion. Mol. Pharmacol. 2015, 88, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.; Kim, S.I.; Park, C.K.; Paek, S.H.; Lee, S.T.; Park, S.H. C-MET overexpression and amplification in gliomas. Int. J. Clin. Exp. Pathol. 2015, 8, 14932–14938. [Google Scholar] [PubMed]
- Testa, J.R.; Bellacosa, A. AKT plays a central role in tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10983–10985. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd. Overcoming acquired resistance to anticancer therapy: Focus on the PI3K/AKT/mTOR pathway. Cancer Chemother. Pharmacol. 2013, 71, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [PubMed]
- Atkins, R.J.; Stylli, S.S.; Luwor, R.B.; Kaye, A.H.; Hovens, C.M. Glycogen synthase kinase-3β (GSK-3β) and its dysregulation in glioblastoma multiforme. J. Clin. Neurosci. 2013, 20, 1185–1192. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients With Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef]
- Klughammer, J.; Kiesel, B.; Roetzer, T.; Fortelny, N.; Nemc, A.; Nenning, K.H.; Furtner, J.; Sheffield, N.C.; Datlinger, P.; Peter, N.; et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med. 2018, 24, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Hu, J.; Gomez, G.G.; Taylor, T.; Villa, G.R.; Pizzo, D.; VandenBerg, S.R.; Thorne, A.H.; Chen, C.C.; Mischel, P.S.; et al. A urokinase receptor-Bim signaling axis emerges during EGFR inhibitor resistance in mutant EGFR glioblastoma. Cancer Res. 2015, 75, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wang, M.; Zhang, G.; Bao, Y.; Wu, Y.; Li, X.; Yang, W.; Cui, H. E2F7-EZH2 axis regulates PTEN/AKT/mTOR signalling and glioblastoma progression. Br. J. Cancer 2020, 123, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sun, Y.; Qi, X.; Gordon, R.E.; O’Brien, J.A.; Yuan, H.; Zhang, J.; Wang, Z.; Zhang, M.; Song, Y.; et al. EZH2 Phosphorylation Promotes Self-Renewal of Glioma Stem-Like Cells Through NF-κB Methylation. Front. Oncol. 2019, 9, 641. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hu, P.; Tang, F.; Lian, H.; Chen, X.; Zhang, Y.; He, X.; Liu, W.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016, 379, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.S.; Hubbert, C.C.; Yao, T.P. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J. Biol. Chem. 2010, 285, 11219–11226. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Williams, K.A.; Zhang, M.; Xiang, S.; Hu, C.; Wu, J.Y.; Zhang, S.; Ryan, M.; Cox, A.D.; Der, C.J.; Fang, B.; et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem. 2013, 288, 33156–33170. [Google Scholar] [CrossRef]
- Ghiaseddin, A.; Reardon, D.; Massey, W.; Mannerino, A.; Lipp, E.S.; Herndon, J.E., 2nd; McSherry, F.; Desjardins, A.; Randazzo, D.; Friedman, H.S.; et al. Phase II Study of Bevacizumab and Vorinostat for Patients with Recurrent World Health Organization Grade 4 Malignant Glioma. Oncologist 2018, 23, 157-e121. [Google Scholar] [CrossRef]
- Friday, B.B.; Anderson, S.K.; Buckner, J.; Yu, C.; Giannini, C.; Geoffroy, F.; Schwerkoske, J.; Mazurczak, M.; Gross, H.; Pajon, E.; et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: A north central cancer treatment group study. Neuro Oncol. 2012, 14, 215–221. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: Results of Alliance N0874/ABTC 02. Neuro Oncol. 