
TNF-Alpha Inhibitor Prevents Cigarette Smoke Extract-Induced Cell Death in Osteoarthritis-Derived Chondrocytes in Culture

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of Chondrocytes from OA Patients
2.2. Generation of CSE
2.3. Assessing Chondrocyte Viability with MTT and PI Staining
2.4. Caspase 3/7 Activity
2.5. Measurement of Transmembrane Mitochondrial Potential (ΔΨm)
2.6. Analysis of ROS
2.7. Protein Detection by Indirect Immunofluorescence
2.8. Evaluation of Genes Involved in Cell Differentiation
2.9. Statistics
3. Results
3.1. Effects of CSE on Chondrocyte Viability and Death
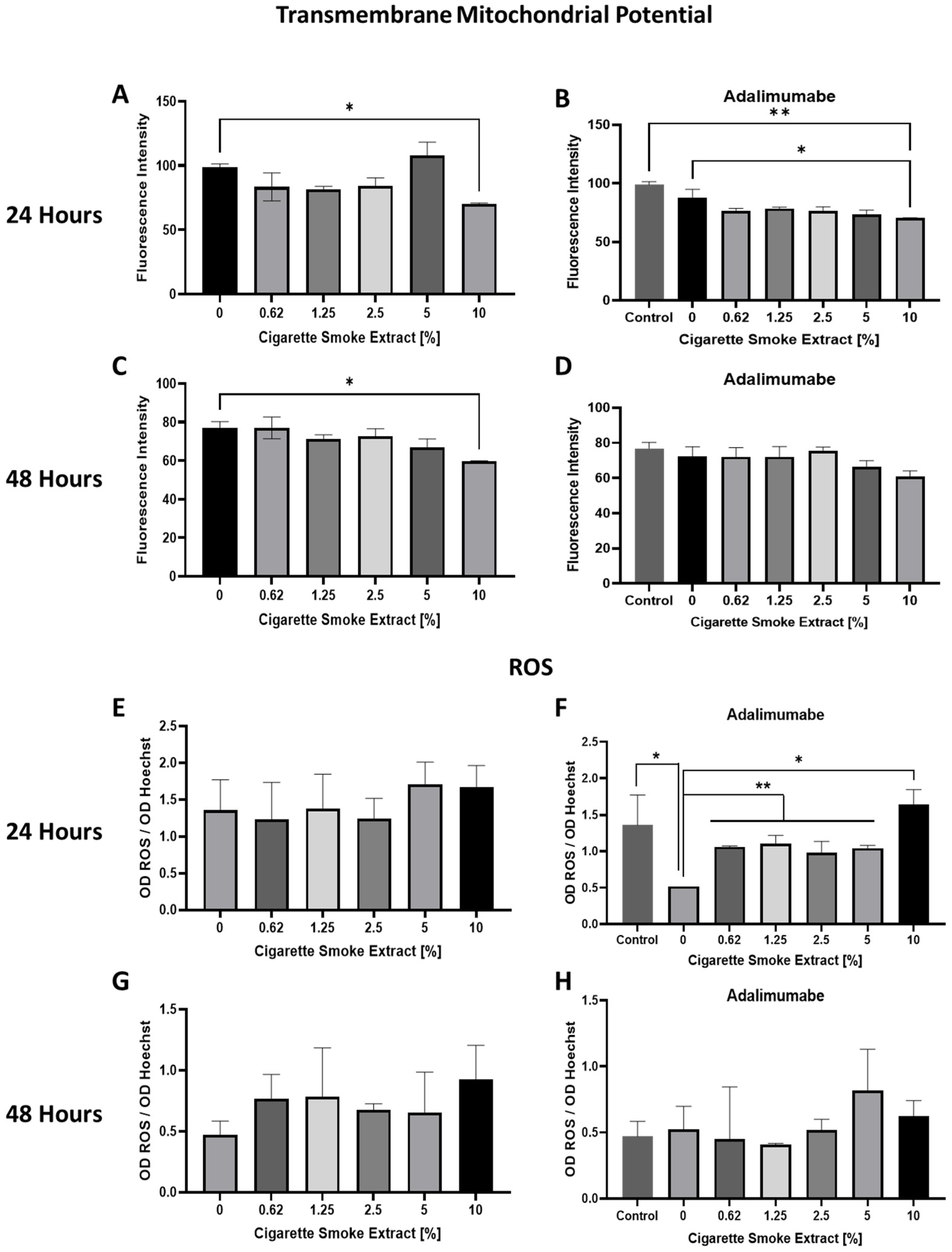
3.2. Mitochondrial Transmembrane Potential (ΔΨm) Changes and ROS Production Promoted by CSE
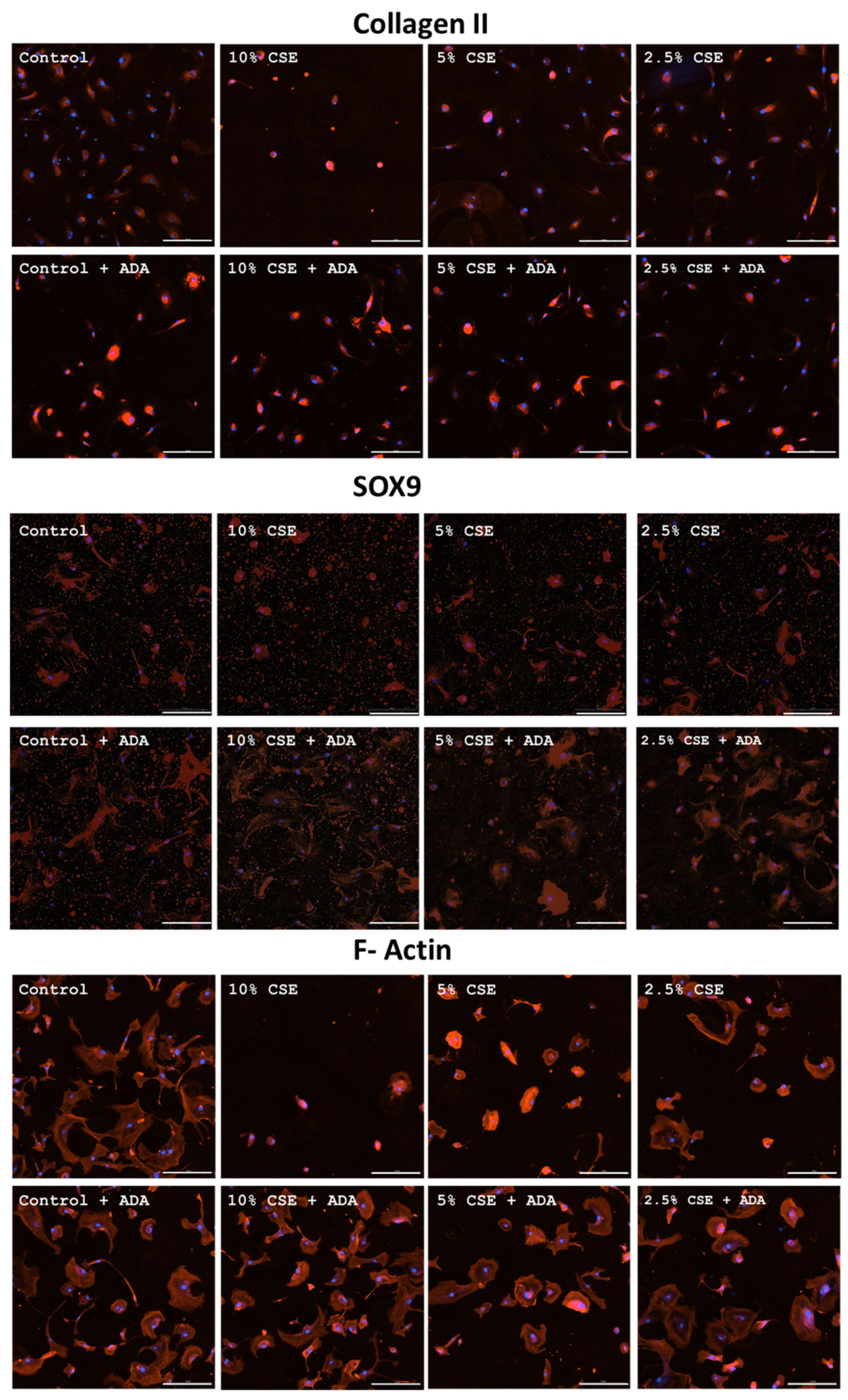
3.3. Effects of CSE on Collagen II, SOX9, and F-Actin
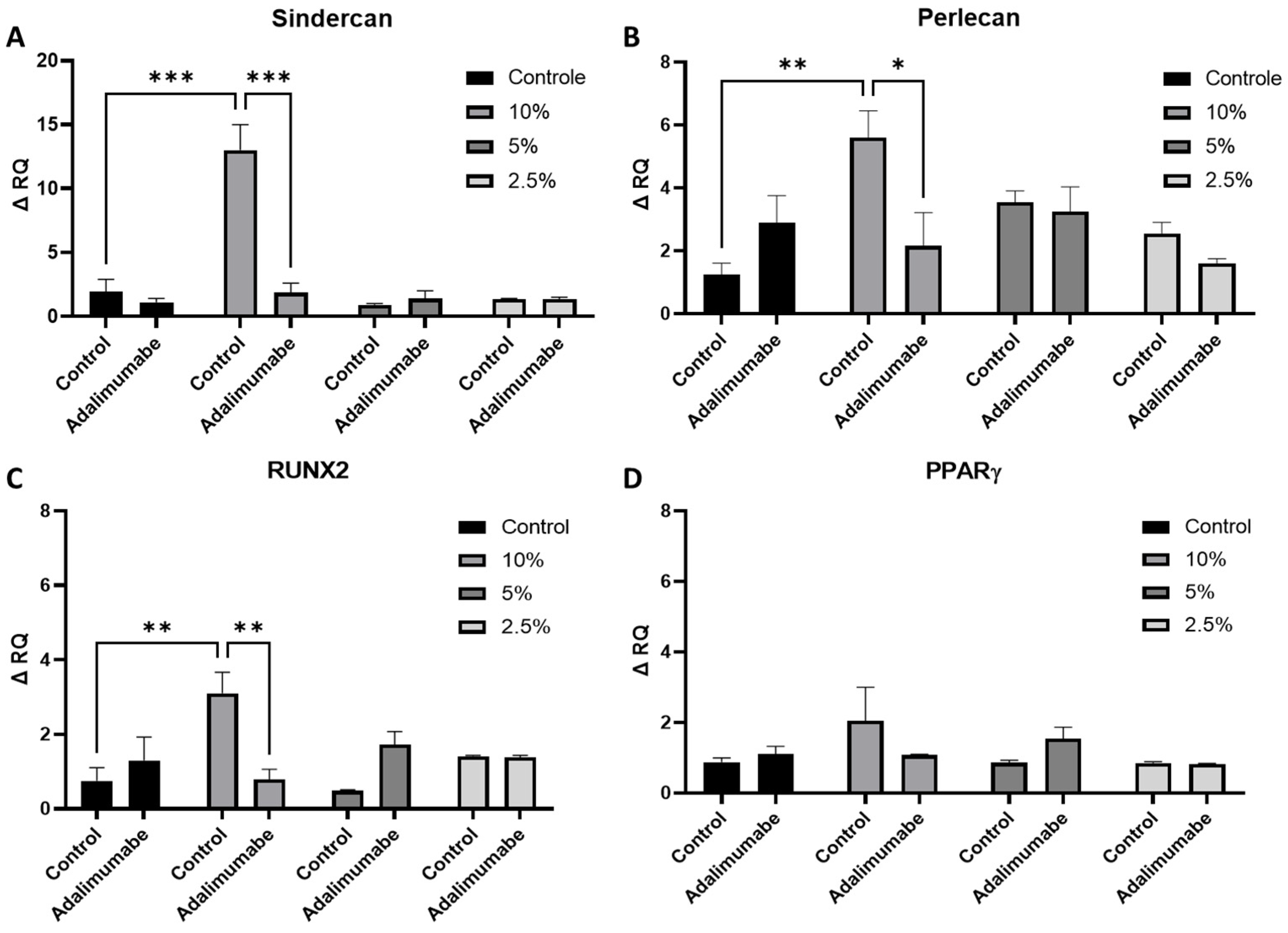
3.4. Analysis of Perlecan and Syndercan-1 Gene Expression
