1. Introduction
The spatial distribution of many nuclear compartments, including proteinaceous bodies and genomic domains, is non-random [
1]. Gene-rich chromosomes, for example, tend to occupy central nuclear regions, whereas gene-poor chromosomes are typically more peripheral [
2]. Similarly, transcription sites and replicating loci, often associated with active chromatin, are generally more centrally localized in the cell nucleus, whereas inactive heterochromatin tend to be positioned at the nuclear periphery or in proximity to nucleoli [
3,
4,
5]. The spatial organization of chromosomes within the nucleus has been suggested to be integral to cellular function, impacting processes such as chromatin accessibility, gene expression, DNA repair, and DNA replication [
1].
Centromeres are specialized regions on each chromosome that are crucial for accurate chromosome segregation. During cell division, centromeres serve as the attachment point for the microtubule spindle via the kinetochore protein complex [
6,
7]. Similar to other nuclear structures, centromeres assume an apparently non-random spatial distribution within the nucleus [
8,
9,
10,
11], and in a cell type-specific manner [
10,
12]. For example, in human stem cells, the majority of centromeres cluster strongly near the nucleoli [
13,
14], and this association is weakened as the stem cells differentiate [
14]. Similarly, the association of centromeres with the nucleolus has also been observed in other non-stem cell types [
15]. Although the proximity of rDNA loci to the centromeres on all five rDNA-containing chromosomes may partly account for this behavior [
16], the spatial proximity of centromeres to the nucleolus may also have functional consequences, as suggested by the finding that transcription from alpha-satellite repeats of centromeres is limited near nucleoli [
17] and by the observation that peripheral chromosomes have a higher rate of mis-segregation compared with centrally positioned ones [
18]. Furthermore, variations in centromere clustering have also been implicated in diverse biological phenomena, including development, cancer progression, and response to cellular stress [
6,
19].
Little is known about the cellular factors and mechanisms that determine spatial centromere distribution. Candidate-based studies in cancer cell lines have demonstrated that alterations in centromere clustering can result from changes in chromatin organization, genomic stability and mitotic fidelity [
20]. Similarly, clustering variations have been associated with perturbations of the NCAPH2 subunit of the condensin II complex [
21], which regulates chromatin compaction and spatial genome organization [
22]. However, the specific molecular mechanisms and processes that determine the spatial organization of centromeres remain unknown.
High-throughput imaging (HTI) assays are a powerful approach to systematically study cellular phenotypes, including centromere distribution, at the single-cell level, and in an unbiased fashion when paired with functional genomics perturbations, such as RNAi or CRISPR-Cas9 gene knockouts [
23]. These assays are ideally suited to perform functional genomics screens to identify regulators of centromeric localization, and they rely on robust metrics to quantify centromere clustering patterns across millions of cells and across thousands of biological conditions. Unfortunately, such metrics are currently missing.
To fill this gap, we conducted extended testing to identify novel analytical tools to quantitatively analyze centromere localization patterns in human cells, including several global and local clustering metrics and spatial distribution modeling approaches. First, we benchmarked six clustering metrics and tested multiple spot generation models to evaluate their utility in measuring different clustering patterns on simulated data. Then, we tested these metrics on HTI experimental data from multiple human cell types. We identified a derivative version of Ripley’s K function [
24] as the most robust indicator of different clustering patterns in single cells. Finally, to extend the applicability of our framework, we also surveyed several modeling approaches to fit the imaging data and recreate realistic centromere distributions in silico. We tested these models on multiple human cell lines, demonstrating their versatility and accuracy in capturing diverse centromere localization patterns in the presence or absence of experimental perturbation of centromere clustering patterns. Our approach establishes quantitative tools for the study of centromere localization and function, and they have potential for wider application in genome research.
2. Methods
2.1. siRNA Oligos Transfection and Immunofluorescence
The image dataset containing four technical replicates of human colon cancer HCT116-Cas9 cells were reverse-transfected with siRNA oligos against the
NCAPH2 gene and a scrambled negative siRNA control in 384-well plates has been described [
25].
2.2. Cell Growth and Centromere Visualization
All cells were grown in 384-well plates (CellVis, cat. No. P384-1.5H-N). The sources of cell lines, culture conditions, media compositions, and relevant references for respective culture protocols are described in
Supplementary Table S1. Cell lines were grown for 72 h after cell seeding before fixation, except for the iPS WTC11 cells that were grown for 3–5 days and fixed before the colony edges merged with each other. All cell lines were fixed with 2% paraformaldehyde (PFA, Electron Microscopy Sciences, Hatfield, PA, USA, cat. No. 15710) solution in media by adding one equal volume of 4% PFA solution in PBS to the cell growth medium in each well for 15 min at room temperature. Fixed cells were then subjected to immunofluorescence (IF) staining using anti-CENP-C antibody (MBL Co., Ltd., Tokyo, Japan, cat. No. PD030) and DAPI (4′,6-diamidino-2-phenylindole) (Thermo Fisher Scientific, Waltham, MA, USA, cat. No. 62248) as previously described [
25]. The total number of cells analyzed from this dataset is as follows: A375, n = 11,925; MDA-MB-231, n = 6347; HFF-hTERT, n = 1322; hTERT-RPE1, n = 3641; A549, n = 7786; WTC11, n = 6071; HAP1, n = 18,359; and HCT116, n = 16,116.
2.3. High-Throughput Image Acquisition
For IF experiments, images were acquired using 405 nm (DAPI channel) or 488 nm (CENP-C channel) excitation lasers and a 405/488/561/640 nm excitation dichroic mirror. A 60× water-immersion objective (NA 1.2) was employed, paired with 445/45 nm (DAPI channel) or 525/50 nm (CENP-C channel) bandpass emission filters. A 16-bit sCMOS camera (2048 × 2048 pixels, 1 × 1 binning, pixel size: 0.108 microns) was used for the capture of image Z-stacks spanning 14 microns in depth, collected at 1-micron intervals and maximally projected on the fly. Images were acquired from 22 fields of view (FOV) per well.
2.4. High-Throughput Image Analysis
The analysis of imaging data was carried out using HiTIPS, a high-throughput image analysis software designed to analyze cell-based assays in fixed and live cells as previously described [
25]. Maximally projected DAPI images were used for nucleus segmentation, while CENP-C images were used for CENP-C spot finding and localization. Specific analysis parameters were selected in HiTIPS tailored to align with the average nucleus size, as well as the size and brightness of the centromere spots observed. The GPU-based Cellpose algorithm for nuclear segmentation [
26] was used in conjunction with the Laplacian of Gaussian method for spot detection. Spot positions were determined as the center of gravity of the segmented spots.
