
Hydrogen Peroxide Modulates the Timely Activation of Jun and Erk in Schwann Cells at the Injury Site and Is Required for Motor Axon Regeneration

 ,
,  , , , ,
, , , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statment
2.2. Antibodies and Reagents
2.3. Sciatic Nerve Compression
2.4. Compound Muscle Action Potential Recordings
2.5. Immunofluorescence of Sciatic Nerves and Muscle Tissues
2.6. Primary Cell Cultures and Treatments
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
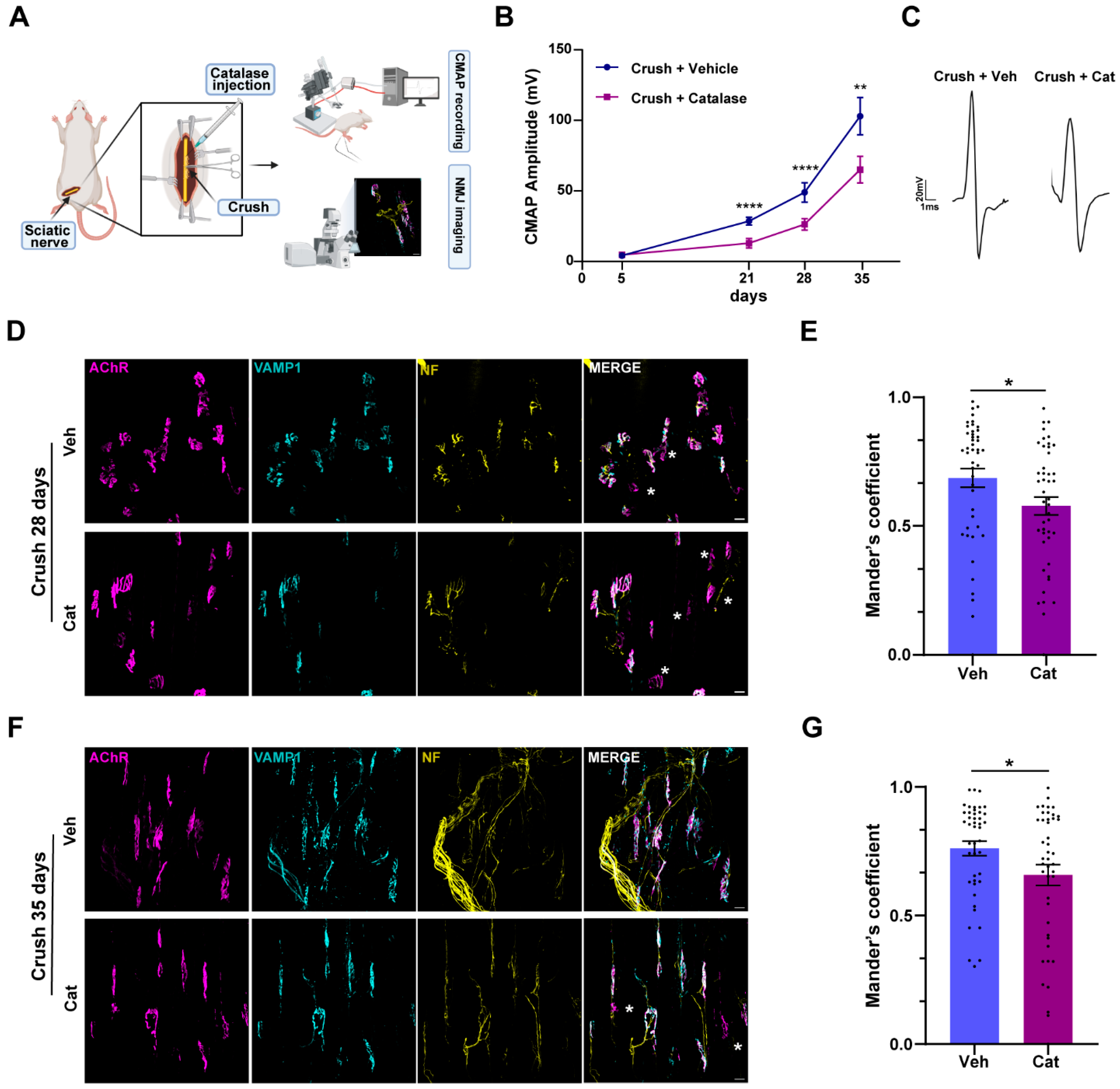
3.1. H2O2 Neutralization Impairs Sciatic Nerve Regeneration and NMJ Recovery of Function Without Affecting Axonal Degeneration
3.2. Injury-Induced H2O2 Triggers ERK Activation in Schwann Cells
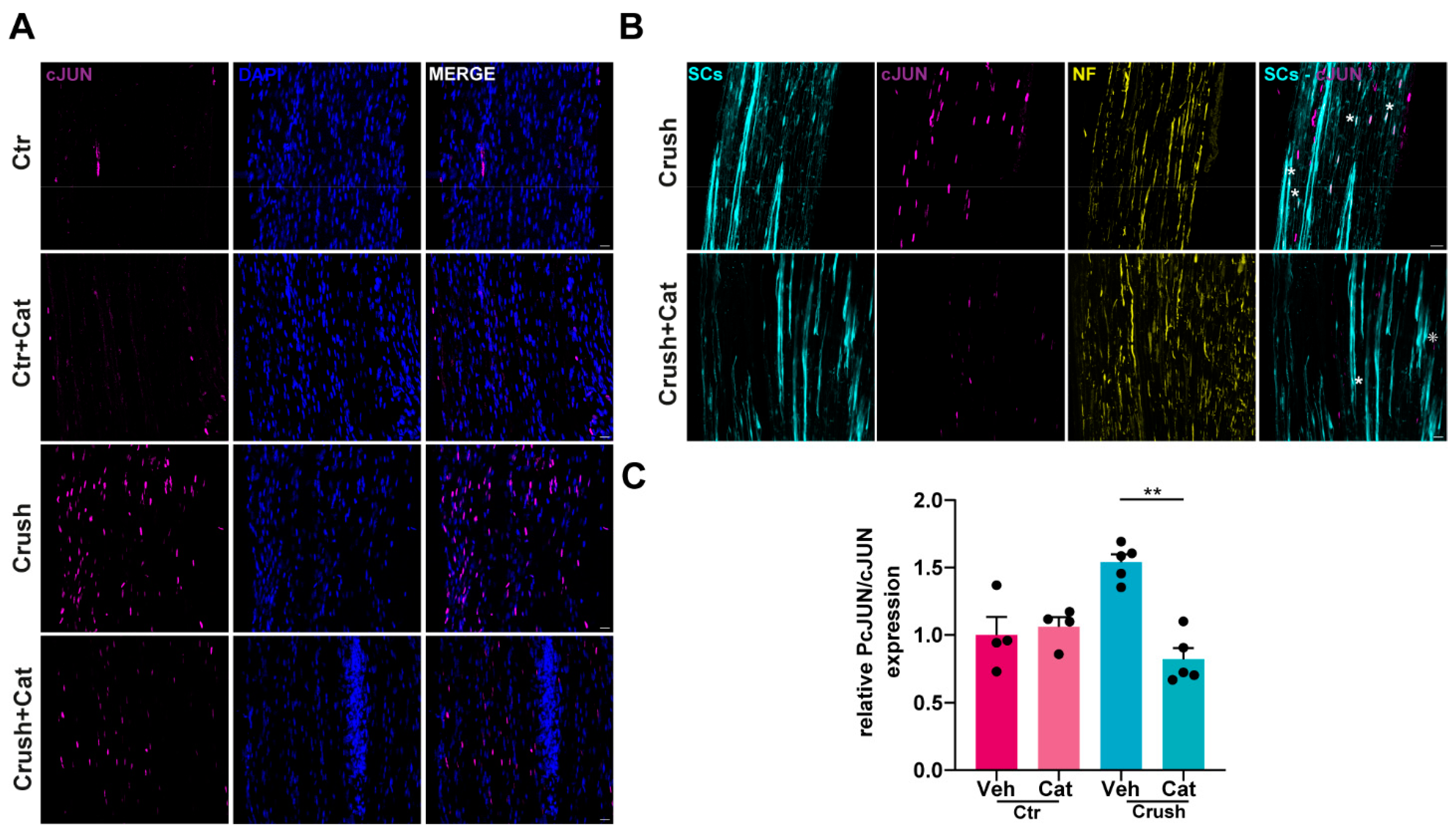
3.3. H2O2-Dependent c-Jun UpRegulation in SCs Following Nerve Injury
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gordon, T. Peripheral Nerve Regeneration and Muscle Reinnervation. Int. J. Mol. Sci. 2020, 21, 8652. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T. Nerve regeneration in the peripheral and central nervous systems. J. Physiol.-Lond. 2016, 594, 3517–3520. [Google Scholar] [CrossRef]
- He, Z.; Jin, Y. Intrinsic Control of Axon Regeneration. Neuron 2016, 90, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Mar, F.; Bonni, A.; Sousa, M. Cell intrinsic control of axon regeneration. EMBO Rep. 2014, 15, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Cattin, A.L.; Lloyd, A.C. The multicellular complexity of peripheral nerve regeneration. Curr. Opin. Neurobiol. 2016, 39, 38–46. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The Success and Failure of the Schwann Cell Response to Nerve Injury. Front. Cell. Neurosci. 2019, 13, 33. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The repair Schwann cell and its function in regenerating nerves. J. Physiol.-Lond. 2016, 594, 3521–3531. [Google Scholar] [CrossRef]
- Jessen, K.R.; Arthur-Farraj, P. Repair Schwann cell update: Adaptive reprogramming, EMT, and stemness in regenerating nerves. Glia 2019, 67, 421–437. [Google Scholar] [CrossRef]
- Duregotti, E.; Negro, S.; Scorzeto, M.; Zornetta, I.; Dickinson, B.C.; Chang, C.J.; Montecucco, C.; Rigoni, M. Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc. Natl. Acad. Sci. USA 2015, 112, E497–E505. [Google Scholar] [CrossRef]