3.5. Analysis of RUNX2 and PPARγ Gene Expression
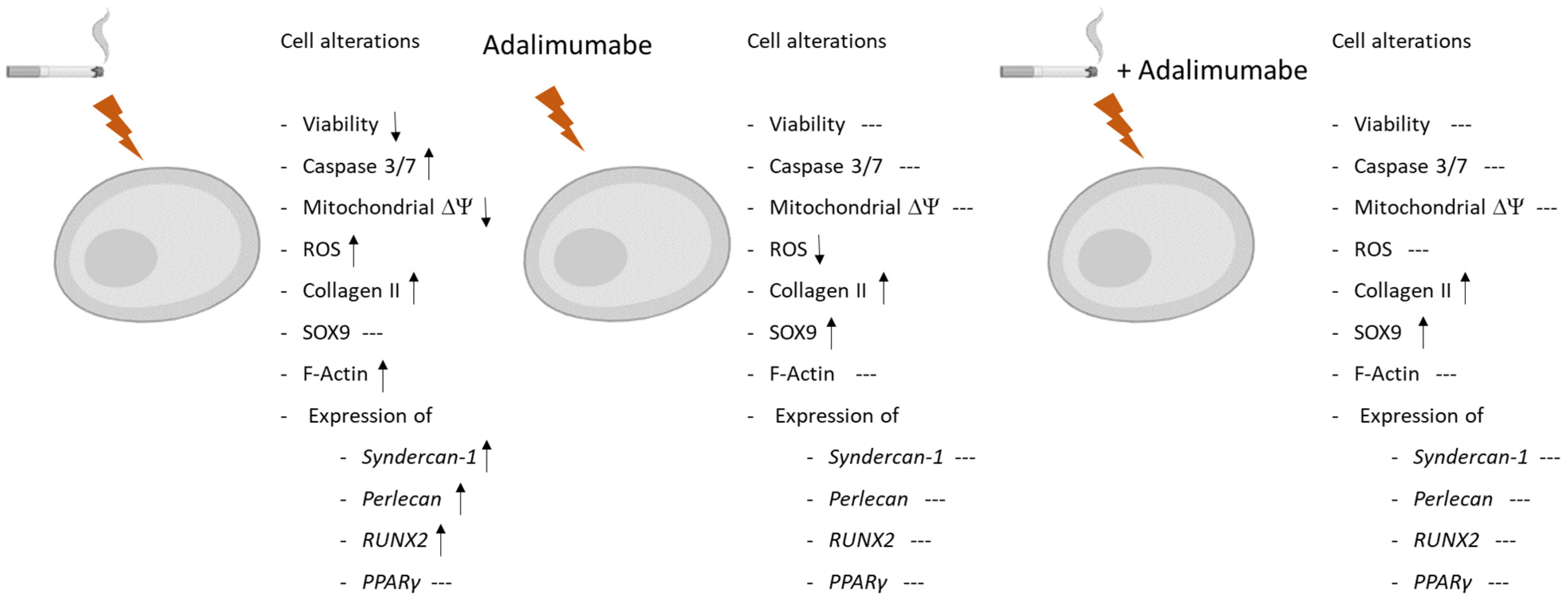
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AO | osteoarthritis |
| TNF-alpha | tumor necrosis factor-alpha |
| CSE | cigarette smoke extract |
| ROS | reactive oxygen species |
| SEM | standard error of the mean |
| FBS | fetal bovine serum |
| OD | optical density |
| MTT | 3-[4,5 dimethylthiazol-2-yl]-2,5 diphenyltetrazolium bromide |
| PI | propidium iodide |
| CT | cycle threshold |
| ΔΨm | mitochondrial transmembrane potential |
| COPD | chronic obstructive pulmonary disease |
| CS | cigarette smoke |
| HS | heparan sulfate |
| PPARγ | peroxisome proliferator-activated receptor gamma |
References
- World Health Organization. Global Report on Tobacco Epidemic 2019; WHO: Geneva, Germany, 2019; Available online: https://www.who.int (accessed on 20 January 2025).
