
Induced Pluripotent Stem Cells-Based Regenerative Therapies in Treating Human Aging-Related Functional Decline and Diseases



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Molecular Mechanisms of Aging Diseases
2.1. Tissue Level Hallmarks of Aging
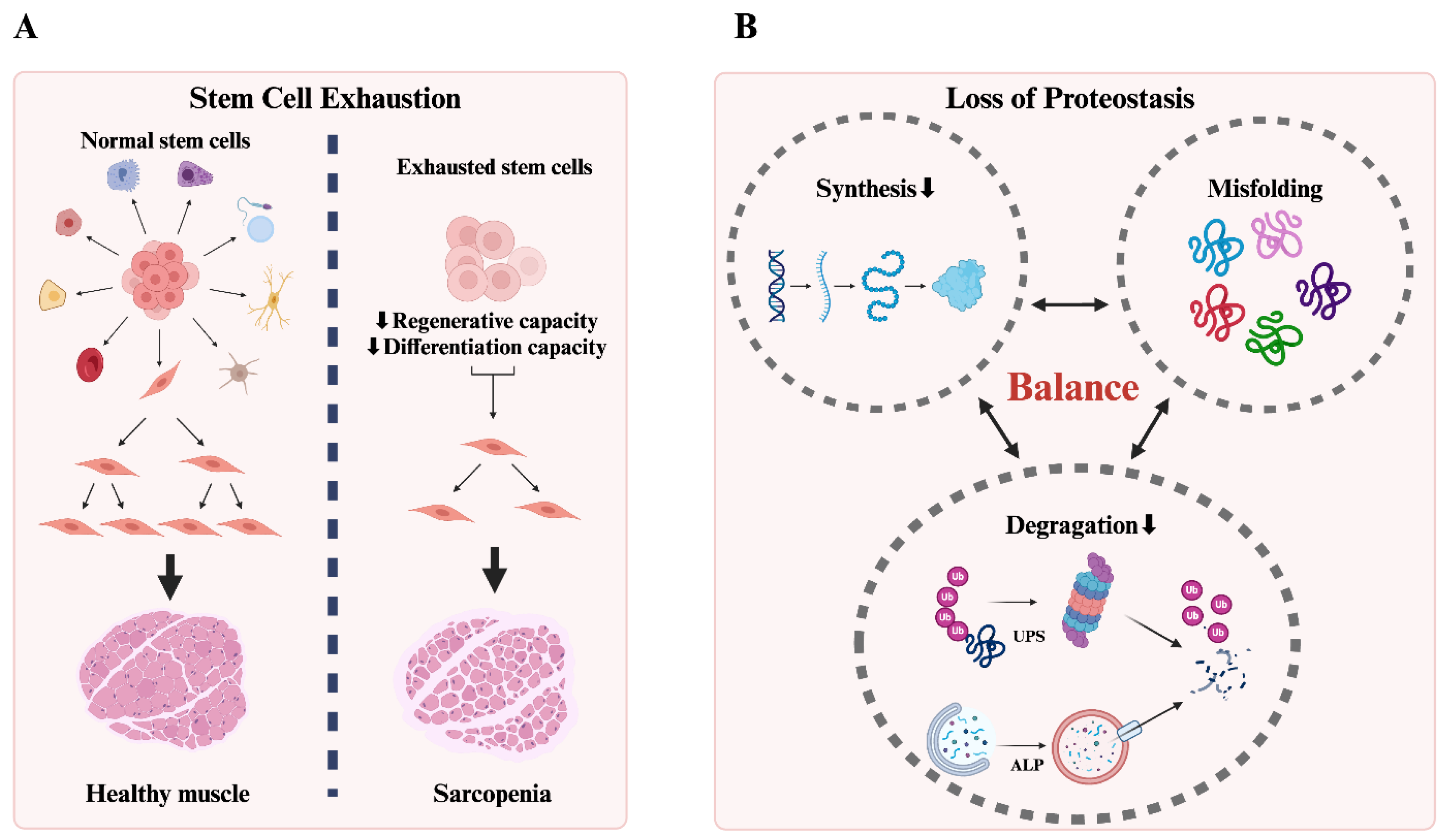
2.1.1. Stem Cell Exhaustion
2.1.2. Loss of Proteostasis
2.2. Hallmarks at the Cellular Level
2.2.1. Genomic Instability
2.2.2. Telomere Attrition
2.2.3. Epigenetic Alterations
2.2.4. Cellular Senescence
2.3. Molecular Level Hallmarks
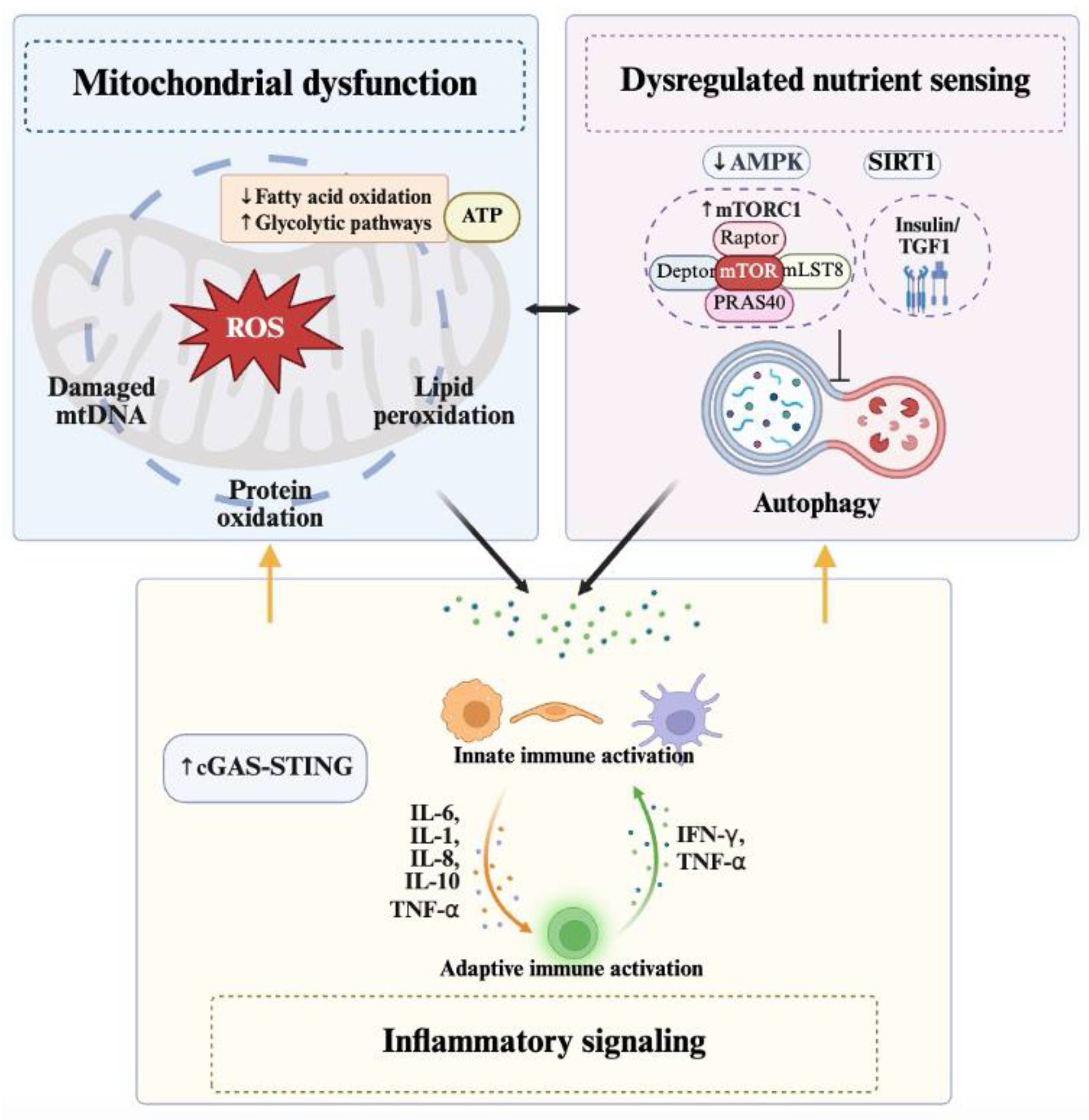
2.3.1. Mitochondrial Dysfunction
2.3.2. Dysregulated Nutrient Sensing
2.3.3. Inflammatory Signaling
3. Cellular Reprogramming for Induced Pluripotent Stem Cell Generation
3.1. Introduction of Reprogramming Factors
3.2. Initiation of Dedifferentiation and Transition to a Pluripotent State
3.3. Verification of Pluripotency
4. Induced Pluripotent Stem Cell-Based Models for Human Aging Diseases Investigation
4.1. Modeling Human Premature Aging Syndromes with iPSCs
4.2. Modeling Human Telomere Dysfunction Disease with iPSCs
4.3. iPSCs-Based Models for Aging-Related Degenerative Diseases
4.3.1. Alzheimer’s Disease (AD)
4.3.2. Parkinson’s Disease (PD)
4.3.3. Huntington’s Disease (HD)
4.3.4. Amyotrophic Lateral Sclerosis (ALS)
4.3.5. Age-Related Macular Degeneration (AMD)
4.4. Modeling Aging-Related Metabolism Diseases with iPSCs
4.4.1. Type 2 Diabetes Mellitus (T2DM)
4.4.2. Diabetic Cardiomyopathy (DCM)
4.4.3. Nonalcoholic Fatty Liver Disease (NAFLD)
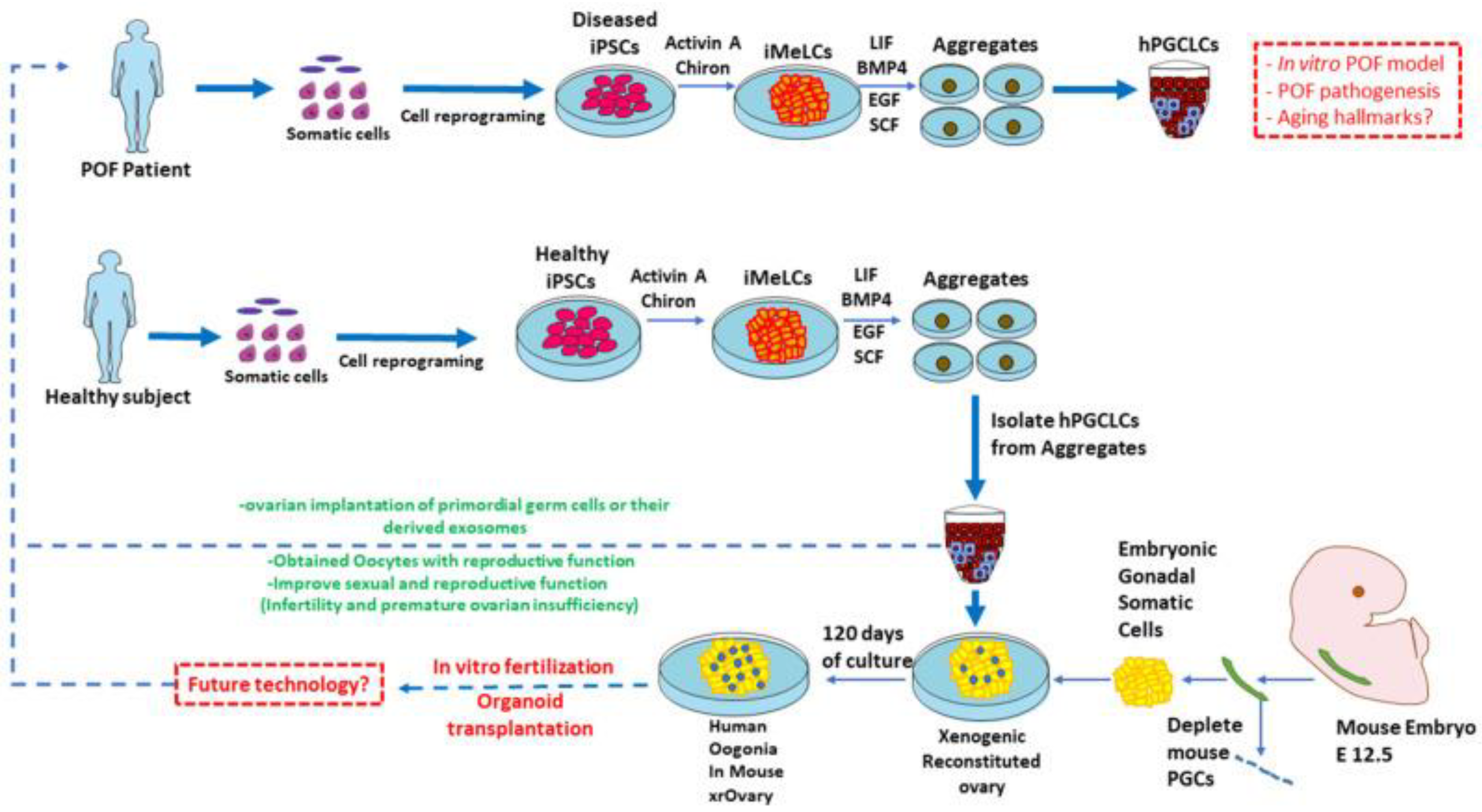
4.5. Modeling Premature Ovarian Aging with iPSC
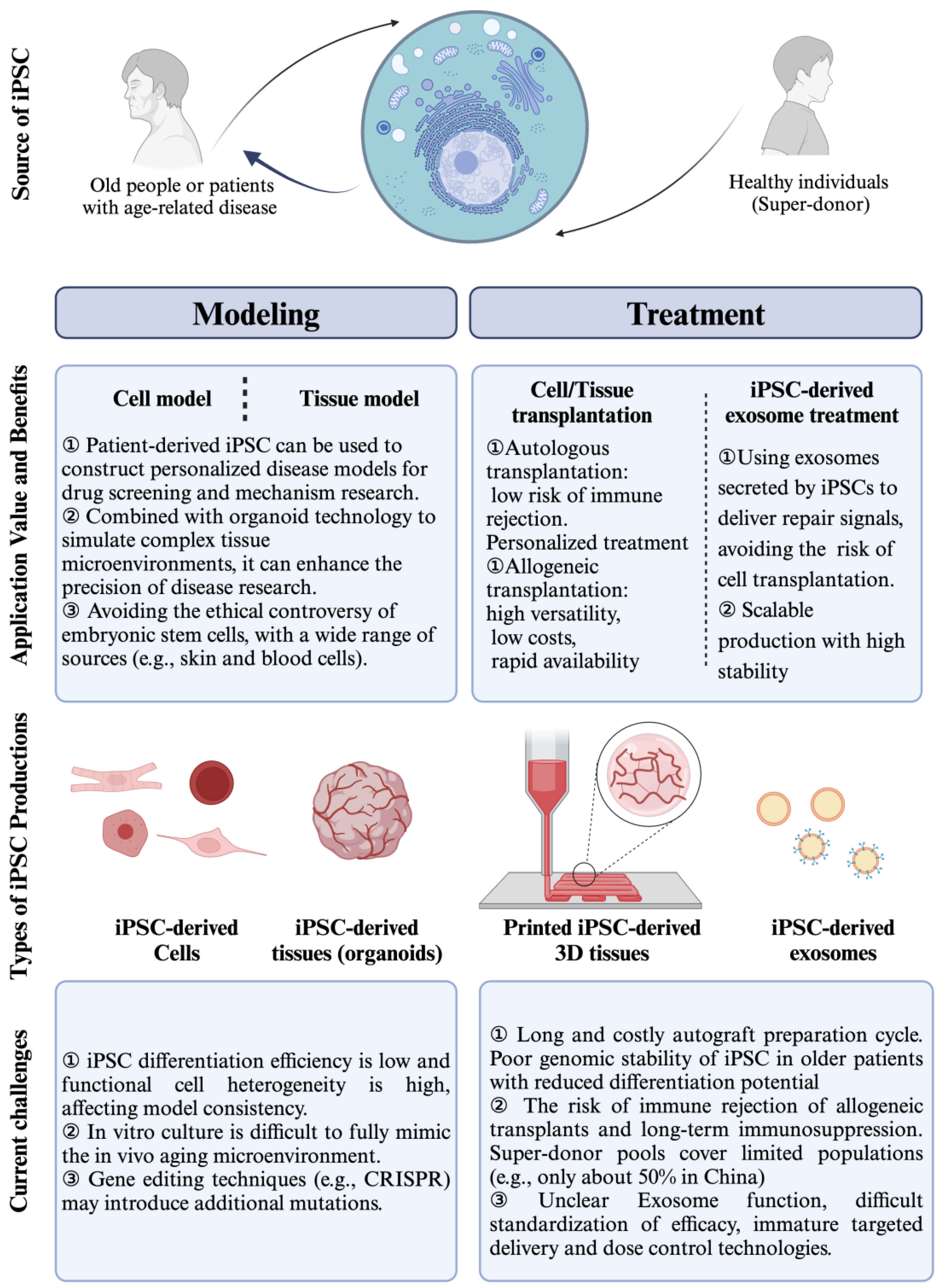
5. Induced Pluripotent Stem Cell-Derived Differentiated Cells for Aging Diseases Therapy
5.1. Patient-Specific Treatment
5.2. Cell Replacement and Regeneration
5.3. Tissue Engineering and Organ Transplantation
5.4. Therapeutic Paracrine Secretion
5.5. iPSC-Induced Immunotherapy for Anti-Aging
6. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
| 3D | Three-dimensional |
| AA | Aplastic anemia |
| AD | Alzheimer’s Disease |
| AEC | Alveolar epithelial cell |
| ALS | Amyotrophic Lateral Sclerosis |
| AMD | Aging-Related Macular Degeneration |
| Apo-E | Apolipoprotein E |
| ATP | Adenosine triphosphate |
| B2M | Beta-2-microglobulin |
| BMF | Bone marrow failure |
| C9ORF72 | Chromosome 9 open reading frame 72 |
| CAR | Chimeric antigen receptors |
| CFH | Complement factor H |
| CS | Cockayne syndrome |
| DC | Dyskeratosis congenita |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| ESCs | Embryonic stem cells |
| FBOs | Forebrain-like organoids |
| HD | Huntington’s Disease |
| HGPS | Hutchinson-Gilford Progeria Syndrome |
| HLA | Human leukocyte antigen |
| IGF1 | Insulin-like growth factor 1 |
| IPF | Idiopathic pulmonary fibrosis |
| iPSC-CM | iPSC-derived cardiomyocyte |
| iPSCs | Induced pluripotent stem cells |
| MSC | Mesenchymal stromal cells |
| mtDNA | Mitochondrial DNA |
| mTOR | The mammalian target of rapamycin |
| NAFLD | Nonalcoholic Fatty Liver Disease |
| NK cell | Natural killer cell |
| POI | Premature ovarian insufficiency |
| PD | Parkinson’s Disease |
| ROS | Reactive oxygen species |
| RPE | Retinal pigment epithelium |
| SASP | Senescence-associated secretory phenotype |
| SIRT1 | NAD-dependent deacetylase sirtuin-1 |
| T2DM | Type 2 diabetes mellitus |
| TDP-43 | TAR DNA binding protein |
| TM cell | Trabecular meshwork cell |
| WS | Werner syndrome |
| α-syn | α-synuclein |
References
- Komp-Leukkunen, K.; Sarasma, J. Social Sustainability in Aging Populations: A Systematic Literature Review. Gerontologist 2024, 64, gnad097. [Google Scholar] [CrossRef]
- Kanev, A.; Kobyakova, O.; Kurakova, N.; Shibalkov, I.I. Population ageing and national healthcare systems sustainability. A review of world practices. Natl. Health Care 2024, 4, 5–13. [Google Scholar] [CrossRef]
- Harper, S. Economic and social implications of aging societies. Science 2014, 346, 587–591. [Google Scholar] [CrossRef]
- Sun, J.; Guo, Y.; Wang, X.; Zeng, Q. mHealth For Aging China: Opportunities and Challenges. Aging Dis. 2016, 7, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Witkowski, J.M.; McElhaney, J.; Loeb, M.; Mitnitski, A.; Pawelec, G. Aging, frailty and age-related diseases. Biogerontology 2010, 11, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Castle, S.C. Clinical relevance of age-related immune dysfunction. Clin. Infect. Dis. 2000, 31, 578–585. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, Y.; Wong, N.D.; Wang, J. Impact of Aging on Cardiovascular Diseases: From Chronological Observation to Biological Insights: JACC Family Series. JACC Asia 2024, 4, 345–358. [Google Scholar] [CrossRef]
- van Onna, M.A.-O.; Boonen, A.A.-O. Challenges in the management of older patients with inflammatory rheumatic diseases. Nat. Rev. Rheumatol. 2022, 18, 326–334. [Google Scholar] [CrossRef]
- Su, S.-Y.; He, C.; Ren, J.; Song, M. Global insights into aging: A multidisciplinary approach to understanding and addressing age-related challenges. Life Med. 2024, 3, lnae029. [Google Scholar] [CrossRef]
- Lin, H. The stem-cell niche theory: Lessons from flies. Nat. Rev. Genet. 2002, 3, 931–940. [Google Scholar] [CrossRef]
- Trosko, J.E.; Chang, C.C. Stem cell theory of carcinogenesis. Toxicol. Lett. 1989, 49, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Kimbrel, E.A.; Lanza, R. Next-generation stem cells—Ushering in a new era of cell-based therapies. Nat. Rev. Drug Discov. 2020, 19, 463–479. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Song, Z.; Ju, Z. Alterations of Systemic Environment Impair Stem Cell Function during Aging. In Molecular Mechanisms of Adult Stem Cell Aging; S.Karger AG: Basel, Switzerland, 2010. [Google Scholar]
- Bueno, V. Stem Cell Exhaustion. In Cellular and Molecular Aspects of Ageing; Bueno, V., Ed.; Springer Nature: Basel, Switzerland, 2024; pp. 77–86. [Google Scholar]
- Ge, Y.; Miao, Y.; Gur-Cohen, S.; Gomez, N.; Yang, H.; Nikolova, M.; Polak, L.; Hu, Y.; Verma, A.; Elemento, O.; et al. The aging skin microenvironment dictates stem cell behavior. Proc. Natl. Acad. Sci. USA 2020, 10, 5339–5350. [Google Scholar] [CrossRef]
- Ganguly, P.; El-Jawhari, J.J.; Giannoudis, P.V.; Burska, A.N.; Ponchel, F.; Jones, E.A. Age-related Changes in Bone Marrow Mesenchymal Stromal Cells: A Potential Impact on Osteoporosis and Osteoarthritis Development. Cell Transplant. 2017, 9, 1520–1529. [Google Scholar] [CrossRef]
- Walter, L.D.; Orton, J.L.; Ntekas, I.; Fong, E.H.H.; Maymi, V.I.; Rudd, B.D.; De Vlaminck, I.; Elisseeff, J.H.; Cosgrove, B.D. Transcriptomic analysis of skeletal muscle regeneration across mouse lifespan identifies altered stem cell states. Nat. Aging 2024, 4, 1862–1881. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, T.; Ikemoto-Uezumi, M.; Yoshimoto, Y.; Minato, K.; Kaji, N.; Chaen, T.; Hase, E.; Minamikawa, T.; Yasui, T.; Horiguchi, K.; et al. Tissue-specific functions of MSCs are linked to homeostatic muscle maintenance and alter with aging. Aging Cell 2024, 23, e14299. [Google Scholar] [CrossRef]
- Santra, M.; Dill, K.A.; de Graff, A.M.R. Proteostasis collapse is a driver of cell aging and death. Proc. Natl. Acad. Sci. USA 2019, 116, 22173–22178. [Google Scholar] [CrossRef]
- Hohn, A.; Tramutola, A.; Cascella, R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5497046. [Google Scholar] [CrossRef]
- Cortés Sanchón, A.; Santhosh Kumar, H.; Mantovani, M.; Osinnii, I.; Mateos, J.A.-O.; Kaech, A.; Shcherbakov, D.; Akbergenov, R.; Böttger, E.A.-O. ER-misfolded proteins become sequestered with mitochondria and impair mitochondrial function. Commun. Biol. 2021, 4, 1350. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.A.-O.; Curley, M.A.-O.; Coleman, Z.; Demontis, F.A.-O. Contribution of proteases to the hallmarks of aging and to age-related neurodegeneration. Aging Cell 2022, 21, e13603. [Google Scholar] [CrossRef]
- Tang, H.; Inoki, K.; Brooks, S.V.; Okazawa, H.; Lee, M.; Wang, J.; Kim, M.; Kennedy, C.L.; Macpherson, P.C.D.; Ji, X.; et al. mTORC1 underlies age-related muscle fiber damage and loss by inducing oxidative stress and catabolism. Aging Cell 2019, 18, e12943. [Google Scholar] [CrossRef]
- Padilla, C.J.; Harrigan, M.E.; Harris, H.; Schwab, J.M.; Rutkove, S.B.; Rich, M.M.; Clark, B.C.; Arnold, W. Profiling age-related muscle weakness and wasting: Neuromuscular junction transmission as a driver of age-related physical decline. Geroscience 2021, 43, 1265–1281. [Google Scholar] [CrossRef]
- Vijg, J.; Suh, Y. Genome instability and aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef]
- Li, W.; Sancar, A. Methodologies for detecting environmentally induced DNA damage and repair. Environ. Mol. Mutagen. 2020, 61, 664–679. [Google Scholar] [CrossRef]