2.5. Methods for Generating Synthetic Spot Patterns
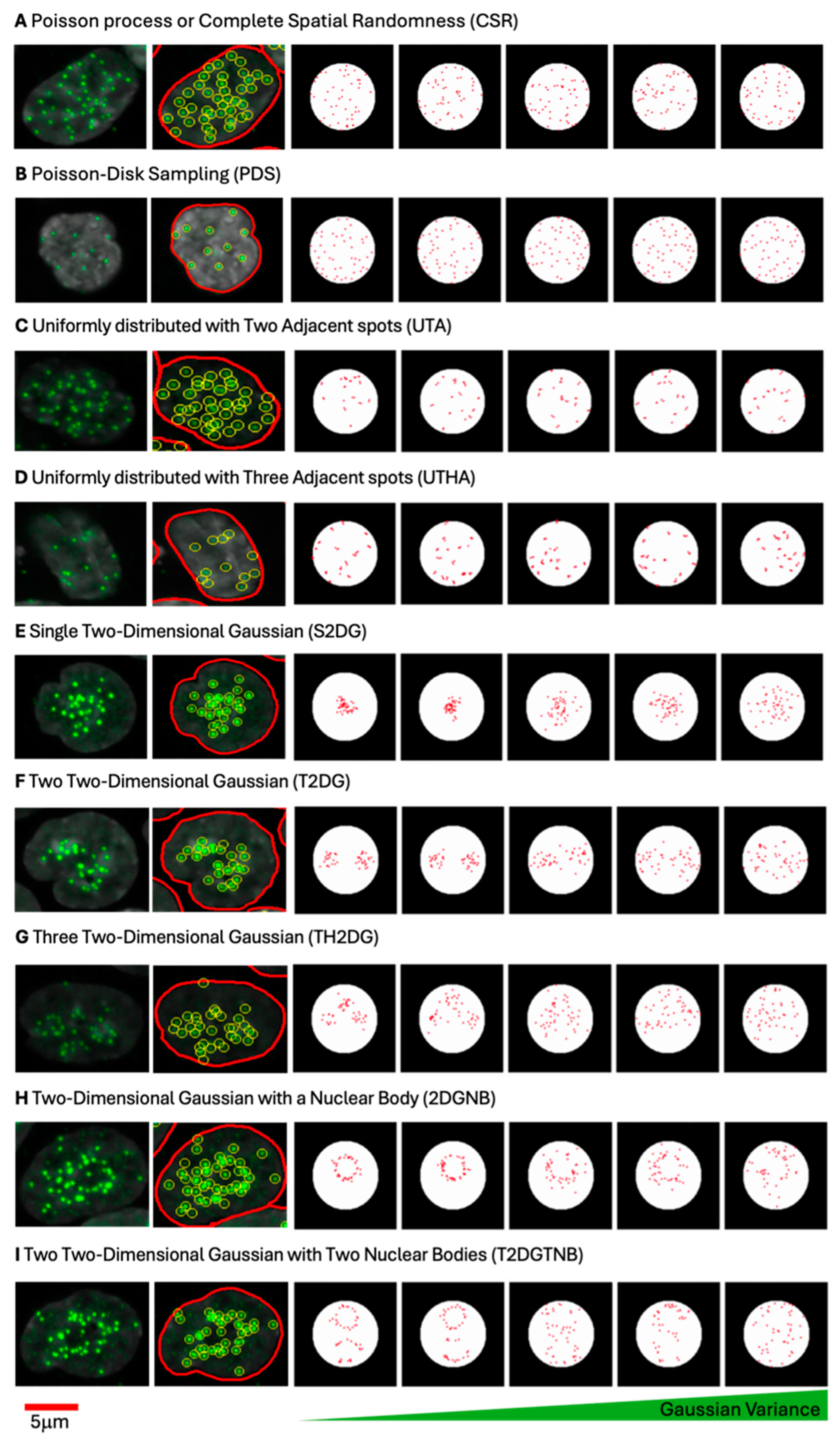
To evaluate centromere spot pattern characterization metrics, synthetic images containing simulated distributions of spots were generated under controlled spatial arrangements within a circular region representing the cell nucleus. For each spatial pattern, 100,000 images were generated. The synthetic spots were generated on an image patch of pixels. A circular region was defined at the center of this patch, with a radius of pixels and center coordinates. All synthetic spots were constrained within this circular region, ensuring consistency across pattern types. The constraint for any spot was . Each distribution was designed with specific statistical properties to mimic potential centromere clustering patterns.
2.5.1. Poisson Process or Complete Spatial Randomness (CSR)
To generate a uniform distribution of spots, random points were sampled according to a uniform distribution within the bounds for and for . Each point was retained only if it lay within the circular region as defined above. For each sample, 46 spots were generated. This uniform sampling, which is also referred to as a Poisson process, provided a baseline distribution to assess other pattern types against a spatially random background.
2.5.2. Single Two-Dimensional Gaussian Distribution (S2DG)
For clustering patterns, spots were generated based on a 2D Gaussian distribution centered at with varying covariance matrices. The covariance matrices systematically varied in size (from 50 to 1000 pixels squared in increments) and orientation (from 0 to radians).
The covariance matrix for each Gaussian distribution was defined as follows:
where
is the rotation matrix for angle
and
and
represent the standard deviations along the rotated principal axes. For each sample, points were drawn from the Gaussian distribution, and only the first 46 spots within the circular boundary were retained. This process provided various levels of spot clustering within the circle.
2.5.3. Two (T2DG) and Three (TH2DG) Two-Dimensional Gaussian Distributions
To model multimodal clustering, Gaussian mixture models (GMMs) [
27] were used with either two or three Gaussian components (modes) centered within the circular region. For the two-mode GMM, the component centers were positioned symmetrically around
at distances of 25 pixels, representing two distinct clusters. For the three-mode GMM, three Gaussian components were similarly spaced 20 pixels apart in a triangular arrangement around
.
Each component’s covariance matrix systematically varied using the same range of sizes and orientations as described for the single Gaussian distribution. Samples from the GMM were filtered to retain only those within the circle. The GMM model used equal weights for each component:
where
is the number of modes (2 or 3) and
is the component weight (0.5 for the 2-mode GMM, 1/3 for the 3-mode GMM).