- Linn, M.S.; Chase, D.C.; Healey, R.M.; Harwood, F.L.; Bugbee, W.D.; Amiel, D. Etanercept enhances preservation of osteochondral allograft viability. Am. J. Sports Med. 2011, 39, 1494–1499. [Google Scholar] [PubMed]
- Aizawa, T.; Kon, T.; Einhorn, T.A.; Gerstenfeld, L.C. Induction of apoptosis in chondrocytes by tumor necrosis factor-alpha. J. Orthop. Res. 2001, 19, 785–796. [Google Scholar] [PubMed]
- Jain, D.; Chaudhary, P.; Varshney, N.; Bin Razzak, K.S.; Verma, D.; Khan Zahra, T.R.; Janmeda, P.; Sharifi-Rad, J.; Daştan, S.D.; Mahmud, S.; et al. Tobacco Smoking and Liver Cancer Risk: Potential Avenues for Carcinogenesis. J. Oncol. 2021, 10, 5905357. [Google Scholar]
- Tarantino, U.; Cariati, I.; Greggi, C.; Gasbarra, E.; Belluati, A.; Ciolli, L.; Maccauro, G.; Momoli, A.; Ripanti, S.; Falez, F.; et al. Skeletal System Biology and Smoke Damage: From Basic Science to Medical Clinic. Int. J. Mol. Sci. 2021, 22, 6629. [Google Scholar] [CrossRef]
- Shen, Z.; Wang, Y.; Xing, X.; Jones, G.; Cai, G. Association of smoking with cartilage loss of knee osteoarthritis: Data from two longitudinal cohorts. BMC Musculoskelet. Disord. 2023, 24, 812. [Google Scholar] [CrossRef]
- Kohler, J.B.; da Silva, A.F.; Farias, W.A.; Sampaio, B.F.C.; Neves, M.A.S.; Lima, L.G.; Lourenço, J.D.; Moreira, A.R.; Barbosa, A.P.; Tibério, I.d.F.L.C.; et al. Smoking induces increased apoptosis in osteoblasts: Changes in bone matrix organic components. Sci. Rep. 2023, 13, 6938. [Google Scholar] [CrossRef]
- Barbosa, A.P.; Lourenço, J.D.; Junqueira, J.J.M.; de França, S.L.E.; Martins, J.S.; Junior, M.C.O.; Begalli, I.; Velosa, A.P.P.; Olivo, C.R.; Bastos, T.B.; et al. The deleterious effects of smoking in bone mineralization and fibrillar matrix composition. Life Sci. 2020, 241, 117132. [Google Scholar]
- Junqueira, J.J.M.; Lourenço, J.D.; da Silva, K.R.; Jorgetti, V.; Vieira, R.P.; de Araujo, A.A.; De Angelis, K.; Correia, A.T.; Alves, L.H.V.; Tibério, I.d.F.L.C.; et al. Increased bone resorption by long-term cigarette smoke exposure in animal model. Heliyon 2021, 7, e08587. [Google Scholar]
- Buckwalter, J.A.; Mankin, H.J.; Grodzinsky, A.J. Articular cartilage and osteoarthritis. Instr. Course Lect. 2005, 54, 465–480. [Google Scholar]
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet. 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Chisari, E.; Yaghmour, K.M.; Khan, W.S. The effects of TNF-alpha inhibition on cartilage: A systematic review of preclinical studies. Osteoarthr. Cartil. 2020, 28, 708–718. [Google Scholar]
- Wang, J. Efficacy and safety of adalimumab by intra-articular injection for moderate to severe knee osteoarthritis: An open-label randomized controlled trial. J. Int. Med. Res. 2018, 46, 326–334. [Google Scholar] [PubMed]
- Aitken, D.; Laslett, L.; Pan, F.; Haugen, I.K.; Otahal, P.; Bellamy, N.; Bird, P.; Jones, G. A randomised double-blind placebo-controlled crossover trial of HUMira (adalimumab) for erosive hand OsteoaRthritis—The HUMOR trial. Osteoarthr. Cartil. 2018, 26, 880–887. [Google Scholar]