- Schumacher, B.A.-O.; Pothof, J.; Vijg, J.A.-O.; Hoeijmakers, J.A.-O. The central role of DNA damage in the ageing process. Nature 2021, 592, 695–703. [Google Scholar] [CrossRef]
- Huang, X.; Tran, T.; Zhang, L.; Hatcher, R.; Zhang, P. DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proc. Natl. Acad. Sci. USA 2005, 102, 1065–1070. [Google Scholar] [CrossRef]
- Chakraborty, U.; Dinh, T.A.; Alani, E.A.-O. Genomic Instability Promoted by Overexpression of Mismatch Repair Factors in Yeast: A Model for Understanding Cancer Progression. Genetics 2018, 209, 439–456. [Google Scholar] [CrossRef]
- Iourov, I.A.-O.; Yurov, Y.B.; Vorsanova, S.G.; Kutsev, S.I. Chromosome Instability, Aging and Brain Diseases. Cells 2021, 10, 1256. [Google Scholar] [CrossRef] [PubMed]
- Lodato, M.A.-O.; Rodin, R.A.-O.; Bohrson, C.A.-O.; Coulter, M.A.-O.X.; Barton, A.A.-O.; Kwon, M.A.-O.X.; Sherman, M.A.-O.; Vitzthum, C.A.-O.; Luquette, L.J.; Yandava, C.N.; et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 2018, 361, 6397. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Lagnado, A.; Leslie, J.; Ruchaud-Sparagano, M.H.; Victorelli, S.; Hirsova, P.; Ogrodnik, M.; Collins, A.L.; Vizioli, M.G.; Habiballa, L.; Saretzki, G.; et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 2021, 40, e106048. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere dysfunction in ageing and age-related diseases. Nat. Cell. Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef]
- Gao, Z.; Santos, R.B.; Rupert, J.; Van Drunen, R.; Yu, Y.; Eckel-Mahan, K.; Kolonin, M.A.-O. Endothelial-specific telomerase inactivation causes telomere-independent cell senescence and multi-organ dysfunction characteristic of aging. Aging Cell 2024, 23, e14138. [Google Scholar] [CrossRef]
- Bloom, S.A.-O.; Liu, Y.; Tucker, J.R.; Islam, M.T.; Machin, D.A.-O.; Abdeahad, H.A.-O.; Thomas, T.G.; Bramwell, R.C.; Lesniewski, L.A.; Donato, A.J. Endothelial cell telomere dysfunction induces senescence and results in vascular and metabolic impairments. Aging Cell 2023, 22, e13875. [Google Scholar] [CrossRef] [PubMed]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight. 2016, 1, e86704. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, L.; Cong, Y.S. Telomere Dysfunction in Idiopathic Pulmonary Fibrosis. Front. Med. 2021, 8, 739810. [Google Scholar] [CrossRef]
- Ren, L.L.; Miao, H.; Wang, Y.N.; Liu, F.; Li, P.; Zhao, Y.Y. TGF-β as A Master Regulator of Aging-Associated Tissue Fibrosis. Aging Dis. 2023, 14, 1633–1650. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Van Groen, T.; Kadish, I.; Li, Y.; Wang, D.; James, S.R.; Karpf, A.R.; Tollefsbol, T.O. Insufficient DNA methylation affects healthy aging and promotes age-related health problems. Clin. Epigenet. 2011, 2, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Soto-Palma, C.; Niedernhofer, L.J.; Faulk, C.D.; Dong, X. Epigenetics, DNA damage, and aging. Epigenetics, DNA damage, and aging. J. Clin. Investig. 2022, 132, e158446. [Google Scholar] [CrossRef] [PubMed]
- Bertucci-Richter, E.M.; Parrott, B.B. The rate of epigenetic drift scales with maximum lifespan across mammals. Nat. Commun. 2023, 14, 7731. [Google Scholar] [CrossRef]
- Singh, K.; Rustagi, Y.; Abouhashem, A.S.; Tabasum, S.; Verma, P.; Hernandez, E.; Pal, D.; Khona, D.K.; Mohanty, S.K.; Kumar, M.; et al. Genome-wide DNA hypermethylation opposes healing in patients with chronic wounds by impairing epithelial-mesenchymal transition. J. Clin. Investig. 2022, 132, e157279. [Google Scholar] [CrossRef]
- Ostanek, B.; Kranjc, T.; Lovšin, N.; Zupan, J.; Marc, J. Chapter 18—Epigenetic Mechanisms in Osteoporosis. In Epigenetics of Aging and Longevity; Moskalev, A., Vaiserman, A.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 365–388. [Google Scholar]
- Ghayor, C.; Weber, F.E. Epigenetic Regulation of Bone Remodeling and Its Impacts in Osteoporosis. Int. J. Mol. Sci. 2016, 17, 1446. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Lin, Z.-J.; Li, C.-C.; Lin, X.; Shan, S.-K.; Guo, B.; Zheng, M.-H.; Li, F.; Yuan, L.-Q.; Li, Z.-H. Epigenetic regulation in metabolic diseases: Mechanisms and advances in clinical study. Signal Transduct. Target. Ther. 2023, 8, 98. [Google Scholar] [CrossRef]
- Castellano-Castillo, D.A.-O.; Ramos-Molina, B.A.-O.; Cardona, F.A.-O.; Queipo-Ortuño, M.A.-O. Epigenetic regulation of white adipose tissue in the onset of obesity and metabolic diseases. Obes. Rev. 2020, 21, e13054. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, H.; Huang, F.; Chen, Y.; Ding, X.; Zhou, C.; Wu, Y.; Zhang, Q.; Ma, X.; Wang, J.; et al. The chromatin remodeling factor Arid1a cooperates with Jun/Fos to promote osteoclastogenesis by epigenetically upregulating Siglec15 expression. J. Bone Miner. Res. 2024, 39, 775–790. [Google Scholar] [CrossRef]
- Hossan, T.; Kundu, S.; Alam, S.S.; Nagarajan, S. Epigenetic Modifications Associated with the Pathogenesis of Type 2 Diabetes Mellitus. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 775–786. [Google Scholar] [CrossRef]
- Tu, P.A.-O.; Huang, B.; Li, M.; Zhang, Y.; Bao, S.; Tu, N.; Yang, Y.; Lu, J. Exendin-4 may improve type 2 diabetes by modulating the epigenetic modifications of pancreatic histone H3 in STZ-induced diabetic C57BL/6 J mice. J. Physiol. Biochem. 2022, 78, 51–59. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, W.; Guo, C.; Wu, J.; Zhang, S.; Shi, H.; Kwon, S.; Chen, J.; Dong, Z. Hypermethylation leads to the loss of HOXA5, resulting in JAG1 expression and NOTCH signaling contributing to kidney fibrosis. Kidney Int. 2024, 106, 98–114. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Carlos Acosta, J.; Adams, P.D.; d’Adda di Fagagna, F.; Baker, D.J.; Bishop, C.L.; Chandra, T.; Collado, M.; Gil, J.; Gorgoulis, V.; et al. Guidelines for minimal information on cellular senescence experimentation. Cell 2024, 187, 4150–4175. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Honda, S.; Ikeda, K.; Urata, R.; Yamazaki, E.; Emoto, N.; Matoba, S. Cellular senescence promotes endothelial activation through epigenetic alteration, and consequently accelerates atherosclerosis. Sci. Rep. 2021, 11, 14608. [Google Scholar] [CrossRef]
- McHugh, D.; Durán, I.; Gil, J. Senescence as a therapeutic target in cancer and age-related diseases. Nat. Rev. Drug Discov. 2024, 24, 57–71. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Chou, L.Y.; Ho, C.T.; Hung, S.C. Paracrine Senescence of Mesenchymal Stromal Cells Involves Inflammatory Cytokines and the NF-kappaB Pathway. Cells 2022, 11, 3324. [Google Scholar] [CrossRef]
- Herbstein, F.A.-O.; Sapochnik, M.; Attorresi, A.; Pollak, C.; Senin, S.; Gonilski-Pacin, D.A.-O.; Ciancio Del Giudice, N.; Fiz, M.; Elguero, B.A.-O.; Fuertes, M.A.-O.; et al. The SASP factor IL-6 sustains cell-autonomous senescent cells via a cGAS-STING-NFκB intracrine senescent noncanonical pathway. Aging Cell 2024, 23, e14258. [Google Scholar] [CrossRef]
- Shang, D.; Liu, H.; Tu, Z. Pro-inflammatory cytokines mediating senescence of vascular endothelial cells in atherosclerosis. Fundam. Clin. Pharmacol. 2023, 37, 928–936. [Google Scholar] [CrossRef]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Cai, B.; Fishkin, N.; Jang, Y.P.; Krane, S.; Vollmer, H.R.; Zhou, J.; Nakanishi, K.; Nakanishi, K. A2E, a fluorophore of RPE lipofuscin: Can it cause RPE degeneration? Adv. Exp. Med. Biol. 2003, 533, 205–211. [Google Scholar]
- Dhirachaikulpanich, D.; Lagger, C.; Chatsirisupachai, K.; de Magalhães, J.P.; Paraoan, L. Intercellular communication analysis of the human retinal pigment epithelial and choroidal cells predicts pathways associated with aging, cellular senescence and age-related macular degeneration. Front. Aging Neurosci. 2022, 14, 1016293. [Google Scholar] [CrossRef]
- Horváth, E.; Sólyom, Á.; Székely, J.; Nagy, E.A.-O. Inflammatory and Metabolic Signaling Interfaces of the Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis. Int. J. Mol. Sci. 2023, 24, 16468. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, W.J.; Hwang, S.C.; Choe, Y.; Kim, S.; Bok, E.; Lee, S.; Kim, S.J.; Kim, H.O.; Ock, S.A.; et al. Chronic inflammation-induced senescence impairs immunomodulatory properties of synovial fluid mesenchymal stem cells in rheumatoid arthritis. Stem Cell Res. Ther. 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, N.; Arnold, R.; Equey, A.; Gandhi, A.; Adams, P.D. The role of the dynamic epigenetic landscape in senescence: Orchestrating SASP expression. NPJ Aging 2024, 10, 48. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Poljsak, B.; Milisav, I.; Milisav, I. Achieving the balance between ROS and antioxidants: When to use the synthetic antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef]
- Jun, Y.A.-O.; Albarran, E.; Wilson, D.L.; Ding, J.; Kool, E.A.-O. Fluorescence Imaging of Mitochondrial DNA Base Excision Repair Reveals Dynamics of Oxidative Stress Responses. Angew. Chem. Int. Ed. Engl. 2022, 61, e202111829. [Google Scholar] [CrossRef]