2018, 20, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Day, B.W.; Harrup, R.; Johns, T.G.; Lwin, Z.; Scott, A.M.; Sim, H.W.; Koh, E.S. Clinical Trials in the Brain Tumour Population: Challenges and Strategies for the Future. Curr. Oncol. Rep. 2023, 25, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, A.V.; Dostdar, S.A.; Attwood, M.M.; Krasilnikova, A.A.; Ilina, A.A.; Nabieva, A.S.; Lisitsyna, A.A.; Chubarev, V.N.; Tarasov, V.V.; Schioth, H.B. Brain Cancer Drug Discovery: Clinical Trials, Drug Classes, Targets, and Combinatorial Therapies. Pharmacol. Rev. 2021, 73, 1172–1203. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining tumor niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; DeCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma stem cells: Driving resilience through chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Orzan, F.; De Bacco, F.; Crisafulli, G.; Pellegatta, S.; Mussolin, B.; Siravegna, G.; D’Ambrosio, A.; Comoglio, P.M.; Finocchiaro, G.; Boccaccio, C. Genetic evolution of glioblastoma stem-like cells from primary to recurrent tumor. Stem Cells 2017, 35, 2218–2228. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Tirosh, I. The glioma stem cell model in the era of single-cell genomics. Cancer Cell 2020, 37, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Jin, X.; Jung, J.-E.; Beck, S.; Kim, H. Cell surface Nestin is a biomarker for glioma stem cells. Biochem. Biophys. Res. Commun. 2013, 433, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; DePinho, R.A. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53, 503–517. [Google Scholar] [CrossRef]
- Annovazzi, L.; Mellai, M.; Caldera, V.; Valente, G.; Schiffer, D. SOX2 expression and amplification in gliomas and glioma cell lines. Cancer Genom. Proteom. 2011, 8, 139–147. [Google Scholar]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; MacSwords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef]
- Son, M.J.; Woolard, K.; Nam, D.-H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef]
- Rasper, M.; Schäfer, A.; Piontek, G.; Teufel, J.; Brockhoff, G.; Ringel, F.; Heindl, S.; Zimmer, C.; Schlegel, J. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro-Oncol. 2010, 12, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Nishimura, M.C.; Bumbaca, S.M.; Kharbanda, S.; Forrest, W.F.; Kasman, I.M.; Greve, J.M.; Soriano, R.H.; Gilmour, L.L.; Rivers, C.S. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.; Leite, S.; Sauvageot, N.; Sarkisjan, D. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Richards, L.M.; Whitley, O.K.; MacLeod, G.; Cavalli, F.M.; Coutinho, F.J.; Jaramillo, J.E.; Svergun, N.; Riverin, M.; Croucher, D.C.; Kushida, M. Gradient of developmental and injury response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer 2021, 2, 157–173. [Google Scholar] [CrossRef]
- Varn, F.S.; Johnson, K.C.; Martinek, J.; Huse, J.T.; Nasrallah, M.P.; Wesseling, P.; Cooper, L.A.; Malta, T.M.; Wade, T.E.; Sabedot, T.S. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 2022, 185, 2184–2199.e2116. [Google Scholar] [CrossRef]
- Larsson, I.; Dalmo, E.; Elgendy, R.; Niklasson, M.; Doroszko, M.; Segerman, A.; Jörnsten, R.; Westermark, B.; Nelander, S. Modeling glioblastoma heterogeneity as a dynamic network of cell states. Mol. Syst. Biol. 2021, 17, e10105. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- de Souza, C.; Sabedot, T.; Malta, T.; Stetson, L.; Morozova, O.; Sokolov, A.; Laird, P.; Wiznerowicz, M.; Iavarone, A.; Snyder, J. A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell. Rep. 2018, 23, 637–651. [Google Scholar] [CrossRef]
- Chaligne, R.; Gaiti, F.; Silverbush, D.; Schiffman, J.S.; Weisman, H.R.; Kluegel, L.; Gritsch, S.; Deochand, S.D.; Gonzalez Castro, L.N.; Richman, A.R. Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat. Genet. 2021, 53, 1469–1479. [Google Scholar] [CrossRef]