- Li, Y.; Mai, Y.; Cao, P.; When, X.; Fan, T.; Wang, X.; Ruan, G.; Tang, S.; Ding, C.; Zhu, Z. Relative Efficacy and Safety of Anti-Inflammatory Biologic Agents for Osteoarthritis: A Conventional and Network Meta-Analysis. J. Clin. Med. 2022, 11, 3958. [Google Scholar] [CrossRef]
- Isyar, M.; Bilir, B.; Yilmaz, I.; Cakmak, S.; Sirin, D.Y.; Guzelant, A.Y.; Mahirogullari, M. Are biological agents toxic to human chondrocytes and osteocytes? J. Orthop. Surg. Res. 2015, 10, 118. [Google Scholar]
- Žigon-Branc, S.; Barlič, A.; Knežević, M.; Jeras, M.; Vunjak-Novakovic, G. Testing the potency of anti-TNF-α and anti-IL-1β drugs using spheroid cultures of human osteoarthritic chondrocytes and donor-matched chondrogenically differentiated mesenchymal stem cells. Biotechnol. Prog. 2018, 34, 1045–1058. [Google Scholar]
- Guzelant, A.Y.; Isyar, M.; Yilmaz, I.; Sirin, D.Y.; Cakmak, S.; Mahirogullari, M. Are chondrocytes damaged when rheumatologic inflammation is suppressed? Drug. Chem. Toxicol. 2017, 40, 13–23. [Google Scholar]
- Vassallo, R.; Tamada, K.; Lau, J.S.; Kroening, P.R.; Chen, L. Cigarette smoke extract suppresses human dendritic cell function leading to preferential induction of Th-2 priming. J. Immunol. 2005, 175, 2684–2691. [Google Scholar]
- Chen, T.; Ehnert, S.; Tendulkar, G.; Zhu, S.; Arnscheid, C.; Aspera-Werz, R.H.; Nussler, A.K. Primary human chondrocytes affected by cigarette smoke—Therapeutic challenges. Int. J. Mol. Sci. 2020, 21, 1901. [Google Scholar] [CrossRef]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar]
- Paz, J.L.; Levy, D.; Oliveira, B.A.; de Melo, T.C.; de Freitas, F.A.; Reichert, C.O.; Rodrigues, A.; Pereira, J.; Bydlowski, S.P. 7-Ketocholesterol Promotes Oxiapoptophagy in Bone Marrow Mesenchymal Stem Cell from Patients with Acute Myeloid Leukemia. Cells 2019, 8, 482. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [PubMed]
- de Freitas, F.A.; Levy, D.; Zarrouk, A.; Lizard, G.; Bydlowski, S.P. Impact of Oxysterols on Cell Death, Proliferation, and Differentiation Induction: Current Status. Cells 2021, 10, 2301. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar]
- Junqueira, J.J.M.; Lourenço, J.D.; da Silva, K.R.; Cervilha, D.A.d.B.; da Silveira, L.K.R.; Correia, A.T.; Silva, L.E.d.F.; Teodoro, W.R.; Tibério, I.d.F.L.C.; Barbosa, A.P.; et al. Decreased Bone Type I Collagen in the Early Stages of Chronic Obstructive Pulmonary Disease (COPD). COPD: J. Chronic Obstr. Pulm. Dis. 2020, 17, 575–586. [Google Scholar]
- Lourenço, J.D.; Teodoro, W.R.; Barbeiro, D.F.; Velosa, A.P.P.; Silva, L.E.F.; Kohler, J.B.; Moreira, A.R.; Aun, M.V.; da Silva, I.C.; Fernandes, F.L.A.; et al. Th17/Treg-Related Intracellular Signaling in Patients with Chronic Obstructive Pulmonary Disease: Comparison between Local and Systemic Responses. Cells 2021, 10, 1569. [Google Scholar] [CrossRef]
- Bazzano, L.A.; He, J.; Muntner, P.; Vupputuri, S.; Whelton, P.K. Relationship between cigarette smoking and novel risk factors for cardiovascular disease in the United States. Ann. Intern. Med. 2003, 138, 891–897. [Google Scholar]
- Lefebvre, V.; Dvir-Ginzberg, M. SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect. Tissue Res. 2017, 58, 2–14. [Google Scholar]