- Hagen, T.M.; Yowe, D.L.; Bartholomew, J.C.; Wehr, C.M.; Do, K.L.; Park, J.Y.; Ames, B.N. Mitochondrial decay in hepatocytes from old rats: Membrane potential declines, heterogeneity and oxidants increase. Proc. Natl. Acad. Sci. USA 1997, 94, 3064–3069. [Google Scholar] [CrossRef]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between mitochondrial membrane potential and ROS formation. Methods Mol. Biol. 2012, 810, 183–205. [Google Scholar] [PubMed]
- Wei, Y.H.; Lee, H.C. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp. Biol. Med. 2002, 227, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Hagen, J.L.; Krause, D.J.; Baker, D.J.; Fu, M.H.; Tarnopolsky, M.A.; Hepple, R.T. Skeletal muscle aging in F344BN F1-hybrid rats: I. Mitochondrial dysfunction contributes to the age-associated reduction in VO2max. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Leija, R.; Arevalo, J.; Brooks, G.; Vazquez-Medina, J.P. Mitochondrial Peroxiredoxin 6 Declines with Aging in Parallel with Increases in Hydrogen Peroxide Generation. Physiology 2024, 39, 2250. [Google Scholar] [CrossRef]
- Cai, R.; Zhang, Y.; Simmering, J.E.; Schultz, J.L.; Li, Y.; Fernandez-Carasa, I.; Consiglio, A.; Raya, A.; Polgreen, P.M.; Narayanan, N.S.; et al. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J. Clin. Investig. 2019, 129, 4539–4549. [Google Scholar] [CrossRef]
- He, M.A.-O.; Zhang, X.; Ran, X.; Zhang, Y.; Nie, X.; Xiao, B.; Lei, L.; Zhai, S.; Zhu, J.; Zhang, J.; et al. Black Phosphorus Nanosheets Protect Neurons by Degrading Aggregative α-syn and Clearing ROS in Parkinson’s Disease. Adv. Mater. 2024, 36, e2404576. [Google Scholar] [CrossRef]
- Keeney, M.A.-O.; Rocha, E.A.-O.; Hoffman, E.A.-O.; Farmer, K.; Di Maio, R.A.-O.; Weir, J.; Wagner, W.G.; Hu, X.; Clark, C.L.; Castro, S.A.-O.; et al. LRRK2 regulates production of reactive oxygen species in cell and animal models of Parkinson’s disease. Sci. Transl. Med. 2024, 16, eadl3438. [Google Scholar] [CrossRef]
- Ruegsegger, G.N.; Creo, A.L.; Cortes, T.M.; Dasari, S.; Nair, K.S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Investig. 2018, 9, 3671–3681. [Google Scholar] [CrossRef]
- Wang, M.; Wang, X.C.; Zhang, Z.Y.; Mou, B.; Hu, R.M. Impaired mitochondrial oxidative phosphorylation in multiple insulin-sensitive tissues of humans with type 2 diabetes mellitus. J. Int. Med. Res. 2010, 38, 769–781. [Google Scholar]
- Zweck, E.; Scheiber, D.; Jelenik, T.; Horn, P.; Albermann, S.; Boeken, U.D.O.; Saeed, D.; Kelm, M.; Roden, M.; Westenfeld, R.; et al. Insulin Resistance and Type 2 Diabetes Mellitus Are Associated with Impaired Mitochondrial Function in Human Ventricular Myocardium. Diabetes 2018, 67, 284-OR. [Google Scholar] [CrossRef]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Turdi, S.; Fan, X.; Li, J.; Zhao, J.; Huff, A.F.; Du, M.; Ren, J. AMP-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell 2010, 9, 592–606. [Google Scholar] [CrossRef]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [PubMed]
- Pande, S.; Raisuddin, S. Molecular and cellular regulatory roles of sirtuin protein. Crit. Rev. Food Sci. Nutr. 2023, 63, 9895–9913. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, F.; He, Z.; Che, L.; Chen, X.; Yuan, Y.; Liu, C. Senescence-Targeted and NAD(+)-Dependent SIRT1-Activated Nanoplatform to Counteract Stem Cell Senescence for Promoting Aged Bone Regeneration. Small 2024, 20, e2304433. [Google Scholar] [CrossRef]
- Duan, J.L.; Ruan, B.; Song, P.; Fang, Z.Q.; Yue, Z.S.; Liu, J.J.; Dou, G.R.; Han, H.; Wang, L. Shear stress-induced cellular senescence blunts liver regeneration through Notch-sirtuin 1-P21/P16 axis. Hepatology 2022, 75, 584–599. [Google Scholar] [CrossRef]
- Roy, C.; Molin, L.; Alcolei, A.; Solyga, M.; Bonneau, B.; Vachon, C.; Bessereau, J.L.; Solari, F.A.-O. DAF-2/insulin IGF-1 receptor regulates motility during aging by integrating opposite signaling from muscle and neuronal tissues. Aging Cell 2022, 21, e13660. [Google Scholar] [CrossRef]
- Roitenberg, N.; Bejerano-Sagie, M.; Boocholez, H.; Moll, L.; Marques, F.C.; Golodetzki, L.; Nevo, Y.; Elami, T.; Cohen, E. Modulation of caveolae by insulin/IGF-1 signaling regulates aging of Caenorhabditis elegans. EMBO Rep. 2018, 19, e45673. [Google Scholar] [CrossRef]
- Anisimov, V.N.; Labunets, I.F.; Popovich, I.G.; Tyndyk, M.L.; Yurova, M.N.; Golubev, A.G. In mice transgenic for IGF1 under keratin-14 promoter, lifespan is decreased and the rates of aging and thymus involution are accelerated. Aging 2019, 11, 2098–2110. [Google Scholar] [CrossRef]
- Li, Q.; Wu, S.; Li, S.Y.; Lopez, F.L.; Du, M.; Kajstura, J.; Anversa, P.; Ren, J. Cardiac-specific overexpression of insulin-like growth factor 1 attenuates aging-associated cardiac diastolic contractile dysfunction and protein damage. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1398–H1403. [Google Scholar] [CrossRef]
- Xu, X.; Hueckstaedt, L.K.; Ren, J. Deficiency of insulin-like growth factor 1 attenuates aging-induced changes in hepatic function: Role of autophagy. J. Hepatol. 2013, 59, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Ashpole, N.M.; Logan, S.; Yabluchanskiy, A.; Mitschelen, M.C.; Yan, H.; Farley, J.A.; Hodges, E.L.; Ungvari, Z.; Csiszar, A.; Chen, S.; et al. IGF-1 has sexually dimorphic, pleiotropic, and time-dependent effects on healthspan, pathology, and lifespan. Geroscience 2017, 39, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Crosby, P.; Hamnett, R.; Putker, M.; Hoyle, N.P.; Reed, M.; Karam, C.J.; Maywood, E.S.; Stangherlin, A.; Chesham, J.E.; Hayter, E.A.; et al. Insulin/IGF-1 Drives PERIOD Synthesis to Entrain Circadian Rhythms with Feeding Time. Cell 2019, 177, 896–909.e820. [Google Scholar] [CrossRef]
- Selman, C.; Partridge, L.; Withers, D.J. Replication of Extended Lifespan Phenotype in Mice with Deletion of Insulin Receptor Substrate 1. PLoS ONE 2011, 6, e16144. [Google Scholar] [CrossRef]
- Gupta, A.; Dey, C.S. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Mol. Biol. Cell 2012, 23, 3882–3898. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.A.-O.; Qian, P.A.-O.; Huang, H.A.-O. Inflammation and aging: Signaling pathways and intervention therapies. Signal Transduct. Target. Ther. 2023, 8, 239. [Google Scholar] [CrossRef]
- Noh, S.G.; Kim, H.W.; Kim, S.; Chung, K.W.; Jung, Y.S.; Yoon, J.H.; Yu, B.P.; Lee, J.; Chung, H.Y. Senoinflammation as the underlying mechanism of aging and its modulation by calorie restriction. Ageing Res. Rev. 2024, 101, 102503. [Google Scholar] [CrossRef]
- Liu, Z.; Liang, Q.; Ren, Y.; Guo, C.; Ge, X.; Wang, L.; Cheng, Q.; Luo, P.; Zhang, Y.; Han, X. Immunosenescence: Molecular mechanisms and diseases. Signal Transduct. Target. Ther. 2023, 8, 200. [Google Scholar] [CrossRef]
- Gulen, M.A.-O.; Samson, N.; Keller, A.; Schwabenland, M.A.-O.; Liu, C.A.-O.; Glück, S.; Thacker, V.A.-O.X.; Favre, L.A.-O.; Mangeat, B.A.-O.; Kroese, L.J.; et al. cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.A.-O.; Gatch, A.J.; Sun, Y.A.-O.; Ding, F.A.-O. Differential Binding and Conformational Dynamics of Tau Microtubule-Binding Repeats with a Preformed Amyloid-β Fibril Seed. ACS Chem. Neurosci. 2023, 14, 1321–1330. [Google Scholar] [CrossRef]
- Gruel, R.; Bijnens, B.; Van Den Daele, J.; Thys, S.; Willems, R.; Wuyts, D.; Van Dam, D.; Verstraelen, P.; Verboven, R.; Roels, J.; et al. S100A8-enriched microglia populate the brain of tau-seeded and accelerated aging mice. Aging Cell 2024, 23, e14120. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Molina, A.; Lebrero-Fernández, C.; Sanz, A.; Calvo-Rubio, M.; Deleyto-Seldas, N.; de Prado-Rivas, L.; Plata-Gómez, A.B.; Fernández-Florido, E.; González-García, P.; Vivas-García, Y.; et al. A mild increase in nutrient signaling to mTORC1 in mice leads to parenchymal damage, myeloid inflammation and shortened lifespan. Nat. Aging 2024, 4, 1102–1120. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Wang, H.; Suttles, J.; Graves, D.T.; Martin, M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates Toll-like receptor 4-mediated inflammatory response via FoxO1. J. Biol. Chem. 2011, 286, 44295–44305. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.A.-O.; Wu, Z.; Stoka, V.; Meng, J.A.-O.; Hayashi, Y.; Peters, C.; Qing, H.; Turk, V.; Nakanishi, H.A.-O. Increased expression and altered subcellular distribution of cathepsin B in microglia induce cognitive impairment through oxidative stress and inflammatory response in mice. Aging Cell 2019, 18, e12856. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef]
- Crook, J.M.; Kravets, L.; Peura, T.; Firpo, M.T. Derivation of Human Embryonic Stem Cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar]
