
Collapsin Response Mediator Protein 2 (CRMP2) Modulates Mitochondrial Oxidative Metabolism in Knock-In AD Mouse Model




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Mouse Oral Gavage
2.3. Neuronal Cell Culture
2.4. Isolation and Purification of Brain Synaptic Mitochondria
2.5. Immunoblotting
2.6. Co-Immunoprecipitation
2.7. Cell Respirometry
2.8. Mitochondrial Membrane Potential in Cultured Neurons
2.9. Mitochondrial Superoxide Anion Production in Cultured Neurons
2.10. Respiration and Membrane Potential in Isolated Mitochondria
2.11. Purification and Reconstitution of the ANT
2.12. Preparation of Recombinant CRMP2 (rCRMP2)
2.13. Evaluation of Adenine Nucleotide Translocase Activity
2.14. Statistics
3. Results
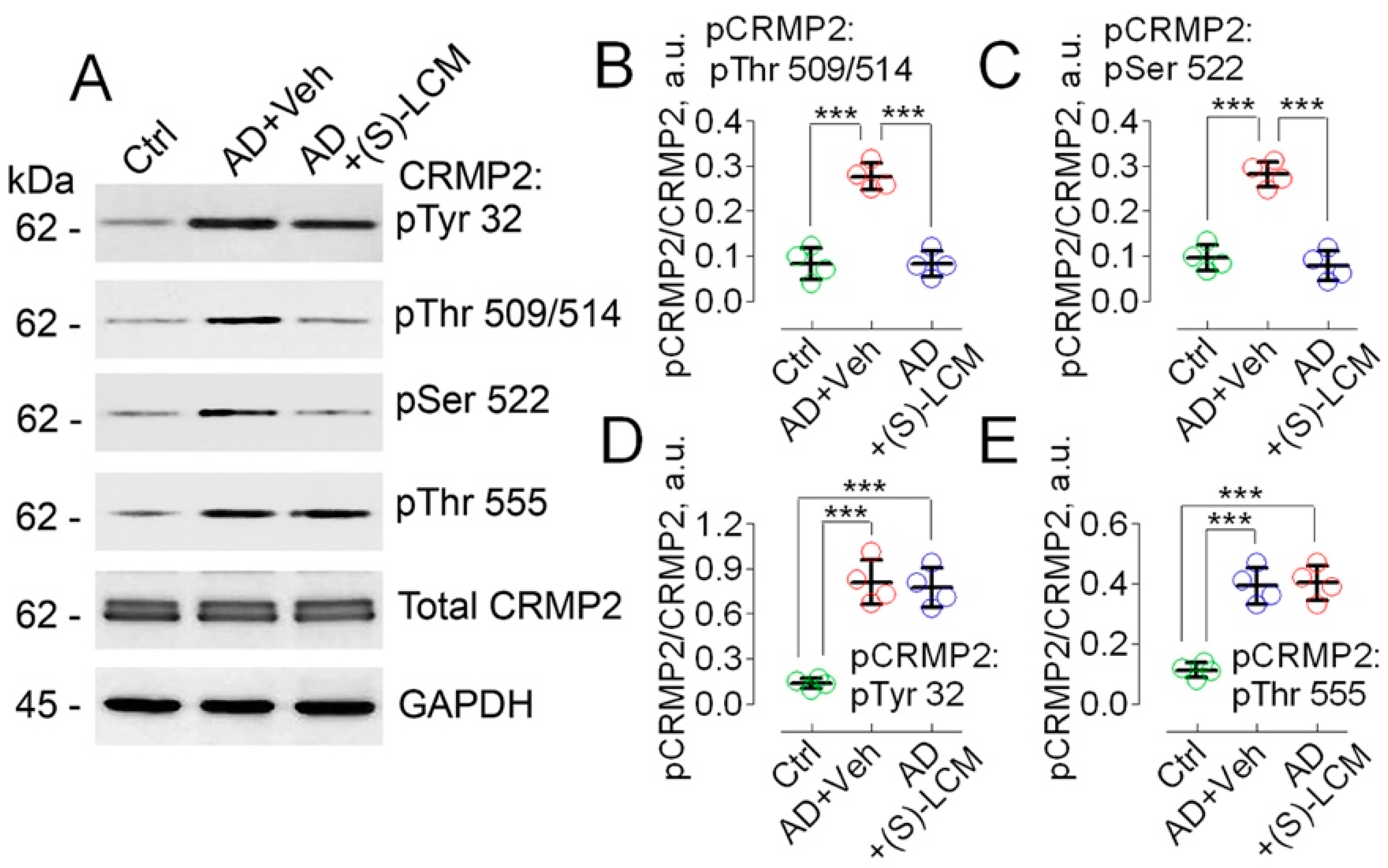
3.1. CRMP2 Phosphorylation
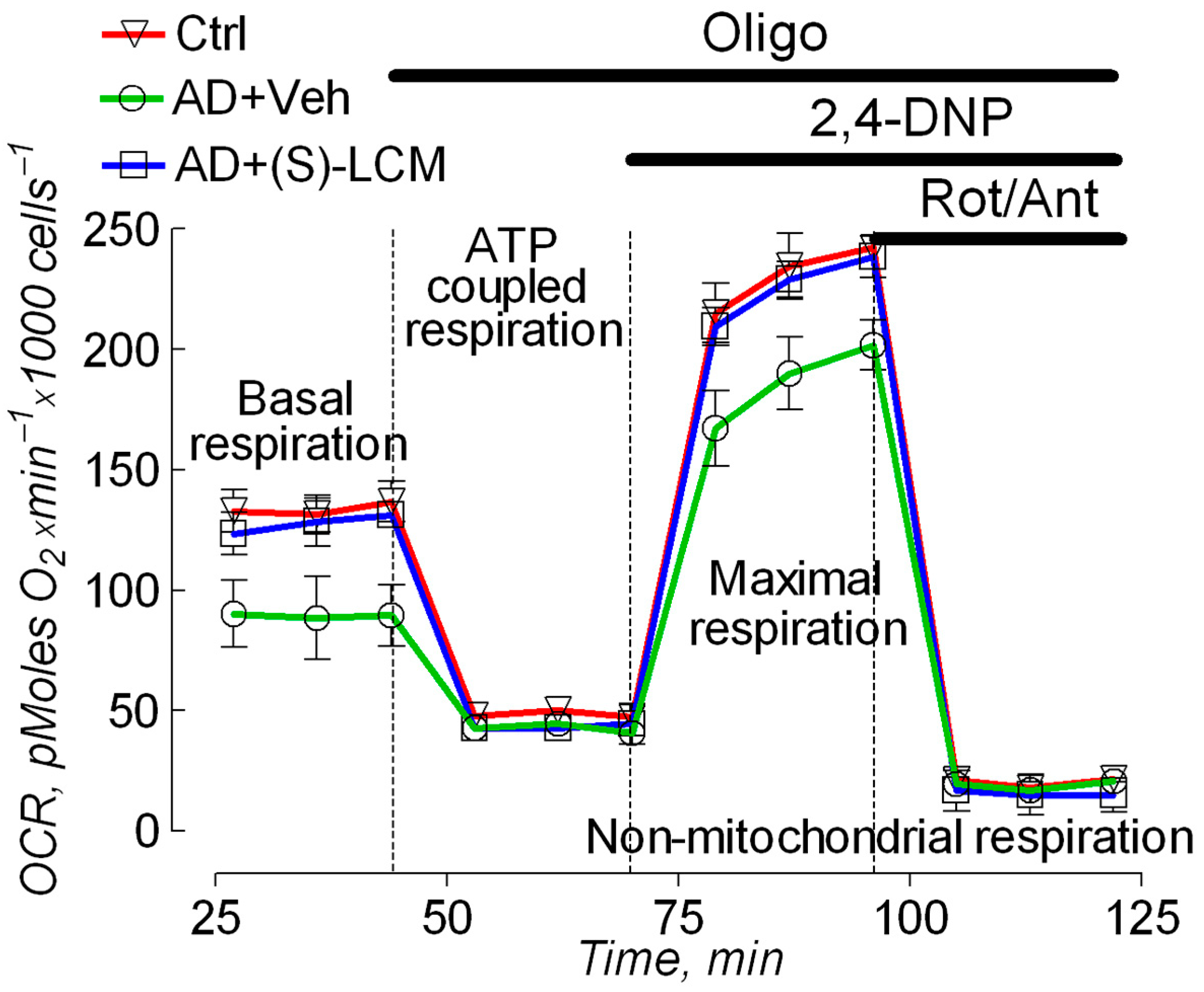
3.2. Cell Respirometry
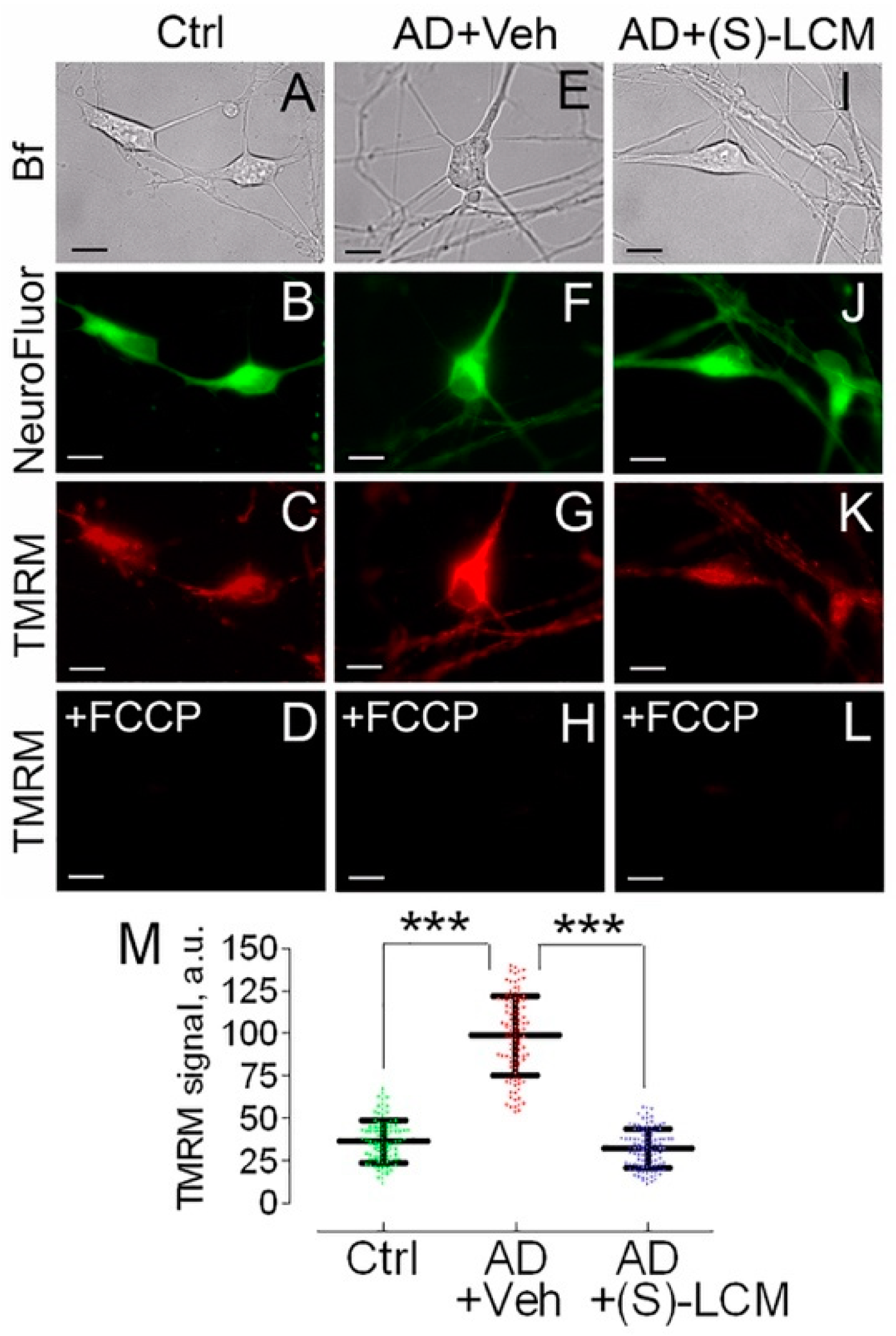
3.3. Mitochondrial Membrane Potential and Superoxide Production in Cultured Neurons

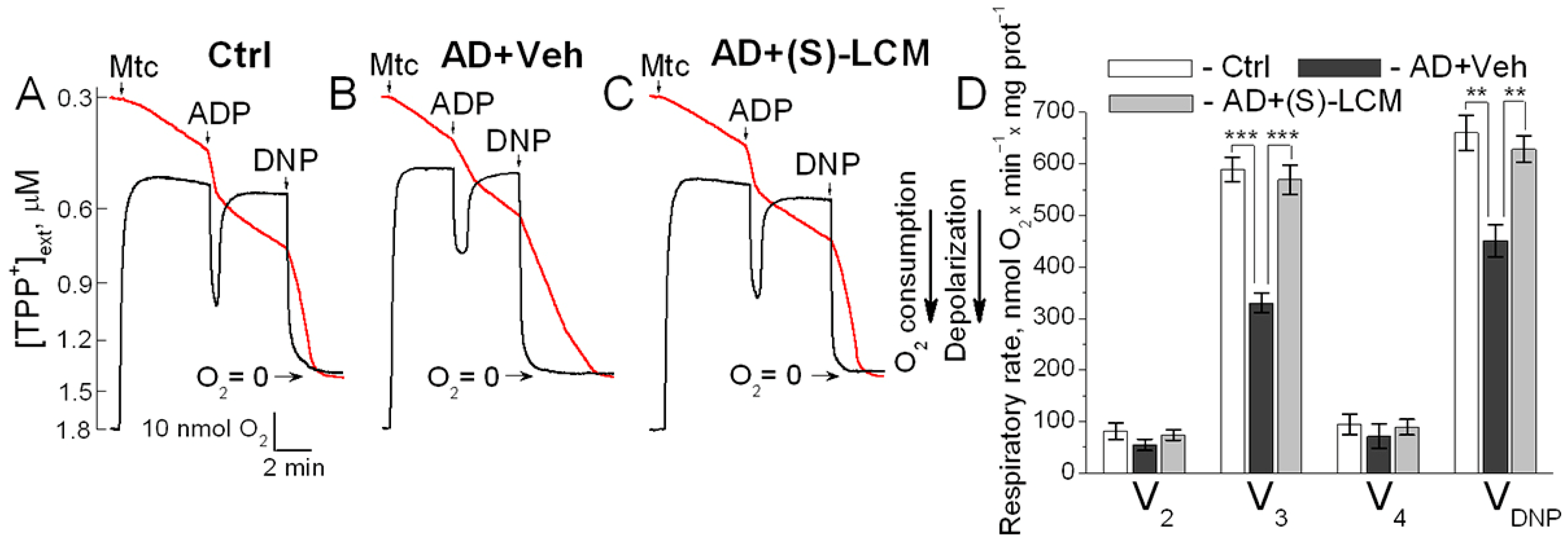
3.4. Respiration and Membrane Potential in Isolated Mitochondria
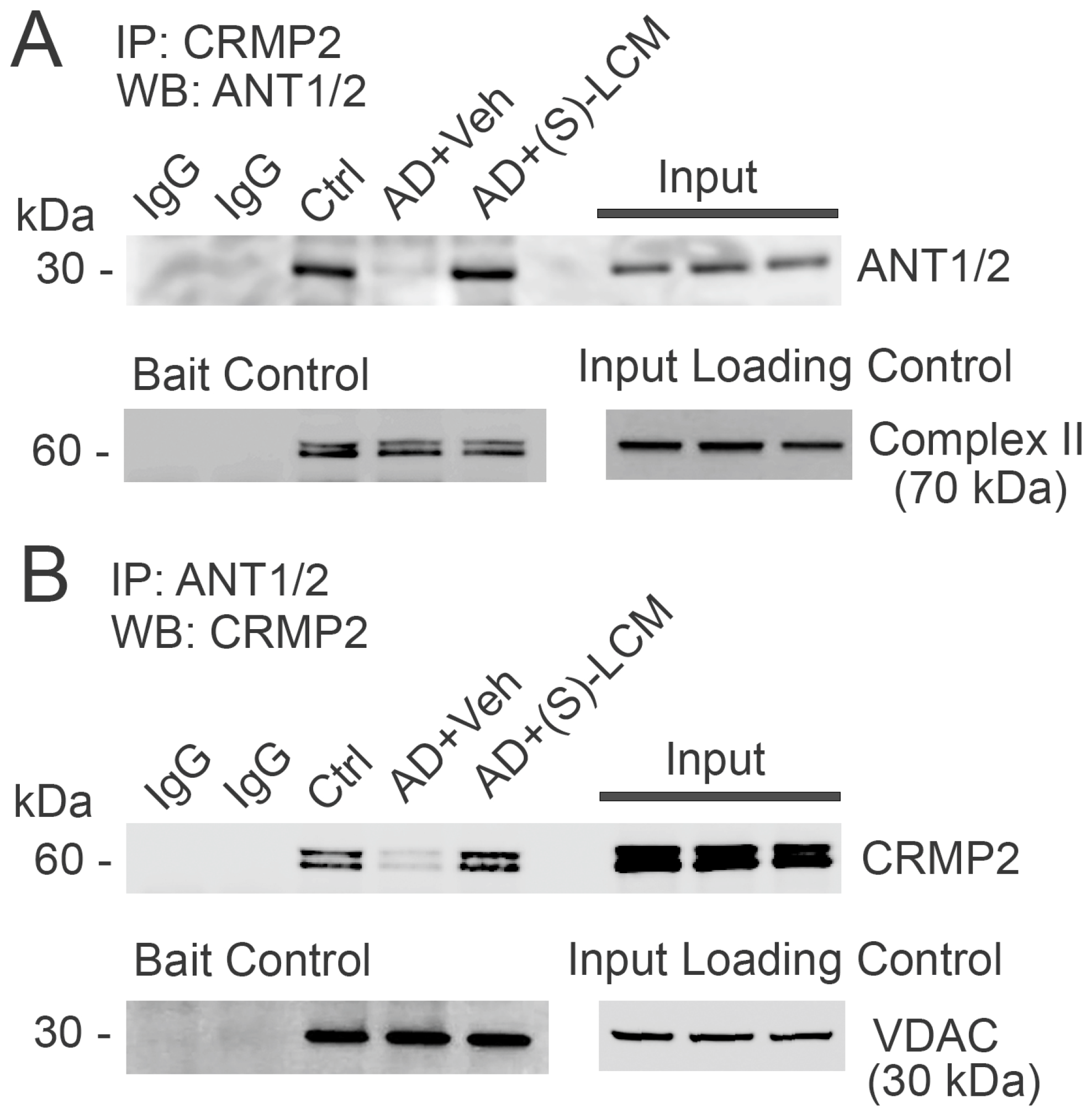
3.5. CRMP2–ANT Co-Immunoprecipitation
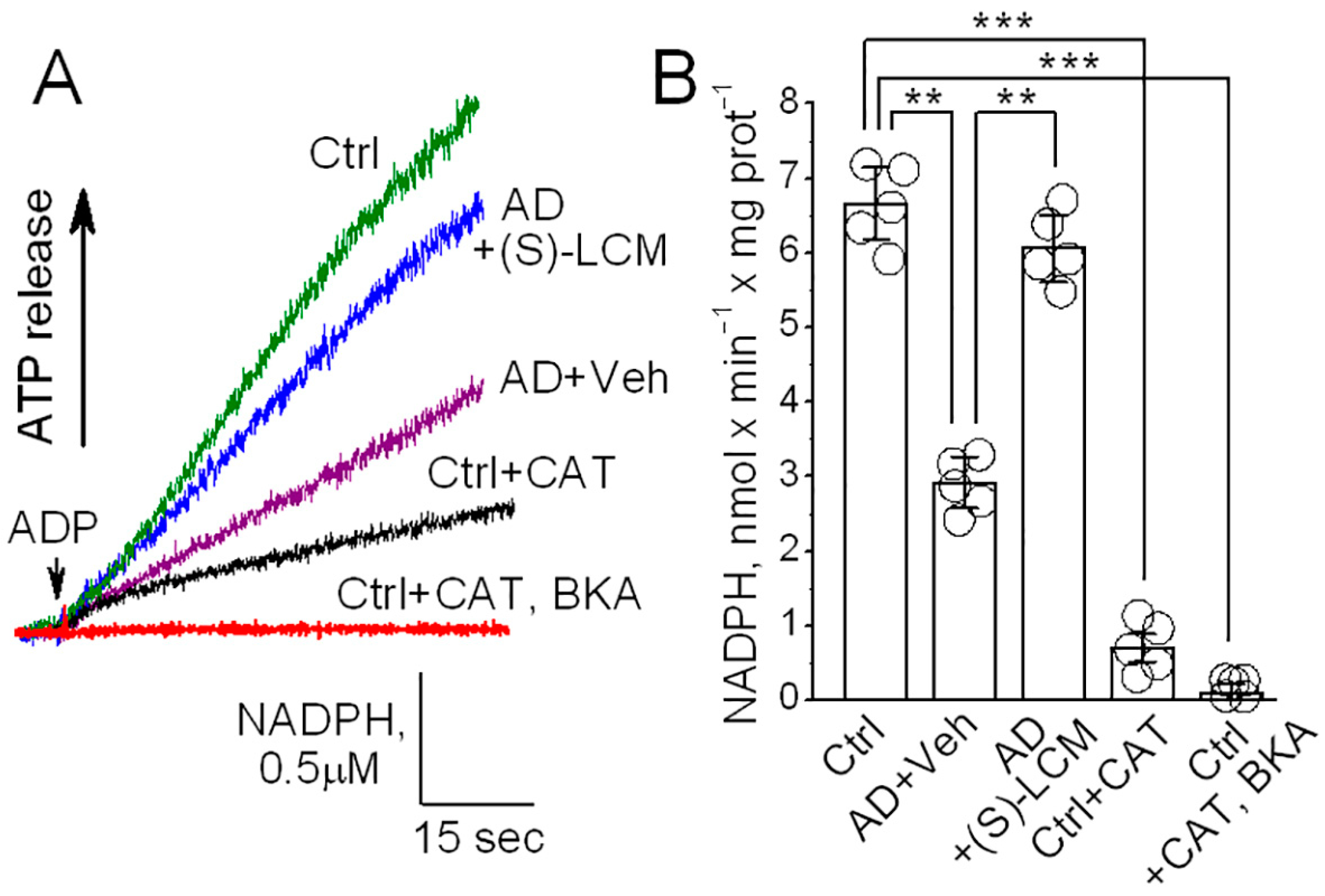
3.6. ANT Activity in Isolated Mitochondria
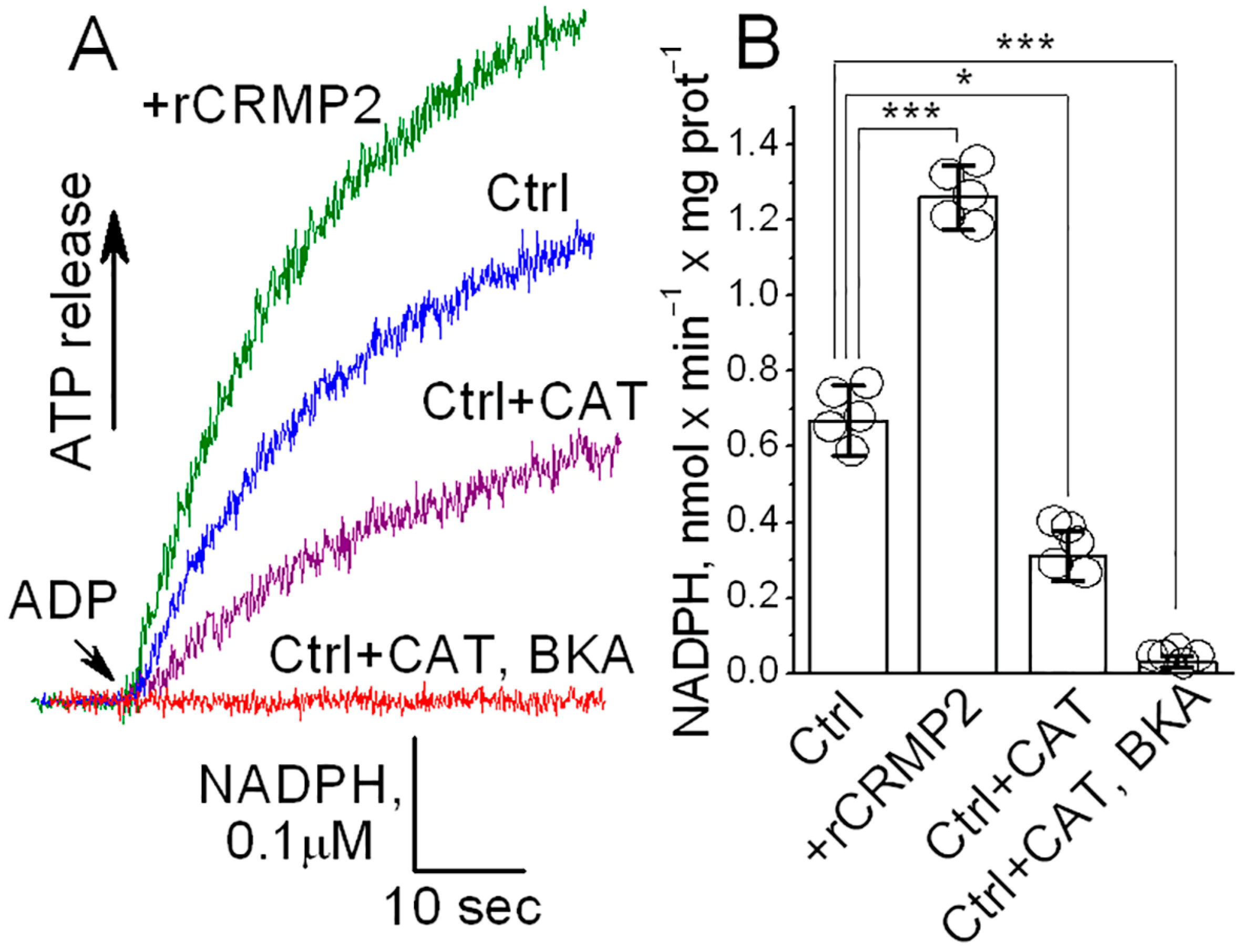
3.7. ANT Activity in ANT-Reconstituted Proteoliposomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Selfridge, J.E.; Lezi, E.; Lu, J.; Swerdlow, R.H. Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis. 2013, 51, 3–12. [Google Scholar] [CrossRef]
- Martins, I.V.; Rivers-Auty, J.; Allan, S.M.; Lawrence, C.B. Mitochondrial Abnormalities and Synaptic Loss Underlie Memory Deficits Seen in Mouse Models of Obesity and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Pedros, I.; Petrov, D.; Allgaier, M.; Sureda, F.; Barroso, E.; Beas-Zarate, C.; Auladell, C.; Pallas, M.; Vazquez-Carrera, M.; Casadesus, G.; et al. Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1556–1566. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. Alzheimer’s disease: Diverse aspects of mitochondrial malfunctioning. Int. J. Clin. Exp. Pathol. 2010, 3, 570–581. [Google Scholar]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement. 2023, 19, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef]
- Parodi-Rullan, R.; Sone, J.Y.; Fossati, S. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 1019–1039. [Google Scholar] [CrossRef] [PubMed]