- Negro, S.; Lauria, F.; Stazi, M.; Tebaldi, T.; D’Este, G.; Pirazzini, M.; Megighian, A.; Lessi, F.; Mazzanti, C.M.; Sales, G.; et al. Hydrogen peroxide induced by nerve injury promotes axon regeneration via connective tissue growth factor. Acta Neuropathol. Commun. 2022, 10, 189. [Google Scholar] [CrossRef]
- Arthur-Farraj, P.J.; Latouche, M.; Wilton, D.K.; Quintes, S.; Chabrol, E.; Banerjee, A.; Woodhoo, A.; Jenkins, B.; Rahman, M.; Turmaine, M.; et al. c-Jun Reprograms Schwann Cells of Injured Nerves to Generate a Repair Cell Essential for Regeneration. Neuron 2012, 75, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Napoli, I.; Noon, L.A.; Ribeiro, S.; Kerai, A.P.; Parrinello, S.; Rosenberg, L.H.; Collins, M.J.; Harrisingh, M.C.; White, I.J.; Woodhoo, A.; et al. A Central Role for the ERK-Signaling Pathway in Controlling Schwann Cell Plasticity and Peripheral Nerve Regeneration In Vivo. Neuron 2012, 73, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Gorza, L.; Schiavo, G.; Schiavo, N.; Scheller, R.H.; Montecucco, C. VAMP/synaptobrevin isoforms 1 and 2 are widely and differentially expressed in nonneuronal tissues. J. Cell Biol. 1996, 132, 167–179. [Google Scholar] [CrossRef]
- Negro, S.; Stazi, M.; Rigoni, M.; Megighian, A. Neurotransmission Recovery by Melatonin Measured by CMAP. Methods Mol. Biol. 2022, 2550, 413–423. [Google Scholar] [CrossRef]
- Sleigh, J.; Tosolini, A.; Schiavo, G. In Vivo Imaging of Anterograde and Retrograde Axonal Transport in Rodent Peripheral Nerves. In Axon Degeneration; Springer: Berlin/Heidelberg, Germany, 2020; Volume 2143, pp. 271–292. [Google Scholar]
- Kalinski, A.; Kar, A.; Craver, J.; Tosolini, A.; Sleigh, J.; Lee, S.; Hawthorne, A.; Brito-Vargas, P.; Miller-Randolph, S.; Passino, R.; et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J. Cell Biol. 2019, 218, 1871–1890. [Google Scholar] [CrossRef]
- Tosolini, A.P.; Villarroel-Campos, D.; Schiavo, G.; Sleigh, J.N. Expanding the Toolkit for In Vivo Imaging of Axonal Transport. J. Vis. Exp. 2021, 178, e63471. [Google Scholar] [CrossRef]
- Stazi, M.; Negro, S.; Megighian, A.; D’Este, G.; Solimena, M.; Jockers, R.; Lista, F.; Montecucco, C.; Rigoni, M. Melatonin promotes regeneration of injured motor axons via MT(1) receptors. J. Pineal Res. 2021, 70, e12695. [Google Scholar] [CrossRef]