- Levenberg, S.; Zoldan, J.; Basevitch, Y.; Langer, R. Endothelial potential of human embryonic stem cells. Blood 2007, 110, 806–814. [Google Scholar] [CrossRef]
- Roubal, I.; Park, S.J.; Kim, Y. Derivation of Neural Precursor Cells from Human Embryonic Stem Cells for DNA Methylomic Analysis. Methods Mol. Biol. 2016, 1341, 345–357. [Google Scholar] [PubMed]
- Modrek, A.S.; Prado, J.; Bready, D.; Dhaliwal, J.; Golub, D.; Placantonakis, D.G. Modeling Glioma with Human Embryonic Stem Cell-Derived Neural Lineages. Methods Mol. Biol. 2018, 1741, 227–237. [Google Scholar]
- Modrek, A.S.; Prado, J.; Bready, D.; Dhaliwal, J.; Golub, D.; Placantonakis, D.G. Immunogenicity of human embryonic stem cells. Cell Tissue Res. 2008, 331, 67–78. [Google Scholar]
- Frederiksen, H.R.; Glantz, A.; Vøls, K.K.; Skov, S.; Tveden-Nyborg, P.; Freude, K.; Doehn, U. CRISPR-Cas9 immune-evasive hESCs are rejected following transplantation into immunocompetent mice. Front. Genome. Ed. 2024, 6, 1403395. [Google Scholar] [CrossRef]
- Yang, X.; Wang, R.; Wang, X.; Cai, G.; Qian, Y.; Feng, S.; Tan, F.; Chen, K.; Tang, K.; Huang, X.; et al. TGFbeta signaling hyperactivation-induced tumorigenicity during the derivation of neural progenitors from mouse ESCs. J. Mol. Cell. Biol. 2018, 10, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Stojkovic, P.; Przyborski, S.; Cooke, M.; Armstrong, L.; Lako, M.; Stojkovic, M. Derivation of human embryonic stem cells from developing and arrested embryos. J. Hepatol. 2013, 59, 308–317. [Google Scholar] [CrossRef]
- Cerneckis, J.; Cai, H.; Shi, Y. Induced pluripotent stem cells (iPSCs): Molecular mechanisms of induction and applications. Signal Transduct. Target. Ther. 2024, 9, 112. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Ruiz, G.A.-O.; Valencia-González, H.A.-O.X.; Pérez-Montiel, D.; Muñoz, F.; Ocadiz-Delgado, R.A.-O.X.; Fernández-Retana, J.; Pérez-Plasencia, C.A.-O.; Reséndis-Antonio, O.A.-O.X.; Gariglio, P.A.-O.; García-Carrancá, A.A.-O. Genes Involved in the Transcriptional Regulation of Pluripotency Are Expressed in Malignant Tumors of the Uterine Cervix and Can Induce Tumorigenic Capacity in a Nontumorigenic Cell Line. Stem Cells Int. 2019, 2019, 7683817. [Google Scholar] [CrossRef]
- Fatma, H.; Siddique, H.R. Pluripotency inducing Yamanaka factors: Role in stemness and chemoresistance of liver cancer. Expert. Rev. Anticancer. Ther. 2021, 21, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.A.-O.; Hong, S.; Zhang, X. Expression and Functional Analysis of core stemness factors OSKM (OCT4, SOX2, KLF4, and MYC) in Pan-cancer. Medicine 2023, 102, e36433. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Lin, Y.C.; Tsai, M.H.; Lin, C.S.; Murayama, Y.; Sato, R.; Yokoyama, K.K. Emerging roles of hypoxia-inducible factors and reactive oxygen species in cancer and pluripotent stem cells. Kaohsiung J. Med. Sci. 2015, 31, 279–286. [Google Scholar] [CrossRef]
- Kisby, T.; de Lázaro, I.; Fisch, S.; Cartwright, E.J.; Cossu, G.; Kostarelos, K. Adenoviral Mediated Delivery of OSKM Factors Induces Partial Reprogramming of Mouse Cardiac Cells In Vivo. Adv. Ther. 2021, 4, 2000141. [Google Scholar] [CrossRef]
- Mao, Y.; Wang, L.; Zhong, B.; Yang, N.; Li, Z.; Cui, T.; Feng, G.; Li, W.A.-O.X.; Zhang, Y.A.-O.; Zhou, Q.A.-O. Continuous expression of reprogramming factors induces and maintains mouse pluripotency without specific growth factors and signaling inhibitors. Cell Prolif. 2021, 54, e13090. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yin, X.; Qin, H.; Zhu, F.; Liu, H.; Yang, W.; Zhang, Q.; Xiang, C.; Hou, P.; Song, Z.; et al. Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell 2008, 3, 475–479. [Google Scholar] [CrossRef]
- Chiew, M.Y.; Wang, E.; Lan, K.C.; Lin, Y.R.; Hsueh, Y.H.; Tu, Y.K.; Liu, S.-F.; Chen, P.-C.; Lu, H.-E.; Chen, W.L. Improving iPSC Differentiation Using a Nanodot Platform. ACS Appl. Mater. Interfaces 2024, 16, 36030–36046. [Google Scholar] [CrossRef]
- Sajedeh Nasr, E.; Agnes, M.R.I.; Xufeng, X.; Samuel Byung-Deuk, L.; Yue, S.; Jianping, F. Micro/nanoengineered technologies for human pluripotent stem cells maintenance and differentiation. Nano Today 2021, 41, 101310. [Google Scholar]
- Dobner, J.; Diecke, S.; Krutmann, J.; Prigione, A.; Rossi, A. Reassessment of marker genes in human induced pluripotent stem cells for enhanced quality control. Nat. Commun. 2024, 15, 8547. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, H.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef]
- Bouma, M.J.; van Iterson, M.; Janssen, B.; Mummery, C.L.; Salvatori, D.C.F.; Freund, C. Differentiation-Defective Human Induced Pluripotent Stem Cells Reveal Strengths and Limitations of the Teratoma Assay and In Vitro Pluripotency Assays. Stem Cell Rep. 2017, 8, 1340–1353. [Google Scholar] [CrossRef]
- Ping, W.; Hu, J.; Hu, G.; Song, Y.; Xia, Q.; Yao, M.; Gong, S.; Jiang, C.; Yao, H. Genome-wide DNA methylation analysis reveals that mouse chemical iPSCs have closer epigenetic features to mESCs than OSKM-integrated iPSCs. Cell Death Dis. 2018, 9, 187. [Google Scholar] [CrossRef]
- Byeongtaek, O.; Wu, Y.-W.; Vishal, S.; Vivek, L.; Jun, D.; Paul, M.G. Modulating the Electrical and Mechanical Microenvironment to Guide Neuronal Stem Cell Differentiation. Adv. Sci. 2021, 8, 2002112. [Google Scholar]
- Tavernier, G.; Wolfrum, K.; Demeester, J.; De Smedt, S.C.; Adjaye, J.; Rejman, J. Activation of pluripotency-associated genes in mouse embryonic fibroblasts by non-viral transfection with in vitro-derived mRNAs encoding Oct4, Sox2, Klf4 and cMyc. Biomaterials 2012, 2, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Manzini, S.; Viiri, L.E.; Marttila, S.; Aalto-Setälä, K. A Comparative View on Easy to Deploy non-Integrating Methods for Patient-Specific iPSC Production. Stem Cell Rev. Rep. 2015, 11, 900–908. [Google Scholar] [CrossRef]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. A biomaterial approach to cell reprogramming and differentiation. J. Mater. Chem. B 2017, 5, 2375–2379. [Google Scholar]
- Miroshnikova, Y.A.; Jorgens, D.M.; Spirio, L.; Auer, M.; Sarang-Sieminski, A.L.; Weaver, V.M. Engineering strategies to recapitulate epithelial morphogenesis within synthetic three-dimensional extracellular matrix with tunable mechanical properties. Phys. Biol. 2011, 8, 026013. [Google Scholar] [CrossRef]
- Chowdhury, M.M.; Zimmerman, S.; Leeson, H.; Nefzger, C.M.; Mar, J.C.; Laslett, A.; Polo, J.M.; Wolvetang, E.; Cooper-White, J.J. Superior Induced Pluripotent Stem Cell Generation through Phactr3-Driven Mechanomodulation of Both Early and Late Phases of Cell Reprogramming. Biomater. Res. 2024, 28, 0025. [Google Scholar] [CrossRef]
- Chang, D.; Sun, C.; Tian, X.; Liu, H.; Jia, Y.; Guo, Z. Regulation of cardiac fibroblasts reprogramming into cardiomyocyte-like cells with a cocktail of small molecule compounds. FEBS. Open Bio 2024, 14, 983–1000. [Google Scholar] [CrossRef]
- Li, S.; Ding, C.; Guo, Y.; Zhang, Y.; Wang, H.; Sun, X.; Zhang, J.; Cui, Z.; Chen, J. Mechanotransduction Regulates Reprogramming Enhancement in Adherent 3D Keratocyte Cultures. Front. Bioeng. Biotechnol. 2021, 9, 709488. [Google Scholar] [CrossRef]
- Alvarez-Palomo, A.B.; Requena-Osete, J.; Delgado-Morales, R.; Moreno-Manzano, V.; Grau-Bove, C.; Tejera, A.M.; Otero, M.J.; Barrot, C.; Santos-Barriopedro, I.; Vaquero, A.; et al. A synthetic mRNA cell reprogramming method using CYCLIN D1 promotes DNA repair, generating improved genetically stable human induced pluripotent stem cells. Stem Cells 2021, 39, 866–881. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wen, C.; Davis, B.; Shi, P.; Abune, L.; Lee, K.; Dong, C.; Wang, Y. Synthetic DNA for Cell Surface Engineering: Experimental Comparison between Click Conjugation and Lipid Insertion in Terms of Cell Viability, Engineering Efficiency, and Displaying Stability. ACS Appl. Mater. Interfaces 2022, 14, 3900–3909. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Harcum, S.W.; Pei, J.; Xie, W. Stochastic biological system-of-systems modelling for iPSC culture. Commun. Biol. 2024, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.A.-O.X.; Rafiei Hashtchin, A.; Manstein, F.; Carvalho Oliveira, M.; Kempf, H.A.-O.; Zweigerdt, R.A.-O.; Lachmann, N.A.-O. Continuous human iPSC-macrophage mass production by suspension culture in stirred tank bioreactors. Nat. Protoc. 2022, 17, 513–539. [Google Scholar] [CrossRef]
- Zhang, T.; Qian, C.A.-O.; Song, M.; Tang, Y.; Zhou, Y.; Dong, G.; Shen, Q.; Chen, W.A.-O.; Wang, A.A.-O.; Shen, S.; et al. Application Prospect of Induced Pluripotent Stem Cells in Organoids and Cell Therapy. Int. J. Mol. Sci. 2024, 25, 2680. [Google Scholar] [CrossRef]
- Durczak, P.M.; Fair, K.L.; Jinks, N.; Cuevas Ocaña, S.; Sainz Zuñiga, C.B.; Hannan, N.R.F. Generation of hiPSC-Derived Intestinal Organoids for Developmental and Disease Modelling Applications. J. Vis. Exp. 2024, 10, 61199. [Google Scholar] [CrossRef]