For each sample, points were drawn from the corresponding distribution, and only the first 46 spots within the circular boundary were retained.
2.5.4. Poisson Disk Sampling (PDS)
A Poisson disk sampling approach [
28] was applied to generate dispersed spots, enforcing a minimum inter-spot distance of 10 pixels. This approach simulated highly dispersed patterns, characteristic of spatially non-clustered arrangements. Candidate points were generated uniformly within the circular boundary, but each new point was retained only if it satisfied the minimum distance requirement from all existing spots. This technique was applied to generate 46 spots per sample while maintaining a minimum spacing constraint.
2.5.5. Uniformly Distributed Centromeres with Two (UTA) and Three (UTHA) Adjacent Spots
To model proximity effects, two and three adjacent spots were generated by perturbing a subset of initial points with a random shift sampled from pixels. For two adjacent spots, the perturbation was applied to 15 spots out of the 46, generating a second, closely placed spot for each. For three adjacent spots, two perturbations were applied to 15 initial spots, creating two adjacent spots per each of the 15. The first 46 points within the circular boundary were retained after applying the perturbation.
2.6. Spot Clustering Metrics
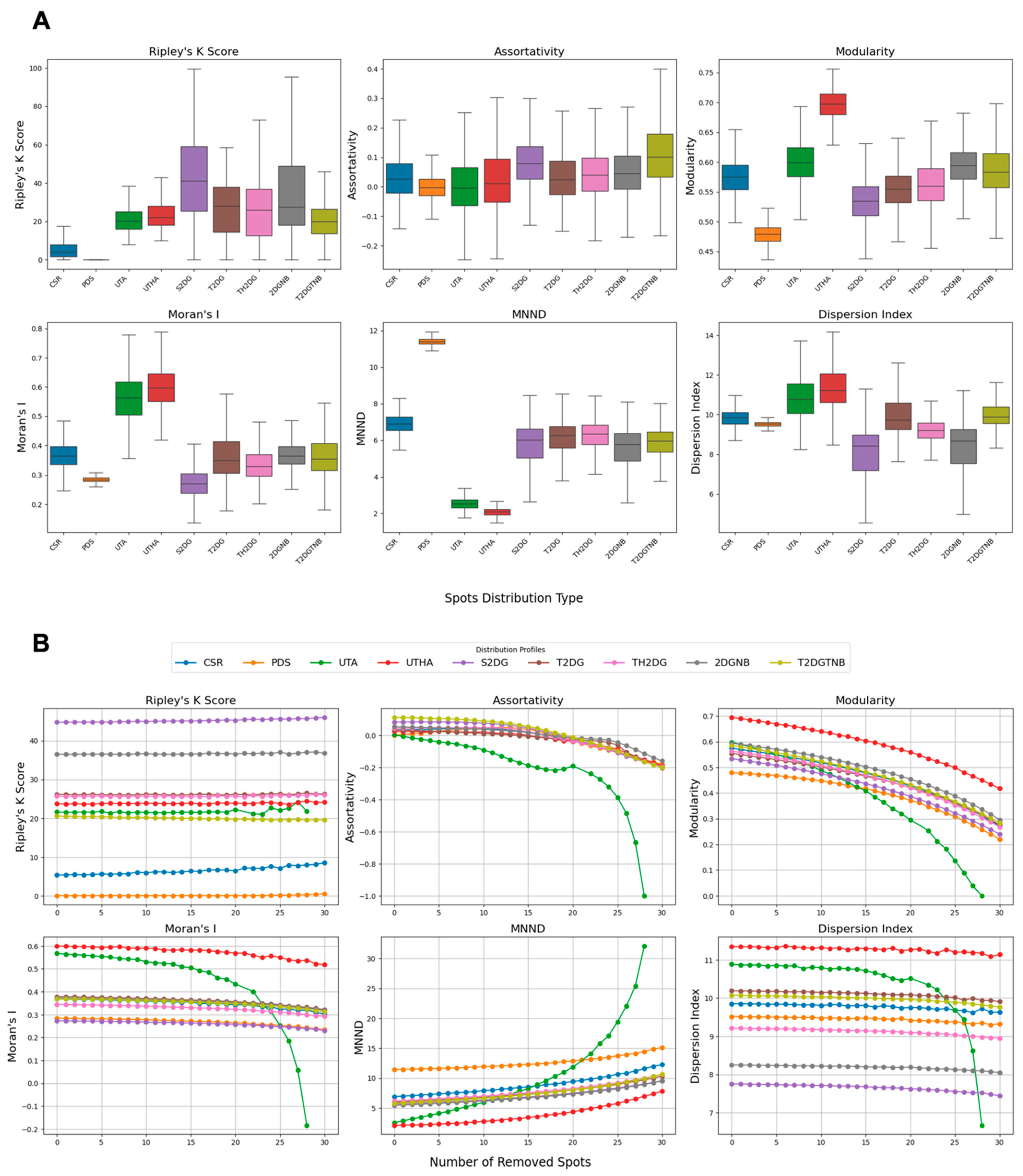
To analyze centromere spot patterns within synthetic distributions, we employed different metrics that capture clustering, modularity, spatial autocorrelation, and dispersion characteristics. Each metric was computed based on specific parameter settings and methodologies, as outlined below.
2.6.1. Ripley’s K Function
Ripley’s K function
[
29] was used to quantify spatial clustering by calculating the difference between the expected number of neighboring points within a radius
of each point, and the expected number of neighboring points within a radius
of each point for a homogeneous spatial Poisson process, or Complete State of Randomness (CSR). Ripley’s K function is maximized when the expected number of spots from any given point is larger than the one for the CSR distribution for a large number of radiuses
. This metric results in higher values for spot patterns from one large cluster such as single 2D Gaussian distribution with and without nuclear bodies. However, for dispersed small clusters, and even two- and three-mode 2D Gaussians distributions, this Ripley’s K function is expected to return significantly lower values. We report the clustering percentage as the fraction of radii where
exceeds the Poisson expectation, indicating clustering. The calculation is as follows:
where
is the area of the region,
is the number of points,
is the distance between points
and
, and
is an indicator function that is 1 if
and 0 otherwise. We calculated Ripley’s K clustering score, or the clustering percentage, as follows:
where
is the expectation for a spatially random distribution.