- Negro, S.; Bergamin, E.; Rodella, U.; Duregotti, E.; Scorzeto, M.; Jalink, K.; Montecucco, C.; Rigoni, M. ATP Released by Injured Neurons Activates Schwann Cells. Front. Cell. Neurosci. 2016, 10, 134. [Google Scholar] [CrossRef]
- Negro, S.; Pirazzini, M.; Rigoni, M. Models and methods to study Schwann cells. J. Anat. 2022, 241, 1235–1258. [Google Scholar] [CrossRef]
- Negro, S.; Stazi, M.; Marchioretto, M.; Tebaldi, T.; Rodella, U.; Duregotti, E.; Gerke, V.; Quattrone, A.; Montecucco, C.; Rigoni, M.; et al. Hydrogen peroxide is a neuronal alarmin that triggers specific RNAs, local translation of Annexin A2, and cytoskeletal remodeling in Schwann cells. Rna 2018, 24, 915–925. [Google Scholar] [CrossRef]
- Magill, C.K.; Tong, A.; Kawamura, D.; Hayashi, A.; Hunter, D.A.; Parsadanian, A.; Mackinnon, S.E.; Myckatyn, T.M. Reinnervation of the tibialis anterior following sciatic nerve crush injury: A confocal microscopic study in transgenic mice. Exp. Neurol. 2007, 207, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Baptista, A.F.; Gomes, J.R.D.; Oliveira, J.T.; Santos, S.M.G.; Vannier-Santos, M.A.; Martinez, A.M.B. A new approach to assess function after sciatic nerve lesion in the mouse—Adaptation of the sciatic static index. J. Neurosci. Methods 2007, 161, 259–264. [Google Scholar] [CrossRef]
- Sheu, J.; Kulhanek, D.; Eckenstein, F. Differential patterns of ERK and STAT3 phosphorylation after sciatic nerve transection in the rat. Exp. Neurol. 2000, 166, 392–402. [Google Scholar] [CrossRef]
- Reth, M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 2002, 3, 1129–1134. [Google Scholar] [CrossRef]
- van der Vliet, A.; Janssen-Heininger, Y. Hydrogen Peroxide as a Damage Signal in Tissue Injury and Inflammation: Murderer, Mediator, or Messenger? J. Cell. Biochem. 2014, 115, 427–435. [Google Scholar] [CrossRef]
- Miller, E.; Dickinson, B.; Chang, C. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef]
- Watanabe, S.; Moniaga, C.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9 facilitates membrane transport of hydrogen peroxide in mammalian cells. Biochem. Biophys. Res. Commun. 2016, 471, 191–197. [Google Scholar] [CrossRef]
- Gough, D.; Cotter, T. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef]