- Puri, D.; Wagner, W. Epigenetic rejuvenation by partial reprogramming. Bioessays 2023, 45, e2200208. [Google Scholar] [CrossRef]
- Elisabeth Tamara, S.; Aalto-Setälä, K.; Kiamehr, M.; Landmesser, U.; Kränkel, N. Age Is Relative—Impact of Donor Age on Induced Pluripotent Stem Cell-Derived Cell Functionality. Front. Cardiovasc. Med. 2018, 5, 4. [Google Scholar]
- Petrini, S.; Borghi, R.; D’Oria, V.; Restaldi, F.; Moreno, S.; Novelli, A.; Bertini, E.; Compagnucci, C. Aged induced pluripotent stem cell (iPSCs) as a new cellular model for studying premature aging. Aging 2017, 5, 1453–1469. [Google Scholar] [CrossRef]
- Shimamoto, A.; Kagawa, H.; Zensho, K.; Sera, Y.; Kazuki, Y.; Osaki, M.; Oshimura, M.; Ishigaki, Y.; Hamasaki, K.; Kodama, Y.; et al. Reprogramming suppresses premature senescence phenotypes of Werner syndrome cells and maintains chromosomal stability over long-term culture. PLoS ONE 2014, 9, e112900. [Google Scholar] [CrossRef] [PubMed]
- Lessel, D.; Kubisch, C. Hereditary Syndromes with Signs of Premature Aging. Dtsch. Arztebl. Int. 2019, 116, 489–496. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Ikram, S.; Bibi, N.; Mir, A.A.-O. Hutchinson-Gilford Progeria Syndrome: A Premature Aging Disease. Mol. Neurobiol. 2018, 55, 4417–4427. [Google Scholar] [CrossRef]
- Zayoud, K.; Chikhaoui, A.; Kraoua, I.; Tebourbi, A.; Najjar, D.; Ayari, S.; Safra, I.; Kraiem, I.; Turki, I.; Menif, S.; et al. Immunity in the Progeroid Model of Cockayne Syndrome: Biomarkers of Pathological Aging. Cells 2024, 13, 402. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Stewart, C.L. Accelerated aging syndromes, are they relevant to normal human aging? Aging 2011, 9, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Matrone, G.A.-O.; Thandavarayan, R.A.; Walther, B.K.; Meng, S.A.-O.; Mojiri, A.; Cooke, J.A.-O. Dysfunction of iPSC-derived endothelial cells in human Hutchinson-Gilford progeria syndrome. Cell Cycle 2019, 18, 2495–2508. [Google Scholar] [CrossRef]
- Monteiro da Rocha, A.; Davis, J.; Hoopingarner, E.; Creech, J.; Campbell, K.; Guerero-Serna, G.; Herron, T.J.; Jalife, J. Abstract 16704: Epigenetic and Morphofunctional Changes in Human Induced Pluripotent Stem Cell-derived Cardiomyocytes Carrying a Mutation Causative of Premature Aging. Circulation 2017, 136, A16704. [Google Scholar]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef]
- Blondel, S.; Egesipe, A.L.; Picardi, P.; Jaskowiak, A.L.; Notarnicola, M.; Ragot, J.; Tournois, J.; Le Corf, A.; Brinon, B.; Poydenot, P.; et al. Drug screening on Hutchinson Gilford progeria pluripotent stem cells reveals aminopyrimidines as new modulators of farnesylation. Cell Death Discov. 2016, 7, e2105. [Google Scholar] [CrossRef]
- Kubben, N.; Brimacombe, K.R.; Donegan, M.; Li, Z.; Misteli, T. A high-content imaging-based screening pipeline for the systematic identification of anti-progeroid compounds. Methods 2016, 96, 46–58. [Google Scholar] [CrossRef]
- Lo Cicero, A.; Jaskowiak, A.L.; Egesipe, A.L.; Tournois, J.; Brinon, B.; Pitrez, P.R.; Ferreira, L.; de Sandre-Giovannoli, A.; Levy, N.; Nissan, X. A High Throughput Phenotypic Screening reveals compounds that counteract premature osteogenic differentiation of HGPS iPS-derived mesenchymal stem cells. Sci. Rep. 2016, 6, 34798. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Z.; Ye, Y.; Li, B.; Liu, T.; Zhang, W.; Liu, G.A.-O.; Zhang, Y.A.; Qu, J.; Xu, D.; et al. Ectopic hTERT expression facilitates reprograming of fibroblasts derived from patients with Werner syndrome as a WS cellular model. Cell Death Dis. 2018, 9, 923. [Google Scholar] [CrossRef]
- Ballew, B.J.; Savage, S.A. Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert. Rev. Hematol. 2013, 6, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, M.; Dokal, I. Dyskeratosis congenita, stem cells and telomeres. Biochim. Biophys. Acta. 2009, 1792, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Paiva, R.M.; Calado, R.T. Telomere dysfunction and hematologic disorders. Prog. Mol. Biol. Transl. Sci. 2014, 125, 133–157. [Google Scholar] [PubMed]
- Babushok, D.V.; Grignon, A.L.; Li, Y.; Atienza, J.; Xie, H.M.; Lam, H.S.; Hartung, H.; Bessler, M.; Olson, T.S. Disrupted lymphocyte homeostasis in hepatitis-associated acquired aplastic anemia is associated with short telomeres. Am. J. Hematol. 2016, 91, 243–247. [Google Scholar] [CrossRef]
- Ahmad, T.; Sundar, I.K.; Tormos, A.M.; Lerner, C.A.; Gerloff, J.; Yao, H.; Rahman, I. Shelterin Telomere Protection Protein 1 Reduction Causes Telomere Attrition and Cellular Senescence via Sirtuin 1 Deacetylase in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2017, 56, 38–49. [Google Scholar] [CrossRef]
- Jothi, D.; Kulka, L.A.M. Strategies for modeling aging and age-related diseases. NPJ Aging 2024, 10, 32. [Google Scholar] [CrossRef]
- Choi, Y.J.; Kim, M.S.; Rhoades, J.H.; Johnson, N.M.; Berry, C.T.; Root, S.; Chen, Q.; Tian, Y.; Fernandez, R.J., 3rd; Cramer, Z.; et al. Patient-Induced Pluripotent Stem Cell-Derived Hepatostellate Organoids Establish a Basis for Liver Pathologies in Telomeropathies. Cell Mol. Gastroenterol. Hepatol. 2023, 16, 451–472. [Google Scholar] [CrossRef]
- Oudrhiri, N.; M’Kacher, R.; Chaker, D.; Colicchio, B.; Borie, C.; Jeandidier, E.; Dieterlen, A.; Griscelli, F.; Bennaceur-Griscelli, A.; Turhan, A.G. Patient-Derived iPSCs Reveal Evidence of Telomere Instability and DNA Repair Deficiency in Coats Plus Syndrome. Genes 2022, 13, 1395. [Google Scholar] [CrossRef] [PubMed]
- Walne, A.J.; Dokal, I. Advances in the understanding of dyskeratosis congenita. Br. J. Haematol. 2009, 145, 164–172. [Google Scholar] [CrossRef]
- Fernandez, R.A.-O.; Gardner, Z.J.G.; Slovik, K.J.; Liberti, D.A.-O.; Estep, K.N.; Yang, W.; Chen, Q.; Santini, G.T.; Perez, J.V.; Root, S.; et al. GSK3 inhibition rescues growth and telomere dysfunction in dyskeratosis congenita iPSC-derived type II alveolar epithelial cells. Elife 2022, 11, e64430. [Google Scholar] [CrossRef]
- Polychronopoulou, S.; Koutroumba, P. Telomere length variation and telomerase activity expression in patients with congenital and acquired aplastic anemia. Acta. Haematol. 2004, 111, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.E.; Gibson, F.M.; Rizzo, S.; Tooze, J.A.; Marsh, J.C.; Gordon-Smith, E.C. Progressive telomere shortening in aplastic anemia. Blood 1998, 91, 3582–3592. [Google Scholar] [CrossRef] [PubMed]
- Melguizo-Sanchis, D.; Xu, Y.; Taheem, D.; Yu, M.; Tilgner, K.; Barta, T.; Gassner, K.; Anyfantis, G.; Wan, T.; Elango, R.; et al. iPSC modeling of severe aplastic anemia reveals impaired differentiation and telomere shortening in blood progenitors. Cell Death Dis. 2018, 9, 128. [Google Scholar] [CrossRef]
- Liu, G.-H.; Suzuki, K.; Li, M.; Qu, J.; Montserrat, N.; Tarantino, C.; Gu, Y.; Yi, F.; Xu, X.; Zhang, W.; et al. Modelling Fanconi anemia pathogenesis and therapeutics using integration-free patient-derived iPSCs. Nat. Commun. 2014, 5, 4330. [Google Scholar] [CrossRef]
- Mulet, A.A.-O.; Signes-Costa, J. Idiopathic Pulmonary Fibrosis and Telomeres. J. Clin. Med. 2022, 11, 6893. [Google Scholar] [CrossRef]
- Ptasinski, V.; Monkley, S.J.; Ost, K.; Tammia, M.; Alsafadi, H.N.; Overed-Sayer, C.; Hazon, P.; Wagner, D.E.; Murray, L.A. Modeling fibrotic alveolar transitional cells with pluripotent stem cell-derived alveolar organoids. Life Sci. Alliance 2023, 6, e202201853. [Google Scholar] [CrossRef]
- Blasiak, J.; Szczepanska, J.; Fila, M.; Pawlowska, E.; Kaarniranta, K. Potential of Telomerase in Age-Related Macular Degeneration-Involvement of Senescence, DNA Damage Response and Autophagy and a Key Role of PGC-1alpha. Int. J. Mol. Sci. 2021, 22, 7194. [Google Scholar] [CrossRef]
- Brümmendorf, T.H.; Pällmann, N.; Preukschas, M.; Steinemann, D.; Hofmann, W.; Gompf, A.; Rudolph, K.L.; Bokemeyer, C.; Koschmieder, S.; Schuppert, A.; et al. BCR-ABL Cooperates With a “Telomere-Associated Secretory Phenotype” (TASP) To Facilitate Malignant Proliferation Of Hematopoietic Stem Cells. Blood 2013, 122, 3976. [Google Scholar] [CrossRef]
- Elitt, M.S.; Barbar, L.; Tesar, P.J. Drug screening for human genetic diseases using iPSC models. Hum. Mol. Genet. 2018, 27, R89–R98. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Ralvenius, W.T.; Tsai, L.-H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Barak, M.; Fedorova, V.; Pospisilova, V.; Raska, J.; Vochyanova, S.; Sedmik, J.; Hribkova, H.; Klimova, H.; Vanova, T.; Bohaciakova, D. Human iPSC-Derived Neural Models for Studying Alzheimer’s Disease: From Neural Stem Cells to Cerebral Organoids. Stem Cell Rev. Rep. 2022, 18, 792–820. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Alatza, A.; Leckey, C.A.; Paterson, R.W.; Zetterberg, H.; Wray, S. Mass spectrometry analysis of tau and amyloid-beta in iPSC-derived models of Alzheimer’s disease and dementia. J. Neurochem. 2021, 159, 305–317. [Google Scholar] [CrossRef]