- Tan, S.-L.; Nishi, M.; Ohtsuka, T.; Matsui, T.; Takemoto, K.; Kamio-Miura, A.; Aburatani, H.; Shinkai, Y.; Kageyama, R. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 2012, 139, 3806–3816. [Google Scholar] [CrossRef]
- Vue, T.Y.; Kollipara, R.K.; Borromeo, M.D.; Smith, T.; Mashimo, T.; Burns, D.K.; Bachoo, R.M.; Johnson, J.E. ASCL1 regulates neurodevelopmental transcription factors and cell cycle genes in brain tumors of glioma mouse models. Glia 2020, 68, 2613–2630. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, I.; Kageyama, R. bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron 2014, 82, 9–23. [Google Scholar] [CrossRef]
- De Silva, M.I.; Stringer, B.W.; Bardy, C. Neuronal and tumourigenic boundaries of glioblastoma plasticity. Trends Cancer 2023, 9, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Osswald, M.; Blaes, J.; Wiestler, B.; Sahm, F.; Schmenger, T.; Solecki, G.; Deumelandt, K.; Kurz, F.T.; Xie, R. Tweety-homolog 1 drives brain colonization of gliomas. J. Neurosci. 2017, 37, 6837–6850. [Google Scholar] [CrossRef]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wißmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917.e2831. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Carcereri de Prati, A.; Boriero, D.; Mariotto, S. Tumor dormancy and interplay with hypoxic tumor microenvironment. Int. J. Mol. Sci. 2019, 20, 4305. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- O’Connor, S.A.; Feldman, H.M.; Arora, S.; Hoellerbauer, P.; Toledo, C.M.; Corrin, P.; Carter, L.; Kufeld, M.; Bolouri, H.; Basom, R. Neural G0: A quiescent-like state found in neuroepithelial-derived cells and glioma. Mol. Syst. Biol. 2021, 17, e9522. [Google Scholar] [CrossRef] [PubMed]
- Antonica, F.; Santomaso, L.; Pernici, D.; Petrucci, L.; Aiello, G.; Cutarelli, A.; Conti, L.; Romanel, A.; Miele, E.; Tebaldi, T. A slow-cycling/quiescent cells subpopulation is involved in glioma invasiveness. Nat. Commun. 2022, 13, 4767. [Google Scholar] [CrossRef]
- Fu, R.Z.; Cottrell, O.; Cutillo, L.; Rowntree, A.; Zador, Z.; Wurdak, H.; Papalopulu, N.; Marinopoulou, E. Identification of genes with oscillatory expression in glioblastoma: The paradigm of SOX2. Sci. Rep. 2024, 14, 2123. [Google Scholar] [CrossRef]
- Robertson, F.L.; O’Duibhir, E.; Gangoso, E.; Bressan, R.B.; Bulstrode, H.; Marqués-Torrejón, M.-Á.; Ferguson, K.M.; Blin, C.; Grant, V.; Alfazema, N. Elevated FOXG1 in glioblastoma stem cells cooperates with Wnt/β-catenin to induce exit from quiescence. Cell Rep. 2023, 42, 112561. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, R.; Wu, M.; Johnson, K.; Kim, H.; Celebre, A.; Shahzad, U.; Graham, M.S.; Kessler, J.A.; Chuang, J.H.; Karamchandani, J. BMP signaling mediates glioma stem cell quiescence and confers treatment resistance in glioblastoma. Sci. Rep. 2019, 9, 14569. [Google Scholar] [CrossRef] [PubMed]
- Uribe, D.; Niechi, I.; Rackov, G.; Erices, J.I.; San Martín, R.; Quezada, C. Adapt to persist: Glioblastoma microenvironment and epigenetic regulation on cell plasticity. Biology 2022, 11, 313. [Google Scholar] [CrossRef]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNα activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A.; Estrada, Y.; Liu, D.; Ossowski, L. ERKMAPK activity as a determinant of tumor growth and dormancy; regulation by p38SAPK. Cancer Res. 2003, 63, 1684–1695. [Google Scholar] [PubMed]