- Niethammer, P.; Grabher, C.; Look, A.; Mitchison, T. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Love, N.; Chen, Y.; Ishibashi, S.; Kritsiligkou, P.; Lea, R.; Koh, Y.; Gallop, J.; Dorey, K.; Amaya, E. Amputation-induced reactive oxygen species are required for successful Xenopus tadpole tail regeneration. Nat. Cell Biol. 2013, 15, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Rieger, S.; Sagasti, A. Hydrogen Peroxide Promotes Injury-Induced Peripheral Sensory Axon Regeneration in the Zebrafish Skin. PLoS Biol. 2011, 9, e1000621. [Google Scholar] [CrossRef]
- Hervera, A.; De Virgiliis, F.; Palmisano, I.; Zhou, L.M.; Tantardini, E.; Kong, G.P.; Hutson, T.; Danzi, M.C.; Perry, R.B.; Santos, C.X.C.; et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat. Cell Biol. 2018, 20, 307–319. [Google Scholar] [CrossRef]
- Rodella, U.; Scorzeto, M.; Duregotti, E.; Negro, S.; Dickinson, B.C.; Chang, C.J.; Yuki, N.; Rigoni, M.; Montecucco, C. An animal model of Miller Fisher syndrome: Mitochondrial hydrogen peroxide is produced by the autoimmune attack of nerve terminals and activates Schwann cells. Neurobiol. Dis. 2016, 96, 95–104. [Google Scholar] [CrossRef]
- Harrisingh, M.; Perez-Nadales, E.; Parkinson, D.; Malcolm, D.; Mudge, A.; Lloyd, A. The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. 2004, 23, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Fontana, X.; Hristova, M.; Da Costa, C.; Patodia, S.; Thei, L.; Makwana, M.; Spencer-Dene, B.; Latouche, M.; Mirsky, R.; Jessen, K.; et al. c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J. Cell Biol. 2012, 198, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, J.A.; Pilch, K.S.; van der Lans, M.; Fazal, S.V.; Benito, C.; Wagstaff, L.J.; Mirsky, R.; Jessen, K.R. After Nerve Injury, Lineage Tracing Shows That Myelin and Remak Schwann Cells Elongate Extensively and Branch to Form Repair Schwann Cells, Which Shorten Radically on Remyelination. J. Neurosci. 2017, 37, 9086–9099. [Google Scholar] [CrossRef]
- Blom, C.; Mårtensson, L.; Dahlin, L. Nerve Injury-Induced c-Jun Activation in Schwann Cells Is JNK Independent. Biomed. Res. Int. 2014, 2014, 392971. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, D.; Bhaskaran, A.; Arthur-Farraj, P.; Noon, L.; Woodhoo, A.; Lloyd, A.; Feltri, M.; Wrabetz, L.; Behrens, A.; Mirsky, R.; et al. c-Jun is a negative regulator of myelination. J. Cell Biol. 2008, 181, 625–637. [Google Scholar] [CrossRef]