- Schofield, M.M.; Rzepski, A.T.; Richardson-Solorzano, S.; Hammerstedt, J.; Shah, S.; Mirack, C.E.; Herrick, M.; Parreno, J. Targeting F-actin stress fibers to suppress the dedifferentiated phenotype in chondrocytes. Eur. J. Cell Biol. 2024, 103, 151424. [Google Scholar]
- González-Cubero, E.; González-Fernández, M.L.; Esteban-Blanco, M.; Pérez-Castrillo, S.; Pérez-Fernández, E.; Navasa, N.; Aransay, A.M.; Anguita, J.; Villar-Suárez, V. The Therapeutic Potential of Adipose-Derived Mesenchymal Stem Cell Secretome in Osteoarthritis: A Comprehensive Study. Int. J. Mol. Sci. 2024, 25, 11287. [Google Scholar] [CrossRef] [PubMed]
- Chanalaris, A.; Clarke, H.; Guimond, S.E.; Vincent, T.L.; Turnbull, J.E.; Troeberg, L. Heparan Sulfate Proteoglycan Synthesis Is Dysregulated in Human Osteoarthritic Cartilage. Am. J. Pathol. 2019, 189, 632–647. [Google Scholar] [PubMed]
- Vincent, T.L. Targeting mechanotransduction pathways in osteoarthritis: A focus on the pericellular matrix. Curr. Opin. Pharmacol. 2013, 13, 449–454. [Google Scholar] [PubMed]
- Tesche, F.; Miosge, N. Perlecan in late stages of osteoarthritis of the human knee joint. Osteoarthr. Cartil. 2004, 12, 852–862. [Google Scholar]
- Gardiner, M.D.; Vincent, T.L.; Driscoll, C. Transcriptional analysis of micro-dissected articular cartilage in post-traumatic murine osteoarthritis. Osteoarthr. Cartil. 2015, 23, 616–628. [Google Scholar]
- Zheru, D.; Peiliang, F.; Yuli, W.; Haishan, W.; Qirong, Q.; Xiaohua, L.; Hui, Z.; Bo, W.; Qiwei, F. Association of PPARγ gene polymorphisms with osteoarthritis in a southeast Chinese population. J. Genet. 2014, 93, 719–723. [Google Scholar]
- Yuan, H.; Yi, N.; Li, D.; Xu, C.; Yin, G.R.; Zhuang, C.; Wang, Y.J.; Ni, S. PPARγ regulates osteoarthritis chondrocytes apoptosis through caspase-3 dependent mitochondrial pathway. Sci. Rep. 2024, 14, 11237. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levy, D.; Calllera, A.F.; Moreira, A.R.; Tibério, I.d.F.L.C.; Giglio, P.N.; Demange, M.K.; Bydlowski, S.P.; Lopes, F.D.T.Q.D.S. TNF-Alpha Inhibitor Prevents Cigarette Smoke Extract-Induced Cell Death in Osteoarthritis-Derived Chondrocytes in Culture. Cells 2025, 14, 489. https://doi.org/10.3390/cells14070489
Levy D, Calllera AF, Moreira AR, Tibério IdFLC, Giglio PN, Demange MK, Bydlowski SP, Lopes FDTQDS. TNF-Alpha Inhibitor Prevents Cigarette Smoke Extract-Induced Cell Death in Osteoarthritis-Derived Chondrocytes in Culture. Cells. 2025; 14(7):489. https://doi.org/10.3390/cells14070489
Chicago/Turabian StyleLevy, Débora, Alexandra Fernandes Calllera, Alyne Riani Moreira, Iolanda de Fátima Lopes Calvo Tibério, Pedro Nogueira Giglio, Marco Kawamura Demange, Sergio Paulo Bydlowski, and Fernanda Degobbi Tenorio Quirino Dos Santos Lopes. 2025. "TNF-Alpha Inhibitor Prevents Cigarette Smoke Extract-Induced Cell Death in Osteoarthritis-Derived Chondrocytes in Culture" Cells 14, no. 7: 489. https://doi.org/10.3390/cells14070489
APA StyleLevy, D., Calllera, A. F., Moreira, A. R., Tibério, I. d. F. L. C., Giglio, P. N., Demange, M. K., Bydlowski, S. P., & Lopes, F. D. T. Q. D. S. (2025). TNF-Alpha Inhibitor Prevents Cigarette Smoke Extract-Induced Cell Death in Osteoarthritis-Derived Chondrocytes in Culture. Cells, 14(7), 489. https://doi.org/10.3390/cells14070489