- Biehs, B.; Dijkgraaf, G.J.; Piskol, R.; Alicke, B.; Boumahdi, S.; Peale, F.; Gould, S.E.; de Sauvage, F.J. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018, 562, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Rajbhandari, N.; Lin, W.-c.; Wehde, B.L.; Triplett, A.A.; Wagner, K.-U. Autocrine IGF1 signaling mediates pancreatic tumor cell dormancy in the absence of oncogenic drivers. Cell Rep. 2017, 18, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Van Den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes. Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells-much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Tobias, A.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.; Ahmed, A. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’avella, D.; Basso, G. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells 2010, 28, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. Abrogation of the Chk1-mediated G2 checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res. 2001, 61, 5843–5849. [Google Scholar] [PubMed]
- Gousias, K.; Theocharous, T.; Simon, M. Mechanisms of cell cycle arrest and apoptosis in glioblastoma. Biomedicines 2022, 10, 564. [Google Scholar] [CrossRef]
- Hsieh, A.; Ellsworth, R.; Hsieh, D. Hedgehog/GLI1 regulates IGF dependent malignant behaviors in glioma stem cells. J. Cell. Physiol. 2011, 226, 1118–1127. [Google Scholar] [CrossRef]
- Nakai, E.; Park, K.; Yawata, T.; Chihara, T.; Kumazawa, A.; Nakabayashi, H.; Shimizu, K. Enhanced MDR1 expression and chemoresistance of cancer stem cells derived from glioblastoma. Cancer Investig. 2009, 27, 901–908. [Google Scholar] [CrossRef]
- Bleau, A.-M.; Huse, J.T.; Holland, E.C. The ABCG2 resistance network of glioblastoma. Cell Cycle 2009, 8, 2937–2945. [Google Scholar] [CrossRef]
- Chua, C.; Zaiden, N.; Chong, K.-H.; See, S.-J.; Wong, M.-C.; Ang, B.-T.; Tang, C. Characterization of a side population of astrocytoma cells in response to temozolomide. J. Neurosurg. 2008, 109, 856–866. [Google Scholar] [CrossRef]
- Chen, L.; Shi, L.; Wang, W.; Zhou, Y. ABCG2 downregulation in glioma stem cells enhances the therapeutic efficacy of demethoxycurcumin. Oncotarget 2017, 8, 43237. [Google Scholar] [CrossRef]
- Tivnan, A.; Zakaria, Z.; O’Leary, C.; Kögel, D.; Pokorny, J.L.; Sarkaria, J.N.; Prehn, J.H. Inhibition of multidrug resistance protein 1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Front. Neurosci. 2015, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. [Google Scholar] [CrossRef] [PubMed]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular mechanisms of radiation-induced cancer cell death: A primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Aoyagi, M.; Ando, N.; Ogishima, T.; Wakimoto, H.; Yamamoto, M.; Ohno, K. Expansion of CD133-positive glioma cells in recurrent de novo glioblastomas after radiotherapy and chemotherapy. J. Neurosurg. 2013, 119, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Aoyagi, M.; Wakimoto, H.; Ando, N.; Nariai, T.; Yamamoto, M.; Ohno, K. Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. J. Neurosurg. 2010, 113, 310–318. [Google Scholar] [CrossRef]
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol. Cancer Res. 2009, 7, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Ahmed, S.U.; Carruthers, R.; Gilmour, L.; Yildirim, S.; Watts, C.; Chalmers, A.J. Selective inhibition of parallel DNA damage response pathways optimizes radiosensitization of glioblastoma stem-like cells. Cancer Res. 2015, 75, 4416–4428. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef]
- Li, S.; Gao, Y.; Tokuyama, T.; Yamamoto, J.; Yokota, N.; Yamamoto, S.; Terakawa, S.; Kitagawa, M.; Namba, H. Genetically engineered neural stem cells migrate and suppress glioma cell growth at distant intracranial sites. Cancer Lett. 2007, 251, 220–227. [Google Scholar] [CrossRef]