2.6.2. Assortativity Coefficient
The assortativity coefficient is a measure of the connectedness of nodes with similar degrees in a graph [
30], and it is computed for a
-nearest neighbor (k-NN) graph created from the centromere points, reflecting the tendency of nodes to connect to others with a similar degree (K = 10). Assortativity is based on degrees and measures the correlation between a node’s degree and the degrees of their neighbors, and it is defined as follows:
where
and
are the degrees of nodes
and
in the graph,
is the mean node degree, and
represents the set of edges in the graph.
Assortativity is reported as a single coefficient between (disassortative) and (assortative).
Since the node degrees are calculated as the inverse of the squared distance of spots from each other, this metric is expected to be maximal when most of the spots surround the nuclear bodies, such as the nucleolus. However, the values are still very close to zero and very closely followed by spots generated using a single 2D Gaussian distribution.
2.6.3. Modularity
Modularity
measures the density of connections within clusters compared with connections between clusters [
31], reflecting how well the graph divides into modules. The Louvain algorithm was used to compute modularity as follows:
where
is the adjacency matrix,
and
are the degrees of nodes
and
,
is the total number of edges, and
is 1 if nodes
and
belong to the same community and 0 otherwise. The modularity index
ranges from
to
, with values closer to 1 indicating stronger modularity. This metric is expected to be maximal in the presence of small clusters dispersed within nuclei.
2.6.4. Moran’s I
Moran’s I statistic was used to evaluate spatial autocorrelation of centromere spots [
32]. Moran’s I measures whether spots are clustered (positive value), dispersed (negative value), or randomly distributed (near zero). Moran’s I is defined as follows:
where
is the number of points,
and
are the coordinates of points
and
,
is the mean coordinate,
is the spatial weight (set to
for
, else 0), and
is the sum of all weights. Moran’s I ranges from
(complete dispersion) to 1 (perfect clustering), with zero indicating no autocorrelation. Moran’s I is expected to be maximized by the presence of local clusters further from the centroid of the spots, such as uniformly distributed centromeres with two and three adjacent spots, and minimized when all the spots are clustered at the center of the nucleus, as generated by a single 2D Gaussian distribution.
2.6.5. Mean Nearest Neighbor Distance (MNND)
The mean nearest neighbor distance (MNND) quantifies the average distance to the nearest neighboring spot [
33], providing a measure of the clustering density of the centromeres. It is calculated as:
where
is the distance between point i and its nearest neighbor and
is the total number of points. A lower MNND indicates tighter clustering, while a higher value suggests more dispersed points. The MNND is expected to be maximized with a hardcore process spot pattern and minimized when each spot has at least one other spot in proximity.
2.6.6. Dispersion Index
The dispersion index quantifies the level of spatial dispersion or clustering by comparing the variance of pairwise distances to their mean distance [
34]. The dispersion index
is defined as follows:
where
is the variance of the pairwise distances between points and
is the mean of those distances. A higher dispersion index indicates greater variability in distances, suggesting spatial clustering, whereas a lower index indicates uniform dispersion. The dispersion index is used to assess the spread of centromeres within the defined circular area, and it is maximized when the average of pairwise distances of the spots is minimum and the variance is high. Both these conditions can occur simultaneously with uniformly distributed centromeres with two and three adjacent spots.
2.7. Centromere Spot Localization Modeling Methods Using Gaussian Distributions
To model the localization of the centromere spots, we used several approaches based on spot location and the distribution of their pairwise distances. Each approach is explained in detail below.
2.7.1. Uniform Distribution of Spots on the Cell Nucleus as a Benchmarking Method
To establish a baseline for centromere spot localization, a uniform distribution of spots was generated within the boundaries of the cell nucleus, also known as Poisson process. This method simulates a completely random spatial arrangement, providing a benchmark for comparing clustering metrics and other spatial pattern models.
2.7.2. Modeling of Centromere Localization Using the Cell Shape
To model centromere localization based on nuclear geometry, an ellipse was fitted to the segmented nucleus. The parameters of the ellipse, specifically the major axis
and minor axis
, were used to define the spatial boundaries for generating a two-dimensional Gaussian distribution. The Gaussian distribution was centered at the nucleus center
, and the probability density function at any point (
was as follows:
where
and
represent the standard deviations along the major and minor axes, respectively, to ensure realistic clustering within the nuclear boundary. The generated spots follow this elliptical Gaussian distribution, accurately reflecting the centromere patterns under the spatial constraints imposed by the nuclear shape.
2.7.3. Radial Distribution and Ripley’s Function Calculation
For this approach, we calculated Ripley’s
function, which describes the expected number of points within a distance
from a randomly chosen point, normalized by the intensity
of the point process. The
function for our radially symmetric Gaussian distribution is defined as follows:
where
is the radial probability density function (PDF) of a 2D Gaussian distribution centered at the origin as follows:
Converting to polar coordinates allows us to express the expected number of points within distance
by integrating
over the radial distance
from the origin. The inner integral over
simplifies due to the independence of
from the angular coordinate, reducing
to the following:
To further evaluate this expression, we used the substitution
, transforming the integral into the following:
which yields the following:
This closed-form solution provides a cumulative description of the expected clustering of points up to a radius , based on a Gaussian spatial distribution of centromere locations.
2.7.4. Bayesian Estimation of Radially Shifted Gaussian Distribution Using Spots Coordinates
Given the observed
and knowing that each
is drawn from a Gaussian distribution with mean
and variance
, the likelihood function
can be expressed as follows:
where each
is calculated as follows:
Thus, the full likelihood function is as follows:
Given that and are themselves normally distributed, we can define the priors:
Prior on
:
, where
and
are the mean and the variance of the prior on
.
Prior on
:
, where
and
are the mean and the variance of the prior on
.
The goal is to find the posterior distribution
, which is proportional to the product of the likelihood and the prior:
Substituting the likelihood and the priors,
This can be expressed more compactly as follows:
2.7.5. Bayesian Estimation of the Radially Shifted Gaussian Distribution Using Pairwise Distances
In polar coordinates, a radially shifted Gaussian distribution can be represented as follows:
where
is the radial distance from the origin,
is the angular coordinate,
is the mean radius of the doughnut shape, and σ is the standard deviation controlling the thickness of the doughnut.
We then convert the polar coordinates
and
into Cartesian coordinates:
The Euclidean distance
between the two points
and
is as follows:
Substituting the expressions for
and
provides the following:
To derive the distribution of pairwise distances, we need to consider the probability distributions of
,
, and
. Assuming
and
are independently drawn from the radially shifted Gaussian distribution and
is uniformly distributed over
, the distribution of
can be obtained by integrating over all possible values of
,
, and
:
Here,
and
are the radial distributions (Gaussian distribution with mean
and variance
) and
is the Dirac delta function that ensures the integration considers only valid pairwise distances, where
and
are Gaussian distributions:
and
is the uniform distribution of the angle difference. Simplifying the angle integral by noting that
is uniform, the following is obtained:
By substituting the variable , we have . The limits of integration for are from −1 to 1.