- Chang, K.H.; Lee-Chen, G.J.; Huang, C.C.; Lin, J.L.; Chen, Y.J.; Wei, P.C.; Lo, Y.S.; Yao, C.F.; Kuo, M.W.; Chen, C.M. Modeling Alzheimer’s Disease by Induced Pluripotent Stem Cells Carrying APP D678H Mutation. Mol. Neurobiol. 2019, 56, 3972–3983. [Google Scholar] [CrossRef]
- Bassil, R.; Shields, K.A.-O.; Granger, K.A.-O.; Zein, I.; Ng, S.A.-O.; Chih, B.A.-O. Improved modeling of human AD with an automated culturing platform for iPSC neurons, astrocytes and microglia. Nat. Commun. 2021, 12, 5220. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e1147. [Google Scholar] [CrossRef]
- Martin, S.A.-O.; Poppe, D.; Olova, N.A.-O.; O’Leary, C.; Ivanova, E.; Pflueger, J.; Dechka, J.; Simmons, R.K.; Cooper, H.M.; Reik, W.; et al. Embryonic Stem Cell-Derived Neurons as a Model System for Epigenome Maturation during Development. Genes 2023, 14, 957. [Google Scholar] [CrossRef]
- Sahlgren Bendtsen, K.A.-O.; Hall, V.A.-O. The Breakthroughs and Caveats of Using Human Pluripotent Stem Cells in Modeling Alzheimer’s Disease. Cells 2023, 12, 420. [Google Scholar] [CrossRef]
- Shimada, H.; Sato, Y.; Sasaki, T.; Shimozawa, A.; Imaizumi, K.; Shindo, T.; Miyao, S.; Kiyama, K.; Kondo, T.; Shibata, S.; et al. A next-generation iPSC-derived forebrain organoid model of tauopathy with tau fibrils by AAV-mediated gene transfer. Cell Rep. Methods 2022, 2, 100289. [Google Scholar] [CrossRef] [PubMed]
- Lomoio, S.A.-O.X.; Pandey, R.A.-O.; Rouleau, N.; Menicacci, B.; Kim, W.; Cantley, W.A.-O.; Haydon, P.A.-O.; Bennett, D.A.; Young-Pearse, T.A.-O.; Carter, G.A.-O.; et al. 3D bioengineered neural tissue generated from patient-derived iPSCs mimics time-dependent phenotypes and transcriptional features of Alzheimer’s disease. Mol. Psychiatry 2023, 12, 5390–5401. [Google Scholar] [CrossRef] [PubMed]
- Nieweg, K.; Andreyeva, A.; van Stegen, B.; Tanriöver, G.; Gottmann, K. Alzheimer’s disease-related amyloid-β induces synaptotoxicity in human iPS cell-derived neurons. Cell Death Dis. 2015, 6, e1709. [Google Scholar] [CrossRef]
- Silva, M.C.; Cheng, C.; Mair, W.; Almeida, S.; Fong, H.; Biswas, M.H.U.; Zhang, Z.; Huang, Y.; Temple, S.; Coppola, G.; et al. Human iPSC-Derived Neuronal Model of Tau-A152T Frontotemporal Dementia Reveals Tau-Mediated Mechanisms of Neuronal Vulnerability. Stem Cell Rep. 2016, 7, 325–340. [Google Scholar] [CrossRef]
- Sullivan, M.A.; Lane, S.D.; McKenzie, A.D.J.; Ball, S.R.; Sunde, M.; Neely, G.G.; Moreno, C.L.; Maximova, A.; Werry, E.L.; Kassiou, M. iPSC-derived PSEN2 (N141I) astrocytes and microglia exhibit a primed inflammatory phenotype. J. Neuroinflammation 2024, 21, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kulas, J.A.; Wang, C.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118. [Google Scholar] [CrossRef]
- Lee, S.E.; Kwon, D.; Shin, N.; Kong, D.; Kim, N.G.; Kim, H.Y.; Kim, M.J.; Choi, S.W.; Kang, K.S. Accumulation of APP-CTF induces mitophagy dysfunction in the iNSCs model of Alzheimer’s disease. Cell Death Discov. 2022, 8, 1. [Google Scholar] [CrossRef]
- Vuidel, A.; Cousin, L.; Weykopf, B.; Haupt, S.; Hanifehlou, Z.; Wiest-Daesslé, N.; Segschneider, M.; Lee, J.; Kwon, Y.J.; Peitz, M.; et al. High-content phenotyping of Parkinson’s disease patient stem cell-derived midbrain dopaminergic neurons using machine learning classification. Stem Cell Rep. 2022, 17, 2349–2364. [Google Scholar] [CrossRef]
- Kim, M.S.; Ra, E.A.; Kweon, S.H.; Seo, B.A.; Ko, H.S.; Oh, Y.; Lee, G. Advanced human iPSC-based preclinical model for Parkinson’s disease with optogenetic alpha-synuclein aggregation. Cell Stem Cell 2023, 7, 973–986. [Google Scholar] [CrossRef]
- Bayati, A.; Ayoubi, R.; Aguila, A.; Zorca, C.; Deyab, G.; Han, C.; Recinto, S.; Nguyen-Renou, E.; Rocha, C.; Maussion, G.; et al. Modeling Parkinson’s disease pathology in human dopaminergic neurons by sequential exposure to α-synuclein fibrils and proinflammatory cytokines. Nat. Neurosci. 2024, 27, 2401–2416. [Google Scholar] [CrossRef]
- Gustavsson, N.; Savchenko, E.; Klementieva, O.; Roybon, L. The intracellular milieu of Parkinson’s disease patient brain cells modulates alpha-synuclein protein aggregation. Acta. Neuropathol. Commun. 2021, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Chedid, J.; Li, Y.; Labrador-Garrido, A.; Abu-Bonsrah, D.; Pavan, C.; Fraser, T.; Ovchinnikov, D.; Zhong, M.; Davis, R.; Strbenac, D.; et al. Dopamine and cortical iPSC-derived neurons with different Parkinsonian mutations show variation in lysosomal and mitochondrial dysfunction: Implications for protein deposition versus selective cell loss. bioRxiv 2024, 617117. [Google Scholar] [CrossRef]
- Gerasimova, T.A.-O.; Poberezhniy, D.A.-O.; Nenasheva, V.A.-O.; Stepanenko, E.A.-O.X.; Arsenyeva, E.; Novosadova, L.; Grivennikov, I.; Illarioshkin, S.A.-O.; Lagarkova, M.A.-O.; Tarantul, V.; et al. Inflammatory Intracellular Signaling in Neurons Is Influenced by Glial Soluble Factors in iPSC-Based Cell Model of PARK2-Associated Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 9621. [Google Scholar] [CrossRef]
- Zhang, N.; Montoro, D.; Ellerby, L.M. Characterization of Human Huntington’s Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr. 2010, 2, RRN1193. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef]
- Cohen-Carmon, D.; Sorek, M.; Lerner, V.; Divya, M.A.-O.; Nissim-Rafinia, M.; Yarom, Y.; Meshorer, E.A.-O.X. Progerin-Induced Transcriptional Changes in Huntington’s Disease Human Pluripotent Stem Cell-Derived Neurons. Mol. Neurobiol. 2020, 57, 1768–1777. [Google Scholar] [CrossRef]
- Cho, I.A.-O.; Yang, B.; Forest, C.; Qian, L.; Chan, A.W.S. Amelioration of Huntington’s disease phenotype in astrocytes derived from iPSC-derived neural progenitor cells of Huntington’s disease monkeys. PLoS ONE 2019, 14, e0214156. [Google Scholar] [CrossRef]
- Wu, G.H.; Smith-Geater, C.A.-O.; Galaz-Montoya, J.G.; Gu, Y.A.-O.; Gupte, S.R.; Aviner, R.; Mitchell, P.A.-O.; Hsu, J.A.-O.; Miramontes, R.; Wang, K.A.-O.; et al. CryoET reveals organelle phenotypes in huntington disease patient iPSC-derived and mouse primary neurons. Nat. Commun. 2023, 14, 692. [Google Scholar] [CrossRef]
- Lépine, S.; Nauleau-Javaudin, A.; Deneault, E.; Chen, C.X.; Abdian, N.; Franco-Flores, A.K.; Haghi, G.; Castellanos-Montiel, M.J.; Maussion, G.; Chaineau, M.; et al. Homozygous ALS-linked mutations in TARDBP/TDP-43 lead to hypoactivity and synaptic abnormalities in human iPSC-derived motor neurons. iScience 2024, 27, 109166. [Google Scholar] [CrossRef]
- Ma, G.-m.; Xia, C.-c.; Lyu, B.-y.; Liu, J.; Luo, F.; Guan, M.-f.; Wang, J.-y.; Sun, L.; Zhang, L.; Chen, Y.; et al. Integrated temporal profiling of iPSCs-derived motor neurons from ALS patients carrying C9orf72, FUS, TARDBP, and SOD1 mutations. bioRxiv 2024, 618602. [Google Scholar] [CrossRef]
- Kaus, A.; Sareen, D. ALS Patient Stem Cells for Unveiling Disease Signatures of Motoneuron Susceptibility: Perspectives on the Deadly Mitochondria, ER Stress and Calcium Triad. Front. Cell. Neurosci. 2015, 9, 448. [Google Scholar] [CrossRef]
- Ritsma, L.; Bsibsi, M.; Popalzij, A.; Sancerni, S.C.; de Kraa, E.; Magnani, D.; Turner, A.; Oosterveen, T.; Fisher, D.F.; Iovino, M.; et al. TDP-43 dysregulation and STMN-2 mis-splicing upon proteasomal inhibition in potential iPSC-derived neuronal ALS model. Alzheimer’s Dement. 2023, 19, e076730. [Google Scholar] [CrossRef]
- Li, J.; Lim, R.G.; Kaye, J.A.; Dardov, V.; Coyne, A.N.; Wu, J.; Milani, P.; Cheng, A.; Thompson, T.G.; Ornelas, L.; et al. An integrated multi-omic analysis of iPSC-derived motor neurons from C9ORF72 ALS patients. iScience 2021, 24, 103221. [Google Scholar] [CrossRef]
- Dennis, K. SBT-272 Improves TDP-43 Pathology in ALS Upper Motor Neurons by Modulating Mitochondrial Integrity, Motility, and Function. Neurobiol. Dis. 2023, 178, 106022. [Google Scholar]
- Nikel, L.M.; Talbot, K.; Vahsen, B.F. Recent insights from human induced pluripotent stem cell models into the role of microglia in amyotrophic lateral sclerosis. Bioessays 2024, 46, e2400054. [Google Scholar] [CrossRef]
- Owen, G.J. Cell-autonomous Immune Dysfunction Driven by Disrupted Autophagy in C9orf72 -ALS Ipsc-Derived Microglia Contributes to Neurodegeneration. Sci. Adv. 2023, 9, eabq0651. [Google Scholar]
- Ruiyun, G.; Yimeng, C.; Jinyu, Z.; Zijing, Z.; Baofeng, F.; Xiaofeng, D.; Xin, L.; Jun, M.; Huixian, C. Neural Differentiation and spinal cord organoid generation from induced pluripotent stem cells (iPSCs) for ALS modelling and inflammatory screening. Mol. Neurobiol. 2024, 61, 4732–4749. [Google Scholar]