- Oishi, T.; Koizumi, S.; Kurozumi, K. Mesenchymal stem cells as therapeutic vehicles for glioma. Cancer Gene Ther. 2024, 31, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Imitola, J.; Raddassi, K.; Park, K.I.; Mueller, F.-J.; Nieto, M.; Teng, Y.D.; Frenkel, D.; Li, J.; Sidman, R.L.; Walsh, C.A. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1α/CXC chemokine receptor 4 pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 18117–18122. [Google Scholar] [CrossRef]
- Hata, N.; Shinojima, N.; Gumin, J.; Yong, R.; Marini, F.; Andreeff, M.; Lang, F.F. Platelet-derived growth factor BB mediates the tropism of human mesenchymal stem cells for malignant gliomas. Neurosurgery 2010, 66, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, D.-S.; Jeong, C.H.; Kim, D.H.; Kim, J.H.; Jeon, H.B.; Kwon, S.-J.; Jeun, S.-S.; Yang, Y.S.; Oh, W. CXC chemokine receptor 1 enhances the ability of human umbilical cord blood-derived mesenchymal stem cells to migrate toward gliomas. Biochem. Biophys. Res. Commun. 2011, 407, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Ryu, C.H.; Kim, S.M.; Lim, J.Y.; Park, S.I.; Jeong, C.H.; Jun, J.; Oh, J.H.; Park, S.H.; Oh, W. CXCR4-transfected human umbilical cord blood-derived mesenchymal stem cells exhibit enhanced migratory capacity toward gliomas. Int. J. Oncol. 2011, 38, 97–103. [Google Scholar]
- Al-Kharboosh, R.; ReFaey, K.; Lara-Velazquez, M.; Grewal, S.S.; Imitola, J.; Quiñones-Hinojosa, A. Inflammatory mediators in glioma microenvironment play a dual role in gliomagenesis and mesenchymal stem cell homing: Implication for cellular therapy. Mayo Clin. Proc. Innov. Qual. Outcomes 2020, 4, 443–459. [Google Scholar] [CrossRef]
- Ceradini, D.J.; Gurtner, G.C. Homing to hypoxia: HIF-1 as a mediator of progenitor cell recruitment to injured tissue. Trends Cardiovasc. Med. 2005, 15, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Najbauer, J.; Garcia, E.; Metz, M.Z.; Gutova, M.; Glackin, C.A.; Kim, S.U.; Aboody, K.S. Neural stem cell tropism to glioma: Critical role of tumor hypoxia. Mol. Cancer Res. 2008, 6, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Bexell, D.; Gunnarsson, S.; Tormin, A.; Darabi, A.; Gisselsson, D.; Roybon, L.; Scheding, S.; Bengzon, J. Bone marrow multipotent mesenchymal stroma cells act as pericyte-like migratory vehicles in experimental gliomas. Mol. Ther. 2009, 17, 183–190. [Google Scholar] [CrossRef]
- Nehama, D.; Woodell, A.S.; Maingi, S.M.; Hingtgen, S.D.; Dotti, G. Cell-based therapies for glioblastoma: Promising tools against tumor heterogeneity. Neuro-Oncol. 2023, 25, 1551–1562. [Google Scholar] [CrossRef]
- Dührsen, L.; Hartfuß, S.; Hirsch, D.; Geiger, S.; Maire, C.L.; Sedlacik, J.; Guenther, C.; Westphal, M.; Lamszus, K.; Hermann, F.G. Preclinical analysis of human mesenchymal stem cells: Tumor tropism and therapeutic efficiency of local HSV-TK suicide gene therapy in glioblastoma. Oncotarget 2019, 10, 6049. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Hou, J.; Zheng, K.; Jin, X.; Xie, Q.; Cheng, L.; Sun, X. Suicide gene therapy against malignant gliomas by the local delivery of genetically engineered umbilical cord mesenchymal stem cells as cellular vehicles. Curr. Gene Ther. 2019, 19, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lee, D.-H.; Kim, H.A.; Choi, S.-A.; Lee, H.J.; Park, C.-K.; Phi, J.H.; Wang, K.-C.; Kim, S.U.; Kim, S.-K. Double suicide gene therapy using human neural stem cells against glioblastoma: Double safety measures. J. Neuro-Oncol. 