The delta function imposes the following condition:
. This, in turn, implies that
. Thus, the integral over
is as follows:
The delta function reduces the integral over
as follows:
where
is the Heaviside step function ensuring that
lies within
.
The final form of
involves evaluating the integral:
For a set of pairwise distances
from a single realization, the likelihood is as follows:
Then, the log-likelihood is as follows:
If (e.g., due to insufficient Monte Carlo samples), a large negative value (e.g., ) is assigned to to avoid numerical issues.
For a cell line with multiple realizations
, the total log-likelihood is as follows:
where each
is as follows:
Thus, the full likelihood function is as follows:
Given that
and
are themselves normally distributed, we can define the priors
and
the same as those defined for Bayesian estimation of a radially shifted Gaussian distribution using spot coordinates. The posterior distribution combines the likelihood and priors:
The log-posterior is as follows:
2.8. MCMC Framework (Metropolis–Hastings Algorithm)
The Metropolis–Hastings algorithm involves iteratively proposing new values for model parameters based on a predefined distribution and calculating an acceptance ratio that compares the likelihood of the new parameters against the current ones. If the new parameters yield a higher probability or meet a random acceptance criterion, they are accepted; otherwise, the current parameters are retained. This process is repeated for a set number of iterations, with each accepted parameter set stored for subsequent analysis. The method ensures exploration of the parameter space while gradually converging on the most probable values based on the observed data.
2.9. Statistical Analysis
2.9.1. Metrics Comparison to the Complete State of Randomness for Different Spot Generation Methods
To evaluate differences between groups for each metric, we conducted statistical testing in a two-step process. First, pairwise comparisons were performed using the Mann–Whitney U test [
35], a rank-based non-parametric method for comparing two independent groups. This test evaluates whether one group tends to have higher or lower values than the other, without assuming normality or equal variances. We compared all groups to a reference group, complete spatial randomness (CSR), for each metric. To control for multiple comparisons and reduce the likelihood of false positives, we applied the Benjamini–Hochberg false discovery rate (FDR) correction [
36] to the raw
p-values. For each comparison, the Mann–Whitney U test results included the test statistic, the raw
p-value, the corrected
p-value, and the significance status (whether the corrected
p-value was below 0.05). Significant results were bolded for presentation in the final dataset. The corrected
p-values were used to determine statistical significance, with values below 0.05 considered indicative of significant differences.
2.9.2. Pairwise Metrics Comparison for Different Spot Generation Methods