- Parodi-Rullan, R.M.; Javadov, S.; Fossati, S. Dissecting the Crosstalk between Endothelial Mitochondrial Damage, Vascular Inflammation, and Neurodegeneration in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Cells 2021, 10, 2903. [Google Scholar] [CrossRef]
- Jang, S.; Chapa-Dubocq, X.R.; Parodi-Rullan, R.M.; Fossati, S.; Javadov, S. Beta-Amyloid Instigates Dysfunction of Mitochondria in Cardiac Cells. Cells 2022, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Budd, S.L.; Nicholls, D.G. Mitochondria in the life and death of neurons. Essays Biochem. 1998, 33, 43–52. [Google Scholar]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Ferguson, S.J. Bioenergetics 4; Academic Press: London, UK, 2013. [Google Scholar]
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef]
- Kunji, E.R.; Aleksandrova, A.; King, M.S.; Majd, H.; Ashton, V.L.; Cerson, E.; Springett, R.; Kibalchenko, M.; Tavoulari, S.; Crichton, P.G.; et al. The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta 2016, 1863, 2379–2393. [Google Scholar] [CrossRef] [PubMed]
- Gauba, E.; Chen, H.; Guo, L.; Du, H. Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol. Dis. 2019, 121, 138–147. [Google Scholar] [CrossRef]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016, 7, 11483. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Amadoro, G.; Bobba, A.; De Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta 2008, 1777, 1289–1300. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; Petragallo, V.A.; Calissano, P.; Atlante, A. Dissecting the molecular mechanism by which NH2htau and Abeta1-42 peptides impair mitochondrial ANT-1 in Alzheimer disease. Biochim. Biophys. Acta 2013, 1827, 848–860. [Google Scholar] [CrossRef]
- Atlante, A.; Valenti, D.; Latina, V.; Amadoro, G. Dysfunction of Mitochondria in Alzheimer’s Disease: ANT and VDAC Interact with Toxic Proteins and Aid to Determine the Fate of Brain Cells. Int. J. Mol. Sci. 2022, 23, 7722. [Google Scholar] [CrossRef]
- Goshima, Y.; Nakamura, F.; Strittmatter, P.; Strittmatter, S.M. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 1995, 376, 509–514. [Google Scholar] [CrossRef]
- Khanna, R.; Wilson, S.M.; Brittain, J.M.; Weimer, J.; Sultana, R.; Butterfield, A.; Hensley, K. Opening Pandora’s jar: A primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future Neurol. 2012, 7, 749–771. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Venkova, K.; Christov, A.; Gunning, W.; Park, J. Collapsin response mediator protein-2: An emerging pathologic feature and therapeutic target for neurodisease indications. Mol. Neurobiol. 