- Gomez-Deza, J.; Slavutsky, A.; Nebiyou, M.; Le Pichon, C. Local production of reactive oxygen species drives vincristine-induced axon degeneration. Cell Death Dis. 2023, 14, 807. [Google Scholar] [CrossRef]
- Wakatsuki, S.; Araki, T. Novel insights into the mechanism of reactive oxygen species-mediated neurodegeneration. Neural Regen. Res. 2023, 18, 746–749. [Google Scholar] [CrossRef]
- Marinho, H.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef]
- Rios, R.; Jablonka-Shariff, A.; Broberg, C.; Snyder-Warwick, A. Macrophage roles in peripheral nervous system injury and pathology: Allies in neuromuscular junction recovery. Mol. Cell. Neurosci. 2021, 111, 103590. [Google Scholar] [CrossRef]
- Clements, M.P.; Byrne, E.; Guerrero, L.F.C.; Cattin, A.L.; Zakka, L.; Ashraf, A.; Burden, J.J.; Khadayate, S.; Lloyd, A.C.; Marguerat, S.; et al. The Wound Microenvironment Reprograms Schwann Cells to Invasive Mesenchymal-like Cells to Drive Peripheral Nerve Regeneration. Neuron 2017, 96, 98–114.e7. [Google Scholar] [CrossRef]
- Wu, L.; He, J.; Shen, N.; Chen, S. Molecular and cellular mechanisms underlying peripheral nerve injury-induced cellular ecological shifts: Implications for neuroregeneration. IBRO Neurosci. Rep. 2025, 18, 120–129. [Google Scholar] [CrossRef]
- Jaramillo, M.; Olivier, M. Hydrogen peroxide induces murine macrophage chemokine gene transcription via extracellular signal-regulated kinaseand cyclic adenosine 5′-monophosphate (cAMP)-dependent pathways:: Involvement of NF-κB, activator protein 1, and cAMP response element binding protein. J. Immunol. 2002, 169, 7026–7038. [Google Scholar]
- Benito, C.; Gomez-Sanchez, J.A.; Davis, C.M.; Meijer, D.; Mirsky, R.; Jessen, K.R. STAT3 controls the long-term survival and phenotype of repair Schwann cells during nerve regeneration. Glia 2017, 65, E107. [Google Scholar] [CrossRef]
- Sun, G.; Li, Z.; Wang, X.; Tang, W.; Wei, Y. Modulation of MAPK and Akt signaling pathways in proximal segment of injured sciatic nerves. Neurosci. Lett. 2013, 534, 205–210. [Google Scholar] [CrossRef]