To assess differences in clustering metrics (e.g., Ripley’s K function, assortativity, modularity, Moran’s I, MNND, and dispersion index) across nine synthetic spot generation models (e.g., CSR, PDS, UTA, UTHA, S2DG, T2DG, TH2DG, 2DGNB, T2DGTNB) relative to complete spatial randomness (CSR), we performed a robust statistical analysis. Initially, the Kruskal–Wallis test [
37] was applied to detect overall differences across all models for each metric, offering a non-parametric assessment of variation without assuming normality. Subsequently, pairwise comparisons were conducted using the Mann–Whitney U test [
35], comparing each model to the CSR reference group to pinpoint specific differences. This rank-based method was selected for its ability to handle non-normal data and unequal variances. To manage the risk of false positives from multiple comparisons, we implemented the Benjamini–Hochberg false discovery rate (FDR) correction [
36] on the raw
p-values. For each pairwise test, we reported the test statistic (U), the raw
p-value, the corrected
p-value, and the significance status (corrected
p-value < 0.05), with significant results bolded in the output. Additionally, the rank-biserial correlation was computed as an effect size to measure the magnitude and direction of differences. Corrected
p-values below 0.05 indicated statistically significant deviations from CSR for the given metric and model.
2.9.3. Metrics for Comparing Distributions
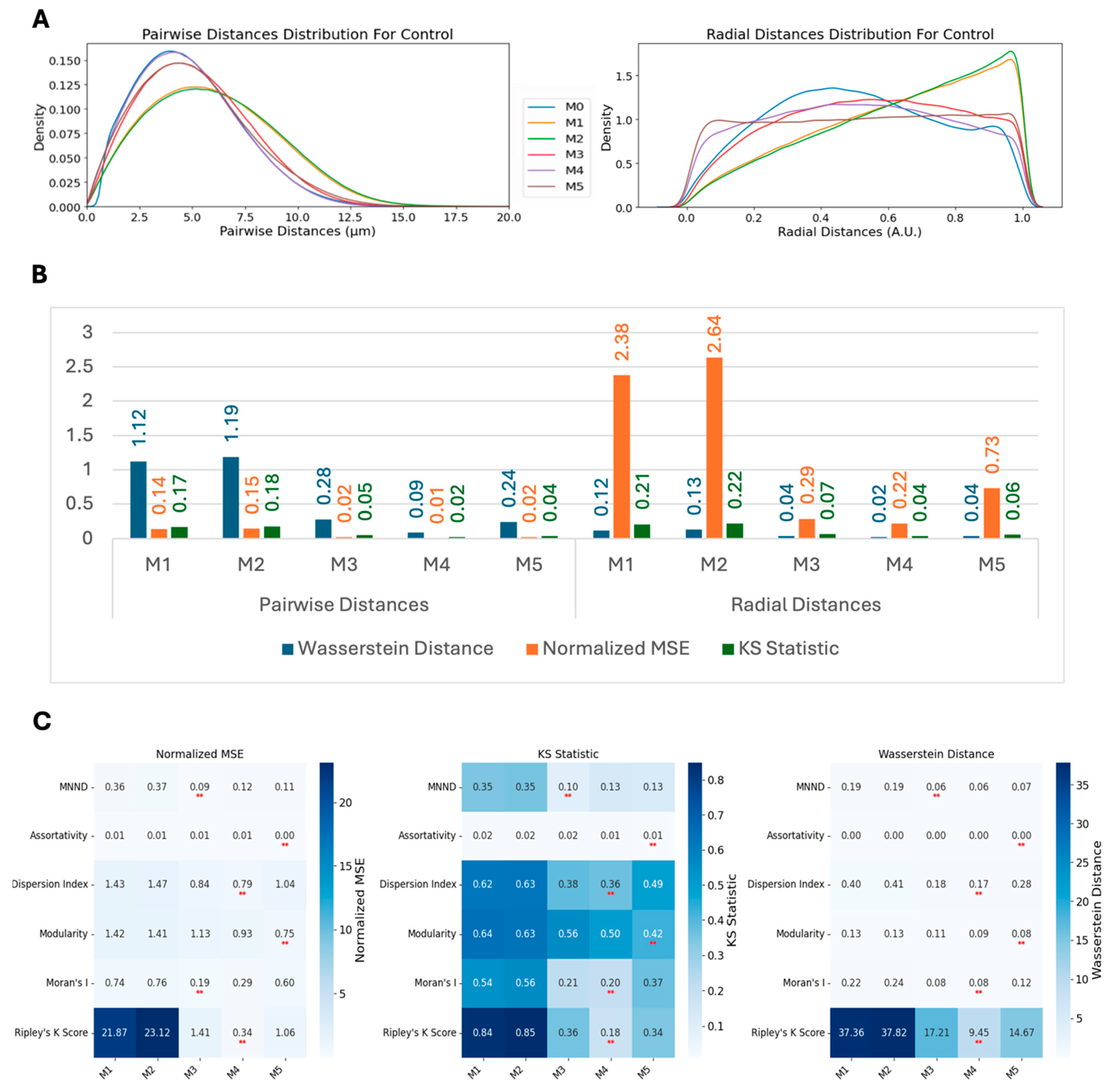
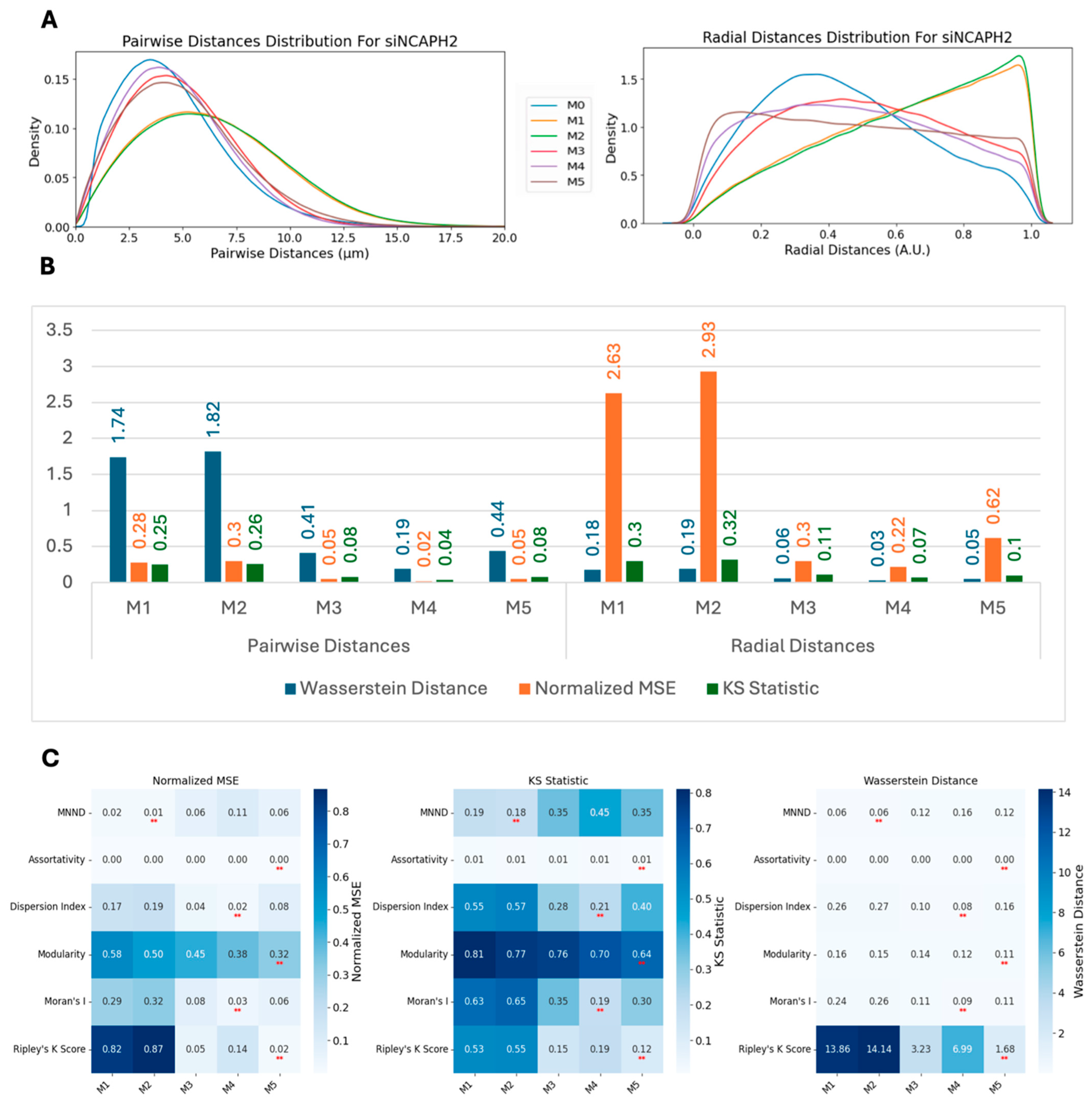
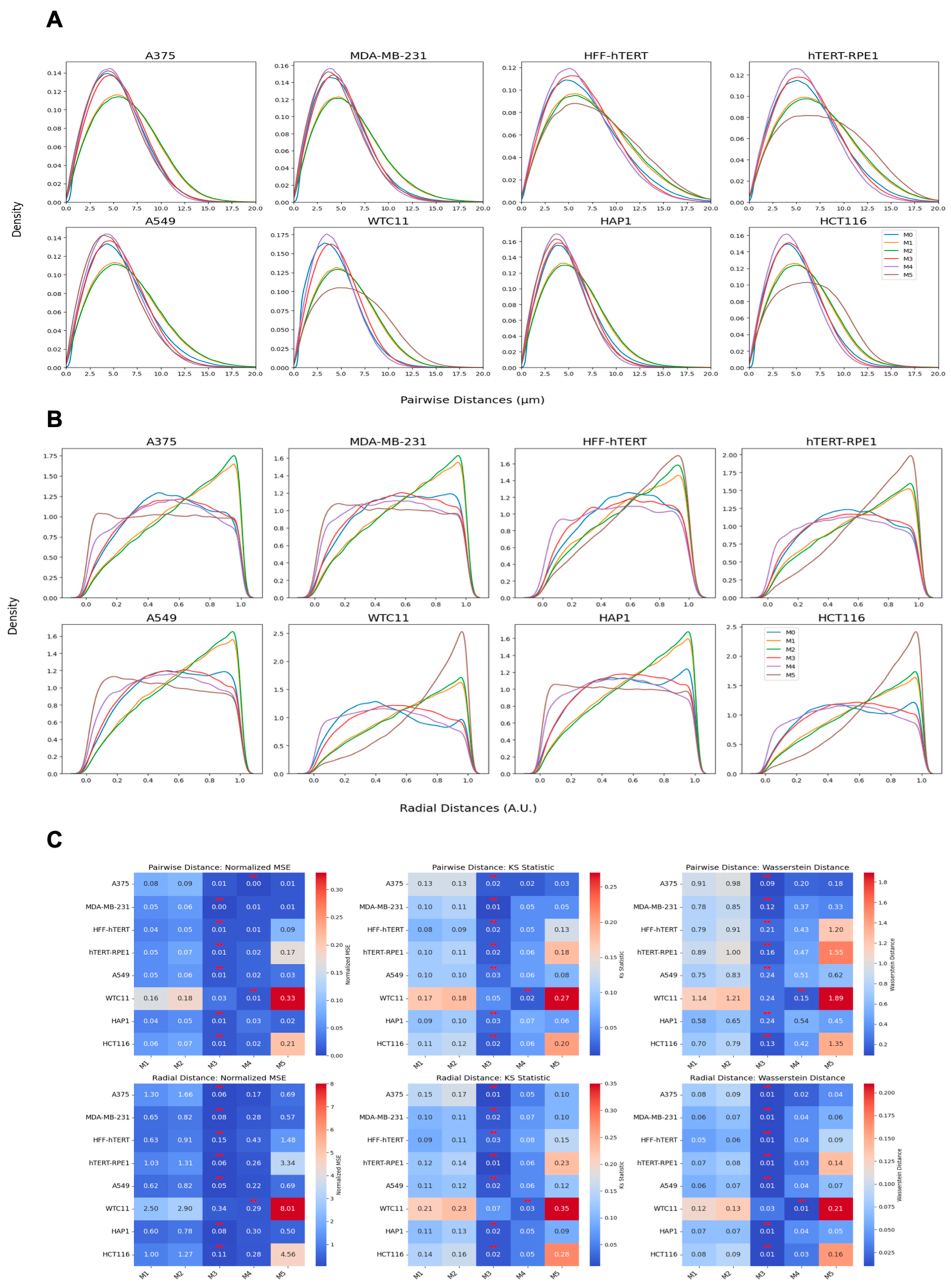
We employed three metrics—the Wasserstein distance, the normalized mean squared error (MSE), and the Kolmogorov–Smirnov (KS) statistic—to compare the similarity between real and synthetic data distributions. These metrics were calculated for multiple methods and across two conditions: scrambled (control) and siNCAPH2 (treated) and for the eight cell line distributions. These metrics assess the level of agreement between two probability density functions (PDFs), enabling us to quantify the fidelity of synthetic data in capturing the characteristics of experimental data.
The Wasserstein distance (WD), also known as the Earth mover’s distance, measures the minimum “cost” of transforming one distribution into another. It is defined for one-dimensional distributions as follows:
where
and
are the cumulative distribution functions (CDFs) of the two datasets. Lower Wasserstein distances indicate greater similarity.
The normalized mean squared error (MSE) quantifies the discrepancy between the real and synthetic PDFs by normalizing the MSE by the variance of the real PDF. It is as follows:
A lower normalized MSE indicates a higher similarity, with values close to zero reflecting strong agreement.
Finally, the Kolmogorov–Smirnov (KS) statistic compares the maximum difference between the CDFs of two distributions as follows:
where
and
are the cumulative distribution functions (CDFs) of the two datasets. Smaller KS values indicate closer alignment between the distributions. Together, these metrics provide a comprehensive assessment of the similarity between real and synthetic data across various methods and experimental conditions.
4. Discussion