- Ruchi, S.; Aman, G.; Malika, N.; Davide, O.; Barbosa-Sabanero, K.; Zoya, Q.; Devika, B.; Roba, D.; Genqing, L.; Qin, W.; et al. Epithelial phenotype restoring drugs suppress macular degeneration phenotypes in an iPSC model. Nat. Commun. 2021, 12, 7293. [Google Scholar]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Mara, C.E.; Zhaohui, G.; Rebecca, J.K.; Heidi, R.; Sandra, R.M.; James, R.D.; Deborah, A.F. Impaired Mitochondrial Function in iPSC-Retinal Pigment Epithelium with the Complement Factor H Polymorphism for Age-Related Macular Degeneration. Cells 2021, 10, 789. [Google Scholar]
- Sujoy, B.; Jinggang, Y.; Weihong, H.; Edward, C. Modeling of mitochondrial bioenergetics and autophagy impairment in MELAS-mutant iPSC-derived retinal pigment epithelial cells. Stem Cell Res. Ther. 2022, 13, 260. [Google Scholar]
- Eunju, K.; Xinjian, W.; Rebecca, T.-H.; Hong, M.; Clifford, D.L.F.; Nuria Marti, G.; Yeonmi, L.; Crystal Van, D.; Riffat, A.; Ying, L.; et al. Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human Ipscs. Cell Stem Cell 2016, 18, 625–636. [Google Scholar]
- Maria, S.; Cristina, C.; Percy, Y.; Timour, B.; Samuel, B.; Cheng, Z.; Christian, A.R.; Lam, D.; Zhong, L.; Simona, G.; et al. ZSCAN10 expression corrects the genomic instability of iPSCs from aged donors. Nat. Cell Biol. 2017, 19, 1037–1048. [Google Scholar]
- Liping, S.; Xiaocen, K.; Sze Jie, L.; Yu, G.; Jean-Paul, K.; Xiaofei, S.; Jianhua, M.; Lei, Y. Diabetic Endothelial Cells Differentiated From Patient iPSCs Show Dysregulated Glycine Homeostasis and Senescence Associated Phenotypes. Front. Cell Dev. Biol. 2021, 9, 667252. [Google Scholar]
- Tingting, T.; Pengwei, D.; Yaqing, W.; Xu, Z.; Yaqiong, G.; Wenwen, C.; Jianhua, Q. Microengineered Multi-Organoid System from hiPSCs to Recapitulate Human Liver-Islet Axis in Normal and Type 2 Diabetes. Adv. Sci. 2022, 9, e2103495. [Google Scholar]
- Yuichi, H.; Anna, W.; Xiaowen, B.; Yasheng, Y.; Wai-Meng, K.; Zeliko, B. Abstract 439: Modeling Diabetic Cardiomyopathy Model Using Human Induced Pluripotent Stem Cell-derived Cardiomyocytes. Circ. Res. 2016, 119, A439. [Google Scholar]
- Ryan, D.C.; Ujang, P.; Marcos, C.-G.; Claudia, N.M.-A.; Anandhakumar, C.; Richard, M.; Maxwell, R.; Charlotte, D.; Francesca, M.B.; Lisa, C.H.; et al. Multiomics-based assessment of 2D and 3D human iPSC-cardiomyocyte models of insulin resistance demonstrate metabolic and contractile dysfunction that recapitulates diabetic cardiomyopathy. bioRxiv 2024, 2024-11. [Google Scholar] [CrossRef]
- Chris, L.H. iPSC-derived hepatocytes from patients with nonalcoholic fatty liver disease display a disease-specific gene expression profile. Gastroenterology 2020, 160, 2591. [Google Scholar]
- Yaqing, W.; Hui, W.; Pengwei, D.; Tingting, T.; Haitao, L.; Shuo, W.; Wenwen, C.; Jianhua, Q. Modeling Human Nonalcoholic Fatty Liver Disease (NAFLD) with an Organoids-on-a-Chip System. ACS Biomater. Sci. Eng. 2020, 6, 5734–5743. [Google Scholar]
- Recognizing the importance of ovarian aging research. Nat. Aging 2022, 2, 1071–1072. [CrossRef]
- Yamashiro, C.A.-O.; Sasaki, K.; Yokobayashi, S.; Kojima, Y.; Saitou, M.A.-O. Generation of human oogonia from induced pluripotent stem cells in culture. Nat. Protoc. 2020, 15, 1560–1583. [Google Scholar] [CrossRef]
- Yamashiro, C.A.-O.; Sasaki, K.; Yabuta, Y.; Kojima, Y.A.-O.; Nakamura, T.A.-O.; Okamoto, I.A.-O.; Yokobayashi, S.; Murase, Y.; Ishikura, Y.; Shirane, K.; et al. Generation of human oogonia from induced pluripotent stem cells in vitro. Science 2018, 362, 356–360. [Google Scholar] [CrossRef]
- Murase, Y.; Yabuta, Y.; Ohta, H.; Yamashiro, C.; Nakamura, T.; Yamamoto, T.; Saitou, M.A.-O. Long-term expansion with germline potential of human primordial germ cell-like cells in vitro. EMBO J. 2020, 39, e104929. [Google Scholar] [CrossRef]
- Li, X.; Zheng, M.; Xu, B.; Li, D.; Shen, Y.; Nie, Y.; Ma, L.; Wu, J. Generation of offspring-producing 3D ovarian organoids derived from female germline stem cells and their application in toxicological detection. Biomaterials 2021, 279, 121213. [Google Scholar] [CrossRef]
- Yoshino, T.A.-O.; Suzuki, T.A.-O.; Nagamatsu, G.A.-O.; Yabukami, H.; Ikegaya, M.; Kishima, M.; Kita, H.; Imamura, T.A.-O.; Nakashima, K.A.-O.; Nishinakamura, R.; et al. Generation of ovarian follicles from mouse pluripotent stem cells. Science 2021, 373, eabe0237. [Google Scholar] [CrossRef]
- Ali, I.; Padhiar, A.A.; Wang, T.; He, L.; Chen, M.; Wu, S.; Zhou, Y.; Zhou, G. Stem Cell-Based Therapeutic Strategies for Premature Ovarian Insufficiency and Infertility: A Focus on Aging. Cells 2022, 23, 3731. [Google Scholar] [CrossRef]
- Eremeev, A.; Pikina, A.; Ruchko, Y.; Bogomazova, A.A.-O. Clinical Potential of Cellular Material Sources in the Generation of iPSC-Based Products for the Regeneration of Articular Cartilage. Int. J. Mol. Sci. 2023, 24, 14408. [Google Scholar] [CrossRef]
- Cheng, Z.; Ito, S.; Nishio, N.; Suganya, T.; Isobe, K.i. Possibility to use iPS-technology in age-related diseases. In Proceedings of the 2012 International Symposium on Micro-NanoMechatronics and Human Science (MHS), Nagoya, Japan, 4–7 November 2012. [Google Scholar]
- Bolton, E.M.; Bradley, J.A. Avoiding Immunological Rejection in Regenerative Medicine. Regen. Med. 2015, 10, 287–304. [Google Scholar] [CrossRef]
- Hammer, Q. Genetic ablation of adhesion ligands effectively averts rejection of allogeneic immune cells. Eur. J. Immunol. 2022, 52, 23. [Google Scholar]
- Lorenzo, P. Engineering of Immune Checkpoints B7-H3 and CD155 Enhances Immune Compatibility of MHC-I−/− Ipscs for Β Cell Replacement. Cell Rep. 2022, 40, 111423. [Google Scholar]
- Norihiro, T.; Tomonori, H.; Yuriko, T.; Kodai, S.; Hidetaka, M.; Naoki, A.; Rie, T.; Sono, W.; Yudai, H.; Eri, K.; et al. Hypoimmunogenic human iPSCs expressing HLA-G, PD-L1, and PD-L2 evade innate and adaptive immunity. Stem Cell Res. Ther. 2024, 15, 193. [Google Scholar]
- Batalov, I.; Feinberg, A.W. Differentiation of Cardiomyocytes from Human Pluripotent Stem Cells Using Monolayer Culture. Biomark. Insights 2015, 10, 71–76. [Google Scholar] [CrossRef]
- Funakoshi, S.; Yoshida, Y. Recent progress of iPSC technology in cardiac diseases. Arch. Toxicol. 2021, 95, 3633–3650. [Google Scholar] [CrossRef]
- Guo, Y.; Zhu, H.; Wang, Y.; Sun, T.; Xu, J.; Wang, T.; Guan, W.; Wang, C.; Liu, C.; Ma, C. Miniature-swine iPSC-derived GABA progenitor cells function in a rat Parkinson’s disease model. Cell Tissue Res. 2023, 391, 425–440. [Google Scholar] [CrossRef]
- Zheng, X.; Han, D.; Liu, W.; Wang, X.; Pan, N.; Wang, Y.; Chen, Z. Human iPSC-derived midbrain organoids functionally integrate into striatum circuits and restore motor function in a mouse model of Parkinson’s disease. Theranostics 2023, 13, 2673–2692. [Google Scholar] [CrossRef]
- Joshi, J.; Xu, B.; Rubart, M.; Chang, Y.; Bao, X.; Chaliki, H.P.; Scott, L.R.; Zhu, W. Optogenetic Control of Engrafted Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes in Live Mice: A Proof-of-Concept Study. Cells 2022, 11, 951. [Google Scholar] [CrossRef]
- Raval, A.N.; Schmuck, E.; Roy, S.; Saito, Y.; Zhou, T.; Conklin, J.; Hacker, T.A.; Koonce, C.; Boyer, M.; Stack, K.; et al. Human iPSC-derived Committed Cardiac Progenitors Generate Cardiac Tissue Grafts in a Swine Ischemic Cardiomyopathy Model without Triggering Ventricular Arrhythmias. bioRxiv 2024, 14, 580375. [Google Scholar] [CrossRef]
- Yi-Hsien, F.; Saprina, P.H.W.; Liao, I.C.; Kuen-Jer, T.; Po-Hsien, H.; Pei-Jung, Y.; Chia-Jui, Y.; Ping-Yen, L.; Yan-Shen, S.; Yen-Wen, L. HLA-Ehigh/HLA-Ghigh/HLA-IIlow Human iPSC-Derived Cardiomyocytes Exhibit Low Immunogenicity for Heart Regeneration. Adv. Healthc. Mater. 2023, 12, 2301186. [Google Scholar]
- Cheng, Y.C.; Hsieh, M.L.; Lin, C.J.; Chang, C.M.C.; Huang, C.Y.; Puntney, R.; Wu Moy, A.; Ting, C.Y.; Herr Chan, D.Z.; Nicholson, M.W.; et al. Combined Treatment of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Endothelial Cells Regenerate the Infarcted Heart in Mice and Non-Human Primates. Circulation 2023, 148, 1395–1409. [Google Scholar] [CrossRef]
- Max, J.C.; Jonas, E.; Amar, J.A.; Nguyen, T.N.V.; Paulus, K.; Andrew, P.H.; Chris, D.; Katja, G. Generation of a human iPSC-derived cardiomyocyte/fibroblast engineered heart tissue model. F1000Research 2023, 12, 1224. [Google Scholar]
- Yike, H.; Wang, T.; López, M.; Minoru, H.; Hasan, A.; Shin, S. Recent advancements of human iPSC derived cardiomyocytes in drug screening and tissue regeneration. Microphysiol. Syst. 2020, 4, 2. [Google Scholar]
- Thomas, D.; Kim, H.; Lopez, N.; Wu, J.C. Fabrication of 3D Cardiac Microtissue Arrays using Human iPSC-Derived Cardiomyocytes, Cardiac Fibroblasts, and Endothelial Cells. J. Vis. Exp. 2021, e61879. [Google Scholar] [CrossRef]
- Jeffrey, S.S.; Bin, S.; Todd, M.H.; Tae-Yoon, P.; Nayeon, L.; Sanghyeok, K.; Jeha, J.; Young, C.; Kyungsang, K.; Quanzheng, L.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar]