2011, 43, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Kursula, P. Collapsin Response Mediator Protein-2 (CRMP2) is a Plausible Etiological Factor and Potential Therapeutic Target in Alzheimer’s Disease: Comparison and Contrast with Microtubule-Associated Protein Tau. J. Alzheimers Dis. 2016, 53, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Quach, T.T.; Moutal, A.; Khanna, R.; Deems, N.P.; Duchemin, A.M.; Barrientos, R.M. Collapsin Response Mediator Proteins: Novel Targets for Alzheimer’s Disease. J. Alzheimers Dis. 2020, 77, 949–960. [Google Scholar] [CrossRef]
- Khanna, R.; Moutal, A.; Perez-Miller, S.; Chefdeville, A.; Boinon, L.; Patek, M. Druggability of CRMP2 for Neurodegenerative Diseases. ACS Chem. Neurosci. 2020, 11, 2492–2505. [Google Scholar] [CrossRef]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 309–317. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, Y.; Zhao, B. Roles of glycogen synthase kinase 3 in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 864–879. [Google Scholar] [CrossRef]
- Tsai, L.H.; Lee, M.S.; Cruz, J. Cdk5, a therapeutic target for Alzheimer’s disease? Biochim. Biophys. Acta 2004, 1697, 137–142. [Google Scholar] [CrossRef]
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 2012, 43, 655–662. [Google Scholar] [CrossRef]
- Gu, Y.; Hamajima, N.; Ihara, Y. Neurofibrillary tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, and Ser-522. Biochemistry 2000, 39, 4267–4275. [Google Scholar] [CrossRef]
- Cole, A.R.; Knebel, A.; Morrice, N.A.; Robertson, L.A.; Irving, A.J.; Connolly, C.N.; Sutherland, C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 2004, 279, 50176–50180. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.R.; Noble, W.; van Aalten, L.; Plattner, F.; Meimaridou, R.; Hogan, D.; Taylor, M.; LaFrancois, J.; Gunn-Moore, F.; Verkhratsky, A.; et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J. Neurochem. 2007, 103, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Soutar, M.P.; Thornhill, P.; Cole, A.R.; Sutherland, C. Increased CRMP2 phosphorylation is observed in Alzheimer’s disease; does this tell us anything about disease development? Curr. Alzheimer Res. 2009, 6, 269–278. [Google Scholar] [CrossRef]
- Mokhtar, S.H.; Kim, M.J.; Magee, K.A.; Aui, P.M.; Thomas, S.; Bakhuraysah, M.M.; Alrehaili, A.A.; Lee, J.Y.; Steer, D.L.; Kenny, R.; et al. Amyloid-beta-dependent phosphorylation of collapsin response mediator protein-2 dissociates kinesin in Alzheimer’s disease. Neural Regen. Res. 2018, 13, 1066–1080. [Google Scholar] [PubMed]