2014, 116, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zhang, M.; Guo, R.; Miao, Y.; Li, B. Bone marrow–derived mesenchymal stem cell–mediated dual-gene therapy for glioblastoma. Hum. Gene Ther. 2019, 30, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Andreou, T.; Williams, J.; Brownlie, R.J.; Salmond, R.J.; Watson, E.; Shaw, G.; Melcher, A.; Wurdak, H.; Short, S.C.; Lorger, M. Hematopoietic stem cell gene therapy targeting TGFβ enhances the efficacy of irradiation therapy in a preclinical glioblastoma model. J. Immunother. Cancer 2021, 9, e001143. [Google Scholar] [CrossRef]
- Ali, S.; Xia, Q.; Muhammad, T.; Liu, L.; Meng, X.; Bars-Cortina, D.; Khan, A.A.; Huang, Y.; Dong, L. Glioblastoma therapy: Rationale for a mesenchymal stem cell-based vehicle to carry recombinant viruses. Stem Cell Rev. Rep. 2022, 18, 523–543. [Google Scholar] [CrossRef]
- Fares, J.; Ahmed, A.U.; Ulasov, I.V.; Sonabend, A.M.; Miska, J.; Lee-Chang, C.; Balyasnikova, I.V.; Chandler, J.P.; Portnow, J.; Tate, M.C. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: A first-in-human, phase 1, dose-escalation trial. Lancet Oncol. 2021, 22, 1103–1114. [Google Scholar] [CrossRef]
- Rempel, S.A.; Dudas, S.; Ge, S.; Gutiérrez, J.A. Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin. Cancer Res. 2000, 6, 102–111. [Google Scholar]
- Adamski, V.; Hattermann, K.; Kubelt, C.; Cohrs, G.; Lucius, R.; Synowitz, M.; Sebens, S.; Held-Feindt, J. Entry and exit of chemotherapeutically-promoted cellular dormancy in glioblastoma cells is differentially affected by the chemokines CXCL12, CXCL16, and CX3CL1. Oncogene 2020, 39, 4421–4435. [Google Scholar] [CrossRef]
- Roboz, G.J.; Ritchie, E.K.; Dault, Y.; Lam, L.; Marshall, D.C.; Cruz, N.M.; Hsu, H.-T.C.; Hassane, D.C.; Christos, P.J.; Ippoliti, C. Phase I trial of plerixafor combined with decitabine in newly diagnosed older patients with acute myeloid leukemia. Haematologica 2018, 103, 1308. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Miyoshi, H.; Imaizumi, K.; Yo, M.; Kase, Y.; Sato, T.; Sato, M.; Morimoto, Y.; Sampetrean, O.; Kohyama, J. Gene therapy using genome-edited iPS cells for targeting malignant glioma. Bioeng. Transl. Med. 2023, 8, e10406. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Miyoshi, H.; Morimoto, Y.; Oishi, Y.; Sampetrean, O.; Iwasawa, C.; Mine, Y.; Saya, H.; Yoshida, K.; Okano, H. Gene therapy using neural stem/progenitor cells derived from human induced pluripotent stem cells: Visualization of migration and bystander killing effect. Hum. Gene Ther. 2020, 31, 352–366. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Drug Name | Molecular Targets | Reference |
|---|---|---|
| Sorafenib | VEGFR, PDGFR, Raf kinase, c-KIT, and FLT3 | [63,64,65,66,67] |
| Cabozantinib | MET, RET, AXL, VEGFR2, FLT3, and c-KIT. | [52] |
| Dasatinib | BCR-ABL, Src, c-KIT, and PDGFR | [53] |
| Nintedanib | PDGFRα + β), VEGFR1-3 and FGFR1-4 | [54,55] |
| Pazopanib | VEGFR, PDGFR and c-KIT. | [56] |
| Pexidartinib | c-KIT, FLT3, and CSF-1/PDGFR | [57,58] |
| Ponatinib | VEGFR, PDGFR, FGFR, c-KIT, RET, and Src | [59] |
| Regorafenib | VEGFR1-3, PDGFR, FGFR, c-KIT, RET, and RAF | [60] |
| Sunitinib | VEGFR, PDGFR, and FLT3 | [61] |
| Vandetanib | RET, VEGFR, and EGFR. | [62] |
| Depatuxizumab mafodotin | EGFR | [68] |
| Erdafitinib | FGFR | [69] |
| Erlotinib | EGFR | [70,71,72,73,74] |
| Dacomitinib | EGFR | [75,76,77] |
| Lapatinib | EGFR | [78] |
| Osimertinib | EGFR | [79,80,81] |
| Pemigatinib | FGFR1-3 | [82] |
| AZD4547 | FGFR1-3 | [83] |
| NVP-BGJ398 (Infigratinib) | FGFR1-4 | [84] |