Understanding the spatial organization of cellular structures is a fundamental question in modern cell biology. Centromeres are prominent cellular features of each chromosome, and the elucidation of their distribution in the human cell nucleus is crucial for deciphering chromosomal behavior and nuclear architecture in both normal and diseased cells. To this end, we generated a systematic framework and specific measurement tools to quantitatively assess the cellular distribution of human centromeres. These new methods will be useful in the investigation of the mechanisms involved in establishing and maintaining centromere localization patterns and their functional implications.
We used this framework to assess multiple centromere spatial distribution types, clustering metrics, and spot generation models. By simulating diverse spatial patterns—including uniform distributions, single Gaussian clusters, multi-modal Gaussian mixtures, and perturbed adjacent spots—these models allow precise comparison and identification of the metrics most suited for specific biological questions. This benchmarking process ensures that the chosen metric aligns with the spatial characteristics of interest, enhancing the rigor and relevance of quantitative assessments.
Based on our analysis, Ripley’s K function emerged as the most sensitive and versatile metric for the measurement of global centromere clustering, with minimal variation across different numbers of detected spots. This property makes it a suitable metric for studying changes in centromere clustering upon experimental perturbation, for example, after the elimination of the condensin protein NCAPH2. Both the sensitivity and robustness of this metric are mainly due to the fact that it is calculated using density-normalized CDFs of all spot distances, irrespective of local features of their distribution. Although the dispersion index is also using the first- and second-order statistics of the same distribution, the compression of the distribution into only two parameters, reduces its effectiveness in separating various spot distributions compared with Ripley’s K function. One limitation of Ripley’s K function may be its inability to detect local clusters dispersed within the nucleus because it is calculated using the overall distance distribution of spots.
To better understand the nature of cellular centromere distribution, we used computational modeling to replicate the localization and clustering behavior of centromeres. Specifically, we applied Bayesian estimation methods to fit a radially shifted Gaussian distribution using three different approaches. The first approach directly fitted the cumulative distribution function (CDF) of pairwise distances between centromeres. The second approach used spot coordinates (approximately 46 spots per cell), while the third relied on pairwise distances (around 1035 values per cell). Although all these models are based on radial Gaussian distributions, the third approach, with its larger number of input values, requires a higher sample size to achieve robust and accurate parameter fitting. This highlights the trade-off between input complexity and the need for larger datasets when modeling spatial distributions.