- Petratos, S.; Li, Q.X.; George, A.J.; Hou, X.; Kerr, M.L.; Unabia, S.E.; Hatzinisiriou, I.; Maksel, D.; Aguilar, M.I.; Small, D.H. The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 2008, 131, 90–108. [Google Scholar] [CrossRef]
- Rembutsu, M.; Soutar, M.P.; van Aalten, L.; Gourlay, R.; Hastie, C.J.; McLauchlan, H.; Morrice, N.A.; Cole, A.R.; Sutherland, C. Novel procedure to investigate the effect of phosphorylation on protein complex formation in vitro and in cells. Biochemistry 2008, 47, 2153–2161. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Khanna, R.; Brustovetsky, N. CRMP2 Is Involved in Regulation of Mitochondrial Morphology and Motility in Neurons. Cells 2021, 10, 2781. [Google Scholar] [CrossRef]
- Xia, D.; Lianoglou, S.; Sandmann, T.; Calvert, M.; Suh, J.H.; Thomsen, E.; Dugas, J.; Pizzo, M.E.; DeVos, S.L.; Earr, T.K.; et al. Novel App knock-in mouse model shows key features of amyloid pathology and reveals profound metabolic dysregulation of microglia. Mol. Neurodegener. 2022, 17, 41. [Google Scholar] [CrossRef]
- Dubinsky, J.M. Intracellular calcium levels during the period of delayed excitotoxicity. J. Neurosci. 1993, 13, 623–631. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Bolshakov, A.; Brustovetsky, N. Calpain activation and Na(+)/Ca(2+) exchanger degradation occur downstream of calcium deregulation in hippocampal neurons exposed to excitotoxic glutamate. J. Neurosci. Res. 2010, 88, 1317–1328. [Google Scholar] [CrossRef]
- Brittain, J.M.; Chen, L.; Wilson, S.M.; Brustovetsky, T.; Gao, X.; Ashpole, N.M.; Molosh, A.I.; You, H.; Hudmon, A.; Shekhar, A.; et al. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J. Biol. Chem. 2011, 286, 37778–37792. [Google Scholar] [CrossRef] [PubMed]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolanos, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death. Differ. 2018, 25, 542–572. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.; Brustovetsky, T.; Sridhar, A.; Pan, Y.; Cummins, T.R.; Meyer, J.S.; Brustovetsky, N. Energy Metabolism and Mitochondrial Superoxide Anion Production in Pre-symptomatic Striatal Neurons Derived from Human-Induced Pluripotent Stem Cells Expressing Mutant Huntingtin. Mol. Neurobiol. 2020, 57, 668–684. [Google Scholar] [CrossRef]