- Woodhoo, A.; Alonso, M.B.D.; Droggiti, A.; Turmaine, M.; D’Antonio, M.; Parkinson, D.B.; Wilton, D.K.; Al-Shawi, R.; Simons, P.; Shen, J.; et al. Notch controls embryonic Schwann cell differentiation, postnatal myelination and adult plasticity. Nat. Neurosci. 2009, 12, 839–U846. [Google Scholar] [CrossRef]
- Ma, K.H.; Hung, H.A.; Svaren, J. Epigenomic Regulation of Schwann Cell Reprogramming in Peripheral Nerve Injury. J. Neurosci. 2016, 36, 9135–9147. [Google Scholar] [CrossRef]
- Nocera, G.; Jacob, C. Mechanisms of Schwann cell plasticity involved in peripheral nerve repair after injury. Cell. Mol. Life Sci. 2020, 77, 3977–3989. [Google Scholar] [CrossRef]
- López-Leal, R.; Alvarez, J.; Court, F. Origin of axonal proteins: Is the axon-schwann cell unit a functional syncytium? Cytoskeleton 2016, 73, 629–639. [Google Scholar] [CrossRef]
- Hung, H.A.; Sun, G.N.; Keles, S.; Svaren, J. Dynamic Regulation of Schwann Cell Enhancers after Peripheral Nerve Injury. J. Biol. Chem. 2015, 290, 6937–6950. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negro, S.; Baggio, C.; Tonellato, M.; Stazi, M.; D’Este, G.; Megighian, A.; Montecucco, C.; Rigoni, M. Hydrogen Peroxide Modulates the Timely Activation of Jun and Erk in Schwann Cells at the Injury Site and Is Required for Motor Axon Regeneration. Cells 2025, 14, 671. https://doi.org/10.3390/cells14090671
Negro S, Baggio C, Tonellato M, Stazi M, D’Este G, Megighian A, Montecucco C, Rigoni M. Hydrogen Peroxide Modulates the Timely Activation of Jun and Erk in Schwann Cells at the Injury Site and Is Required for Motor Axon Regeneration. Cells. 2025; 14(9):671. https://doi.org/10.3390/cells14090671
Chicago/Turabian StyleNegro, Samuele, Chiara Baggio, Marika Tonellato, Marco Stazi, Giorgia D’Este, Aram Megighian, Cesare Montecucco, and Michela Rigoni. 2025. "Hydrogen Peroxide Modulates the Timely Activation of Jun and Erk in Schwann Cells at the Injury Site and Is Required for Motor Axon Regeneration" Cells 14, no. 9: 671. https://doi.org/10.3390/cells14090671
APA StyleNegro, S., Baggio, C., Tonellato, M., Stazi, M., D’Este, G., Megighian, A., Montecucco, C., & Rigoni, M. (2025). Hydrogen Peroxide Modulates the Timely Activation of Jun and Erk in Schwann Cells at the Injury Site and Is Required for Motor Axon Regeneration. Cells, 14(9), 671. https://doi.org/10.3390/cells14090671