- Wei, Z.; Oliver, W.G.; Lauren, L.; Ankur, J.; Val, C.S.; Jeffrey, M.T.; Budd, A.T.; Markus, H.K. Transplantation of iPSC-derived TM cells rescues glaucoma phenotypes in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, E3492–E3500. [Google Scholar]
- Tanaka, N.; Kato, H.; Tsuda, H.; Sato, Y.; Muramatsu, T.; Iguchi, A.; Nakajima, H.; Yoshitake, A.; Senbonmatsu, T. Development of a High-Efficacy Reprogramming Method for Generating Human Induced Pluripotent Stem (iPS) Cells from Pathologic and Senescent Somatic Cells. Int. J. Mol. Sci. 2020, 21, 6764. [Google Scholar] [CrossRef]
- Elias, K.M.; Ng, N.W.; Dam, K.U.; Milne, A.; Disler, E.R.; Gockley, A.; Holub, N.; Seshan, M.L.; Church, G.M.; Ginsburg, E.S.; et al. Fertility restoration in mice with chemotherapy induced ovarian failure using differentiated iPSCs. EBioMedicine 2023, 94, 104715. [Google Scholar] [CrossRef]
- Frank, P.L. In Vitro and in Vivo Evaluation of 3D Constructs Engineered with Human Ipsc-Derived Chondrocytes in Gelatin Methacryloyl Hydrogel. Biotechnol. Bioeng. 2022, 119, 2950–2963. [Google Scholar]
- Takanori, T.; Keisuke, S.; Masahiro, E.; Hiroyuki, K.; Masaki, K.; Takunori, O.; Ran-Ran, Z.; Yasuharu, U.; Yun-Wen, Z.; Naoto, K.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar]
- Saeed, A.; Sahab, B.; Reza, K.-E.; Mazidi, Z.; Yichao, S.; Jacob, W.; Cabral, J.; Langer, R.; Traverso, G.; Baharvand, H. Continuous Production of Highly Functional Vascularized Hepatobiliary Organoids from Human Pluripotent Stem Cells using a Scalable Microfluidic Platform. Adv. Func. Mater. 2023, 33, 2210233. [Google Scholar]
- Kojima, H.; Yagi, H.A.-O.; Kushige, H.; Toda, Y.; Takayama, K.; Masuda, S.; Morisaku, T.; Tsuchida, T.; Kuroda, K.; Hirukawa, K.; et al. Decellularized Organ-Derived Scaffold Is a Promising Carrier for Human Induced Pluripotent Stem Cells-Derived Hepatocytes. Cells 2022, 11, 1258. [Google Scholar] [CrossRef]
- Minami, T.; Ishii, T.; Yasuchika, K.; Fukumitsu, K.; Ogiso, S.; Miyauchi, Y.; Kojima, H.; Kawai, T.; Yamaoka, R.; Oshima, Y.; et al. Novel hybrid three-dimensional artificial liver using human induced pluripotent stem cells and a rat decellularized liver scaffold. Regen. Ther. 2019, 10, 127–133. [Google Scholar] [CrossRef]
- Dongzhi, W.; Yibing, G.; Jiacheng, Z.; Fang, L.; Yan, X.; Yan, H.; Biwen, Z.; Di, W.; Haopeng, P.; Tiancheng, G.; et al. Hyaluronic acid methacrylate/pancreatic extracellular matrix as a potential 3D printing bioink for constructing islet organoids. Acta Biomater. 2023, 165, 86–101. [Google Scholar]
- Daniel, N.; Christian, P.; Débora, C.C.-H.; Doris, W.; Thomas, M.; Martin, H.; Judith, H.; Michael, J.A. 3D bioprinted, vascularized neuroblastoma tumor environment in fluidic chip devices for precision medicine drug testing. Biofabrication 2022, 14, ac5fb7. [Google Scholar]
- Anna, N.; Daiki, M.; Ryota, F.; Sakura, T.; Sanae, N.; Makoto, I.; Junya, T.; Koichi, N. Bio-3D printing iPSC-derived human chondrocytes for articular cartilage regeneration. Biofabrication 2021, 13, 044103. [Google Scholar]
- Spitzhorn, L.S.; Megges, M.; Wruck, W.; Rahman, M.S.; Otte, J.; Degistirici, Ö.; Meisel, R.; Sorg, R.V.; Oreffo, R.O.C.; Adjaye, J. Human iPSC-derived MSCs (iMSCs) from aged individuals acquire a rejuvenation signature. Stem Cell Res. Ther. 2019, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Santoso, M.R.; Ikeda, G.; Tada, Y.; Jung, J.H.; Vaskova, E.; Sierra, R.G.; Gati, C.; Goldstone, A.B.; von Bornstaedt, D.; Shukla, P.; et al. Exosomes From Induced Pluripotent Stem Cell-Derived Cardiomyocytes Promote Autophagy for Myocardial Repair. J. Am. Heart Assoc. 2020, 9, e014345. [Google Scholar] [CrossRef]
- Hongmei, L.; Fenfang, W.; Guangrui, H.; Di, W.; Ting, W.; Xiashuang, W.; Kai, W.; Yuyin, F.; Anlong, X. Cardiomyocytes induced from hiPSCs by well-defined compounds have therapeutic potential in heart failure by secreting PDGF-BB. Signal Transduct. Target. Ther. 2022, 7, 253. [Google Scholar]
- Zhu, Y.; Wang, Y.; Zhao, B.; Niu, X.; Hu, B.; Li, Q.; Zhang, J.; Ding, J.; Chen, Y.; Wang, Y. Comparison of exosomes secreted by induced pluripotent stem cell-derived mesenchymal stem cells and synovial membrane-derived mesenchymal stem cells for the treatment of osteoarthritis. Stem Cell Res. Ther. 2017, 9, 64. [Google Scholar] [CrossRef]
- Manai, F.; Smedowski, A.; Kaarniranta, K.; Comincini, S.; Amadio, M. Extracellular vesicles in degenerative retinal diseases: A new therapeutic paradigm. J. Control Release 2024, 365, 448–468. [Google Scholar] [CrossRef]
- Zhou, J.; Flores-Bellver, M.; Pan, J.; Benito-Martin, A.; Shi, C.; Onwumere, O.; Mighty, J.; Qian, J.; Zhong, X.; Hogue, T.; et al. Human retinal organoids release extracellular vesicles that regulate gene expression in target human retinal progenitor cells. Sci. Rep. 2021, 11, 21128. [Google Scholar] [CrossRef]
- Kurzawa-Akanbi, M.; Whitfield, P.; Burté, F.; Bertelli, P.M.; Pathak, V.; Doherty, M.; Hilgen, B.; Gliaudelytė, L.; Platt, M.; Queen, R.; et al. Retinal pigment epithelium extracellular vesicles are potent inducers of age-related macular degeneration disease phenotype in the outer retina. J. Extracell. Vesicles 2022, 11, 12295. [Google Scholar] [CrossRef]
- Ding, C.; Qian, C.; Hou, S.; Lu, J.; Zou, Q.; Li, H.; Huang, B. Exosomal miRNA-320a Is Released from hAMSCs and Regulates SIRT4 to Prevent Reactive Oxygen Species Generation in POI. Mol. Ther. Nucleic. Acids 2020, 21, 37–50. [Google Scholar] [CrossRef]
- Yang, Z.; Du, X.; Wang, C.; Zhang, J.; Liu, C.; Li, Y.; Jiang, H. Therapeutic effects of human umbilical cord mesenchymal stem cell-derived microvesicles on premature ovarian insufficiency in mice. Stem Cell Res. Ther. 2019, 10, 250. [Google Scholar] [CrossRef]
- Huang, B.A.-O.; Lu, J.; Ding, C.; Zou, Q.; Wang, W.; Li, H. Exosomes derived from human adipose mesenchymal stem cells improve ovary function of premature ovarian insufficiency by targeting SMAD. Stem Cell Res. Ther. 2018, 9, 216. [Google Scholar] [CrossRef]
- Amor, C.; Fernández-Maestre, I.; Chowdhury, S.; Ho, Y.J.; Nadella, S.; Graham, C.; Carrasco, S.E.; Nnuji-John, E.; Feucht, J.; Hinterleitner, C.; et al. Prophylactic and long-lasting efficacy of senolytic CAR T cells against age-related metabolic dysfunction. Nat. Aging 2024, 4, 336–349. [Google Scholar] [CrossRef]
- Amor, C.A.-O.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.A.-O.X.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Chang, Y.; Syahirah, R.; Wang, X.; Jin, G.; Torregrosa-Allen, S.; Elzey, B.D.; Hummel, S.N.; Wang, T.; Li, C.; Lian, X.; et al. Engineering chimeric antigen receptor neutrophils from human pluripotent stem cells for targeted cancer immunotherapy. Cell Rep. 2022, 40, 111128. [Google Scholar] [CrossRef]
- Jin, G.; Chang, Y.; Bao, X. Generation of chimeric antigen receptor macrophages from human pluripotent stem cells to target glioblastoma. Immunooncol. Technol. 2023, 20, 100409. [Google Scholar] [CrossRef]
- Chang, Y.; Jin, G.; Luo, W.; Luo, Q.; Jung, J.; Hummel, S.N.; Torregrosa-Allen, S.; Elzey, B.D.; Low, P.S.; Lian, X.L.; et al. Engineered human pluripotent stem cell-derived natural killer cells with PD-L1 responsive immunological memory for enhanced immunotherapeutic efficacy. Bioact. Mater. 2023, 27, 168–180. [Google Scholar] [CrossRef]
- Ueda, T.; Shiina, S.; Iriguchi, S.; Terakura, S.; Kawai, Y.; Kabai, R.; Sakamoto, S.; Watanabe, A.; Ohara, K.; Wang, B.; et al. Optimization of the proliferation and persistency of CAR T cells derived from human induced pluripotent stem cells. Nat. Biomed. Eng. 2023, 7, 24–37. [Google Scholar] [CrossRef]
- Ando, M.; Kinoshita, S.; Furukawa, Y.; Ando, J.; Nakauchi, H.; Brenner, M.K. Chapter 3—Improving the safety of iPSC-derived T cell therapy. In Molecular Players in iPSC Technology; Birbrair, A., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 95–115. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, P.; Liu, B.; Dong, C.; Chang, Y. Induced Pluripotent Stem Cells-Based Regenerative Therapies in Treating Human Aging-Related Functional Decline and Diseases. Cells 2025, 14, 619. https://doi.org/10.3390/cells14080619
Yu P, Liu B, Dong C, Chang Y. Induced Pluripotent Stem Cells-Based Regenerative Therapies in Treating Human Aging-Related Functional Decline and Diseases. Cells. 2025; 14(8):619. https://doi.org/10.3390/cells14080619
Chicago/Turabian StyleYu, Peijie, Bin Liu, Cheng Dong, and Yun Chang. 2025. "Induced Pluripotent Stem Cells-Based Regenerative Therapies in Treating Human Aging-Related Functional Decline and Diseases" Cells 14, no. 8: 619. https://doi.org/10.3390/cells14080619
APA StyleYu, P., Liu, B., Dong, C., & Chang, Y. (2025). Induced Pluripotent Stem Cells-Based Regenerative Therapies in Treating Human Aging-Related Functional Decline and Diseases. Cells, 14(8), 619. https://doi.org/10.3390/cells14080619