- Polster, B.M.; Nicholls, D.G.; Ge, S.X.; Roelofs, B.A. Use of potentiometric fluorophores in the measurement of mitochondrial reactive oxygen species. Methods Enzymol. 2014, 547, 225–250. [Google Scholar]
- Kamo, N.; Muratsugu, M.; Hongoh, R.; Kobatake, Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J. Membr. Biol. 1979, 49, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Brustovetsky, T. Techniques to Investigate Mitochondrial Fucntion in Neurons; Strack, S., Usachev, Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 199–210. [Google Scholar]
- Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. The effect of mitochondrial calcium uniporter and cyclophilin D knockout on resistance of brain mitochondria to Ca(2+)-induced damage. J. Biol. Chem. 2021, 296, 100669. [Google Scholar] [CrossRef]
- Gawaz, M.; Douglas, M.G.; Klingenberg, M. Structure-function studies of adenine nucleotide transport in mitochondria. II. Biochemical analysis of distinct AAC1 and AAC2 proteins in yeast. J. Biol. Chem. 1990, 265, 14202–14208. [Google Scholar] [CrossRef]
- Brustovetsky, N.; Klingenberg, M. The reconstituted ADP/ATP carrier can mediate H+ transport by free fatty acids, which is further stimulated by mersalyl. J. Biol. Chem. 1994, 269, 27329–27336. [Google Scholar] [CrossRef]
- Brittain, J.M.; Piekarz, A.D.; Wang, Y.; Kondo, T.; Cummins, T.R.; Khanna, R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem. 2009, 284, 31375–31390. [Google Scholar] [CrossRef]
- Dustrude, E.T.; Perez-Miller, S.; Francois-Moutal, L.; Moutal, A.; Khanna, M.; Khanna, R. A single structurally conserved SUMOylation site in CRMP2 controls NaV1.7 function. Channels 2017, 11, 316–328. [Google Scholar] [CrossRef]
- Passarella, S.; Ostuni, A.; Atlante, A.; Quagliariello, E. Increase in the ADP/ATP exchange in rat liver mitochondria irradiated in vitro by helium-neon laser. Biochem. Biophys. Res. Commun. 1988, 156, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Lustorff, J.; Schlimme, E. Does an inhibitor of mitochondrial adenylate kinase also affect oxidative phosphorylation? Experientia 1976, 32, 298–299. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, M.; Grebe, K.; Scherer, B. The binding of atractylate and carboxy-atractylate to mitochondria. Eur. J. Biochem. 1975, 52, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, M.; Buchholz, M. On the mechanism of bongkrekate effect on the mitochondrial adenine-nucleotide carrier as studied through the binding of ADP. Eur. J. Biochem. 1973, 38, 346–358. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Khanna, R.; Brustovetsky, N. CRMP2 participates in regulating mitochondrial morphology and motility in Alzheimer’s disease. Cells 2023, 12, 1287. [Google Scholar] [CrossRef]