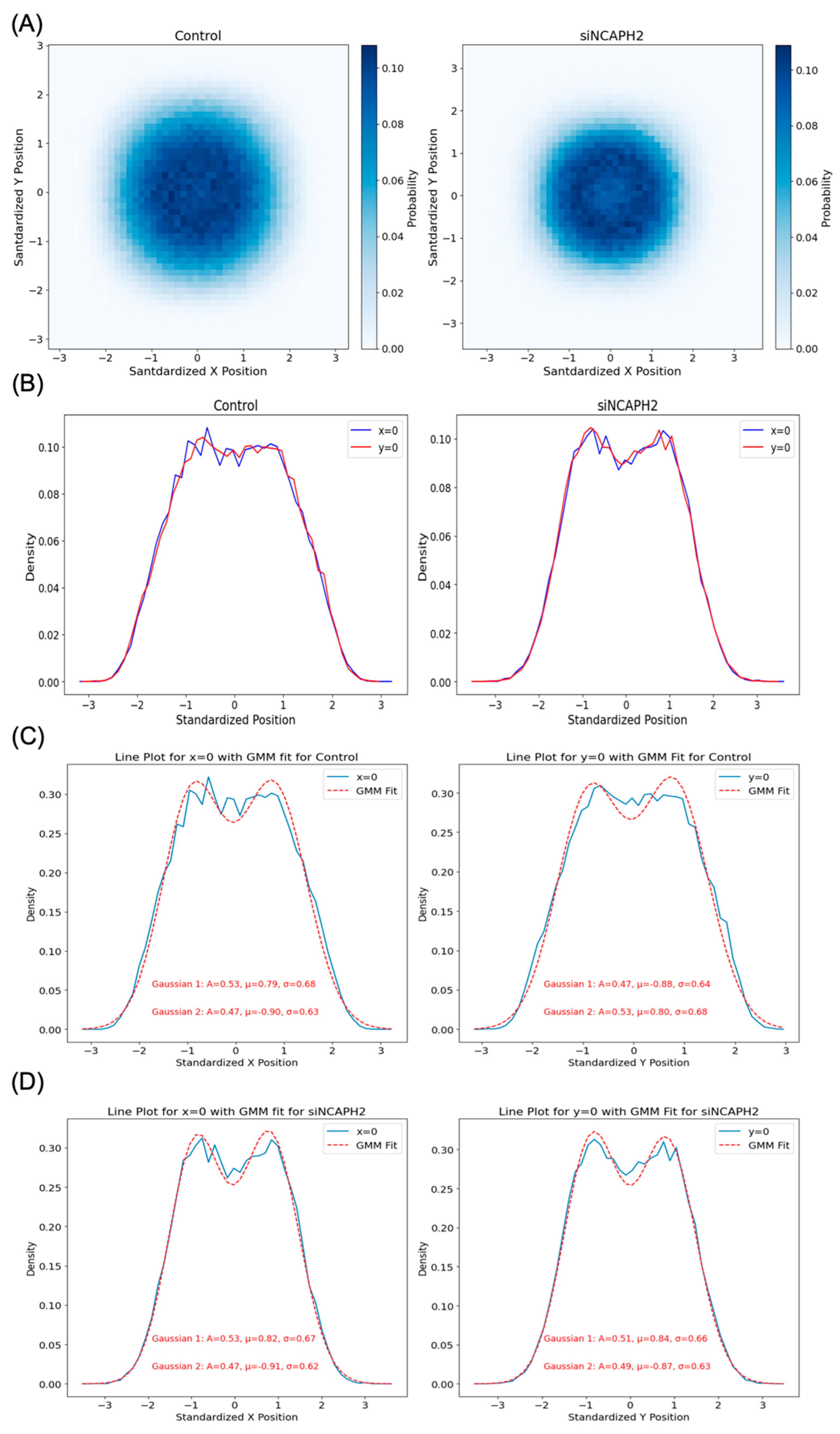
One of our findings, based on mapping the location of several thousand centromeres by imaging, is that centromeres are distributed in a doughnut-like distribution within the cell nucleus. This pattern was evident when mapping individual centromeres across cell populations and was reproduced in our modeling approaches. Despite the noted discrepancies in fitting radial distance distributions due to differences in calculation methods and spot localization variability, our models effectively captured the essential centromere-to-centromere distance patterns critical for biological interpretation. This finding aligns well with previous studies on the radial organization of nuclear structures, such as the observation of a lower density of chromatin and chromosomes in the nuclear interior [
2,
11]. These studies highlight the influence of biological constraints, such as gene density and chromatin architecture, as the key drivers of spatial nuclear organization.
The analytical tools we generated have practical applications. These analysis methods can be used to quantitatively describe changes in centromere distribution in response to a specific experimental perturbation, for example, loss of the cohesin component NCAPH2 as shown here, or during physiological or pathological processes such as differentiation, development, and in disease such as cancer. We also anticipate that the methods are applicable to analysis of tissue samples, provided nuclei can be segmented accurately. Probably more importantly, our analysis approaches will now enable the execution of large-scale functional genomic screen, such as CRISPR screens, which will allow the identification of entirely novel modulators of centromere localization in an unbiased fashion.
The combination of advanced imaging techniques with robust computational analysis, as demonstrated here, is not limited to centromeres, but can also be extended to study the spatial organization of other cellular structures. For instance, nuclear bodies such as nucleoli, Cajal bodies, and PML nuclear bodies exhibit distinct spatial patterns influenced by chromatin interactions and nuclear architecture [
45,
46,
47]. Probabilistic and spatial modeling approaches, such as those described here, have already been applied to chromosomal territories and transcription factories, revealing the roles of gene density and transcriptional activity in nuclear organization [
2,
11,
48]. Additionally, approaches using spatial modeling or clustering metrics can be tailored to study other biological structures, such as cytoplasmic structures, for example, stress granules [
49], focal adhesions [
50] or mitochondrial networks [
51], to better understand their distribution and functional implications. By adopting and adapting these methods, the principles underlying spatial organization and its impact on cellular processes across a wide range of subcellular structures can be explored using quantitative measures.
Taken together, this study presents a framework for characterizing the cellular distribution of centromeres using a combination of spatial metrics and computational modeling. By systematically benchmarking metrics with synthetic spot generation models, we demonstrate the capability of combining data-driven analysis with computational tools to capture centromere clustering patterns with potential application to other cellular structures.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}