| Futibatinib (TAS-120) | FGFR1-4 | [85] |
| E-7090 | FGFR1-3 | [86] |
| Roblitinib (FGF-401) | FGF4 | [87] |
| H3B-6527 | FGFR4 | [88] |
| Fisogatinib (BLU-554) | FGFR4 | [89] |
| LY2874455 | Pan FGFR | [90] |
| FIIN-1 (FGFR irreversible inhibitor-1) | FGFR1 | [91] |
| PD173074 | FGFR1 | [92] |
| V4-015 | FGFR4 | [93] |
| BLU-9931 | FGFR4 | [94] |
| Capmatinib | HGFR | [95], NCT02386826 |
| Crizotinib | ALK and c-MET | [96,97] |
| Dactolisib | PI3K/mTOR | [98,99], NCT02430363 |
| Voxtalisib | PI3K/mTOR | [100] |
| Paxalisib | PI3K/mTOR | [101,102,103,104] |
| Buparlisib (BKM120) | PI3K | [105,106] |
| Pilaralisib | PI3K | [100] |
| Pictisilib | PI3K | [107] |
| Sonolisib (PX-866) | PI3K | [108] |
| NCT01458067 | PIK3K (p110β isoform-specific) | NCT01458067 |
| Everolimus | mTORC | [109] |
| Temsirolimus | mTORC | [110,111] |
| Decitabine | DNMT | [112,113] |
| 5-azacytidine | DNMT | [114] |
| Balinostat | HDAC | [115] |
| Panobinostat | HDAC | [116] |
| Vorinostat | HDAC | [117] |
| Valproic acid | HDACI/IIa | [118,119,120] |
| KA2507 | HDAC6 | NCT03008018 [121] |
| JBI-802 | HDAC6 | NCT05268666 |
| Melatonin | ABCG2/BCRP | [122] |
| Perifosine | BRCA1 | [123] |
| GSI-1 | NOTCH | [124] |
| Cyclopamine | SMO/SHH | [124] |
| RI-1 | RAD51 | [125] |
| B02 | RAD51 | [125] |
| Cell Type | Targeting Cells | Drug | Mechanism | Targeted Tumors | Clinical Phase (Status) | Study ID |
|---|---|---|---|---|---|---|
| NSCs | HB1.F3.CD NSCs | 5-FC (Prodrug) | Inhibits TS (Suicide gene therapy) | Recurrent GBM | I (Completed) | NCT02015819 |
| HB1.F3.CD NSCs | 5-FC (Prodrug) | Inhibits TS (Suicide gene therapy) | Recurrent GBM | I (Completed) | NCT01172964 | |
| NSC-CRAd-S-pk7 | CRAd-S-pk7 virus | Oncolytic adenovirus (Virotherapy) | Newly diagnosed GBM | I (Completed) | NCT03072134 | |
| NSC-CRAd-S-pk7 | CRAd-S-pk7 virus | Oncolytic adenovirus (Virotherapy) | Recurrent GBM | I (Recruiting) | NCT05139056 | |
| hCE1m6-NSCs | Irinotecan (Prodrug) | Inhibits Topo IA (Suicide Gene therapy) | Recurrent GBM | I (Active, not recruiting) | NCT02192359 | |
| MSCs | BM-MSCs | Ad5-DNX-2401 | Oncolytic adenovirus (Virotherapy) | Recurrent GBM | I (Recruiting) | NCT03896568 |
| A-MSCs | / | Dose-escalation study of A-MSCs | Recurrent GBM | I (Recruiting) | NCT05789394 | |
| MSC-CD | 5-FC (Prodrug) | Inhibits Thymidine Synthase (Suicide gene therapy) | Recurrent GBM | I/II (Completed) | NCT04657315 | |
| HSPCs | HPCs—P140K MGMT Lentiviral Vector | O6-benzylguanine | MGMT inhibition (Tumor chemosensitization) | GBM | II (Recruiting) | NCT05052957 |
| PBSCs—P140K MGMT Lentiviral Vector | O6-benzylguanine | MGMT inhibition (Tumor chemosensitization) | GBM | I (Completed) | NCT01269424 | |
| HSPCs—Interferon-α2 | / | Enhance immune response (Immunomodulation) | GBM | I/II (Recruiting) | NCT03866109 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahdi, A.; Aittaleb, M.; Tissir, F. Targeting Glioma Stem Cells: Therapeutic Opportunities and Challenges. Cells 2025, 14, 675. https://doi.org/10.3390/cells14090675
Mahdi A, Aittaleb M, Tissir F. Targeting Glioma Stem Cells: Therapeutic Opportunities and Challenges. Cells. 2025; 14(9):675. https://doi.org/10.3390/cells14090675
Chicago/Turabian StyleMahdi, Asma, Mohamed Aittaleb, and Fadel Tissir. 2025. "Targeting Glioma Stem Cells: Therapeutic Opportunities and Challenges" Cells 14, no. 9: 675. https://doi.org/10.3390/cells14090675
APA StyleMahdi, A., Aittaleb, M., & Tissir, F. (2025). Targeting Glioma Stem Cells: Therapeutic Opportunities and Challenges. Cells, 14(9), 675. https://doi.org/10.3390/cells14090675