- Moutal, A.; Francois-Moutal, L.; Perez-Miller, S.; Cottier, K.; Chew, L.A.; Yeon, S.K.; Dai, J.; Park, K.D.; Khanna, M.; Khanna, R. (S)-Lacosamide Binding to Collapsin Response Mediator Protein 2 (CRMP2) Regulates CaV2.2 Activity by Subverting Its Phosphorylation by Cdk5. Mol. Neurobiol. 2015, 53, 1959–1976. [Google Scholar] [CrossRef]
- Moutal, A.; Chew, L.A.; Yang, X.; Wang, Y.; Yeon, S.K.; Telemi, E.; Meroueh, S.; Park, K.D.; Shrinivasan, R.; Gilbraith, K.B.; et al. (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain 2016, 157, 1448–1463. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Brittain, M.K.; Sheets, P.L.; Cummins, T.R.; Pinelis, V.; Brustovetsky, N. KB-R7943, an inhibitor of the reverse Na+ /Ca2+ exchanger, blocks N-methyl-D-aspartate receptor and inhibits mitochondrial complex I. Br. J. Pharmacol. 2011, 162, 255–270. [Google Scholar] [CrossRef]
- Hamilton, J.; Pellman, J.J.; Brustovetsky, T.; Harris, R.A.; Brustovetsky, N. Oxidative metabolism in YAC128 mouse model of Huntington’s disease. Hum. Mol. Genet. 2015, 24, 4862–4878. [Google Scholar] [CrossRef]
- Wang, S.; Ichinomiya, T.; Savchenko, P.; Devulapalli, S.; Wang, D.; Beltz, G.; Saito, T.; Saido, T.C.; Wagner, S.L.; Patel, H.H.; et al. Age-Dependent Behavioral and Metabolic Assessment of App (NL-G-F/NL-G-F) Knock-in (KI) Mice. Front Mol. Neurosci. 2022, 15, 909989. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Vercesi, A.E.; Kowaltowski, A.J.; Grijalba, M.T.; Meinicke, A.R.; Castilho, R.F. The role of reactive oxygen species in mitochondrial permeability transition. Biosci. Rep. 1997, 17, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 2017, 16, 943–955. [Google Scholar] [CrossRef]
- Kent, A.C.; El Baradie, K.B.Y.; Hamrick, M.W. Targeting the Mitochondrial Permeability Transition Pore to Prevent Age-Associated Cell Damage and Neurodegeneration. Oxidative Med. Cell. Longev. 2021, 2021, 6626484. [Google Scholar] [CrossRef] [PubMed]
- Morinaka, A.; Yamada, M.; Itofusa, R.; Funato, Y.; Yoshimura, Y.; Nakamura, F.; Yoshimura, T.; Kaibuchi, K.; Goshima, Y.; Hoshino, M.; et al. Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci. Signal. 2011, 4, ra26. [Google Scholar] [CrossRef]
- Nicholls, D.G. Fluorescence measurement of mitochondrial membrane potential changes in cultured cells. Methods Mol. Biol. 2012, 810, 119–133. [Google Scholar]
- Wojtala, A.; Bonora, M.; Malinska, D.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Methods to monitor ROS production by fluorescence microscopy and fluorometry. Methods Enzymol. 2014, 542, 243–262. [Google Scholar]
- Bertholet, A.M.; Natale, A.M.; Bisignano, P.; Suzuki, J.; Fedorenko, A.; Hamilton, J.; Brustovetsky, T.; Kazak, L.; Garrity, R.; Chouchani, E.T.; et al. Mitochondrial uncouplers induce proton leak by activating AAC and UCP1. Nature 2022, 606, 180–187. [Google Scholar] [CrossRef]
- Hamilton, J.; Brustovetsky, T.; Rysted, J.E.; Lin, Z.; Usachev, Y.M.; Brustovetsky, N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J. Biol. Chem. 2018, 293, 15652–15663. [Google Scholar] [CrossRef]
- Brustovetsky, N.; Brustovetsky, T.; Jemmerson, R.; Dubinsky, J.M. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J. Neurochem. 2002, 80, 207–218. [Google Scholar] [CrossRef]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohshima, T.; Sasaki, Y.; Suzuki, H.; Yanai, S.; Yamashita, N.; Nakamura, F.; Takei, K.; Ihara, Y.; Mikoshiba, K.; et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: Implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 2005, 10, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hawkes, C.; Qureshi, H.Y.; Kar, S.; Paudel, H.K. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry 2006, 45, 3134–3145. [Google Scholar] [CrossRef]
- Toba, J.; Nikkuni, M.; Ishizeki, M.; Yoshii, A.; Watamura, N.; Inoue, T.; Ohshima, T. PPARgamma agonist pioglitazone improves cerebellar dysfunction at pre-Abeta deposition stage in APPswe/PS1dE9 Alzheimer’s disease model mice. Biochem. Biophys. Res. Commun. 2016, 473, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kuboyama, T.; Tohda, C. A Systematic Strategy for Discovering a Therapeutic Drug for Alzheimer’s Disease and Its Target Molecule. Front Pharmacol. 2017, 8, 340. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Khanna, R.; Brustovetsky, N. Involvement of CRMP2 in Regulation of Mitochondrial Morphology and Motility in Huntington’s Disease. Cells 2021, 10, 3172. [Google Scholar] [CrossRef]
- Chang, K.L.; Wong, L.R.; Pee, H.N.; Yang, S.; Ho, P.C. Reverting Metabolic Dysfunction in Cortex and Cerebellum of APP/PS1 Mice, a Model for Alzheimer’s Disease by Pioglitazone, a Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma) Agonist. Mol. Neurobiol. 2019, 56, 7267–7283. [Google Scholar] [CrossRef]
- Isono, T.; Yamashita, N.; Obara, M.; Araki, T.; Nakamura, F.; Kamiya, Y.; Alkam, T.; Nitta, A.; Nabeshima, T.; Mikoshiba, K.; et al. Amyloid-beta(2)(5)(-)(3)(5) induces impairment of cognitive function and long-term potentiation through phosphorylation of collapsin response mediator protein 2. Neurosci. Res. 2013, 77, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Ohshima, T.; Nakamura, F.; Kolattukudy, P.; Honnorat, J.; Mikoshiba, K.; Goshima, Y. Phosphorylation of CRMP2 (collapsin response mediator protein 2) is involved in proper dendritic field organization. J. Neurosci. 2012, 32, 1360–1365. [Google Scholar] [CrossRef]
- Belzacq, A.S.; Vieira, H.L.; Kroemer, G.; Brenner, C. The adenine nucleotide translocator in apoptosis. Biochimie 2002, 84, 167–176. [Google Scholar] [CrossRef]
- Held, P. Determination of NADH concentrations with the SynergyTM 2 multi-detection microplate reader using fluorescence or absorbance. Agil. Appl. Note 2021, 1–6. [Google Scholar]
- Rego, A.C.; Vesce, S.; Nicholls, D.G. The mechanism of mitochondrial membrane potential retention following release of cytochrome c in apoptotic GT1-7 neural cells. Cell Death Differ. 2001, 8, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, B.; Koshenov, Z.; Malli, R.; Graier, W.F. Implications of mitochondrial membrane potential gradients on signaling and ATP production analyzed by correlative multi-parameter microscopy. Sci. Rep. 2024, 14, 14784. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brustovetsky, T.; Khanna, R.; Brustovetsky, N. Collapsin Response Mediator Protein 2 (CRMP2) Modulates Mitochondrial Oxidative Metabolism in Knock-In AD Mouse Model. Cells 2025, 14, 647. https://doi.org/10.3390/cells14090647
Brustovetsky T, Khanna R, Brustovetsky N. Collapsin Response Mediator Protein 2 (CRMP2) Modulates Mitochondrial Oxidative Metabolism in Knock-In AD Mouse Model. Cells. 2025; 14(9):647. https://doi.org/10.3390/cells14090647
Chicago/Turabian StyleBrustovetsky, Tatiana, Rajesh Khanna, and Nickolay Brustovetsky. 2025. "Collapsin Response Mediator Protein 2 (CRMP2) Modulates Mitochondrial Oxidative Metabolism in Knock-In AD Mouse Model" Cells 14, no. 9: 647. https://doi.org/10.3390/cells14090647
APA StyleBrustovetsky, T., Khanna, R., & Brustovetsky, N. (2025). Collapsin Response Mediator Protein 2 (CRMP2) Modulates Mitochondrial Oxidative Metabolism in Knock-In AD Mouse Model. Cells, 14(9), 647. https://doi.org/10.3390/cells14090647